

TEXT
The flow of genetic information from the genome through RNA and finally into protein has numerous regulatory steps. Recent studies have shed light upon the complexity of the human genome, especially in cancer, where genomic rearrangements, mutation, amplifications, and deletions can deregulate the transcription rates and/or RNA stability of specific genes (1, 2). This often leads to the overexpression of mRNAs, which, when translated into protein, can deregulate a multitude of cellular processes, ultimately leading to tumorigenesis. In a simplistic view of translation's impact on the cell, it is possible for mass action (i.e., an increase in mRNA number) to drive an increase in corresponding protein levels. This is generally controlled at the level of translation initiation. However, several studies have shown that the transcriptional landscape does not necessarily reflect the protein levels of those mRNAs. This indicates that there is an additional level of posttranscriptional control that selects which mRNAs are allowed access to the translational machinery of the cell (3). One mechanism of posttranscriptional regulation of mRNA translation that has gained considerable attention in recent years is microRNAs (miRNAs) (4). Generally, these small, 22-nucleotide noncoding RNAs target specific sequences in the 3′ untranslated regions (3′UTR) of mRNAs to inhibit their translation. Given the ever-expanding number of identified miRNAs in mammals, a complex network of selective translation might be achieved for each cellular mRNA. However, miRNAs need not be the only level of mRNA translational regulation. Indeed, several mRNAs rely on specific RNA binding proteins as a mechanism for altering their translation efficiency.
The study presented by Kawagishi et al. (5) in this issue characterizes the function of one such RNA binging protein, HuR (Fig. 1). A ubiquitously expressed RNA binding protein, HuR has been shown to play a significant role in all facets of RNA biology, including splicing, stability, localization, and translation (6). The canonical role of HuR is to bind to the 3′UTR of mRNAs that contain AU-rich elements (AREs). The functional outcome of the HuR-RNA interaction appears to be context specific. In most cases, HuR stabilizes the target mRNA. However, HuR has also been shown to regulate the binding of miRNAs to their targets, both promoting and inhibiting miRNA binding. This argues that HuR is part of a larger functional complex whose assembly is directed by the HuR targeting and surrounding mRNA sequence.
Fig 1.
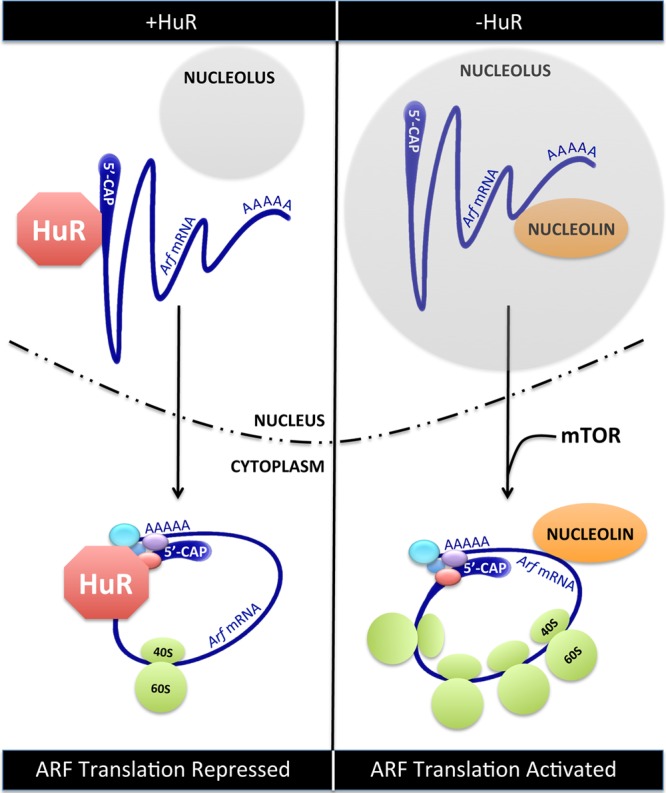
Translation of existing Arf mRNAs. (Left) In young cells or when the level of HuR protein is elevated, HuR binds to the 5′UTR of Arf mRNAs in the nucleus. HuR-Arf mRNA complexes are exported to the cytoplasm but are translationally repressed by HuR. (Right) In aging cells or when HuR is diminished, nucleolin binds to the 3′UTR of Arf mRNAs in the nucleolus. Nucleolin-Arf mRNA complexes are exported to the cytoplasm, where they are actively translated in response to appropriate signals from mTOR.
Biologically, loss of HuR expression is associated with the senescence of human diploid fibroblasts (7). HuR binds to and destabilizes the Ink4a mRNA in young fibroblasts, but as the fibroblasts age, HuR levels decrease, thus allowing an accumulation of Ink4a mRNA. Increased translation of Ink4a mRNA into protein ensues, leading to the inhibition of retinoblastoma (Rb) phosphorylation and ultimately to cell cycle arrest. Interestingly, destabilization of the Ink4a mRNA by HuR in young human diploid fibroblasts involves miRNA-independent targeting of the RISC complex to Ink4a mRNA. HuR's regulation of cellular senescence is not a cell type-specific event, as HuR levels fall in numerous types of aging human tissues. Finally, HuR is also required for embryonic development, as HuR-null mice die by embryonic day 19.5 due to splenic and skeletal defects (8).
In their study, Kawagishi and colleagues (5) examine the effects of HuR loss on senescence in mouse embryo fibroblasts (MEFs). Senescence in MEFs can be achieved through induction of ARF as cells are passaged in culture (9). This study demonstrates that aging MEFs express reduced levels of HuR protein, as previously shown in human diploid fibroblasts. Knockdown of HuR accelerates cellular senescence in wild-type MEFs by increasing ARF protein levels. Induction of ARF in this setting is achieved through enhanced translation of existing Arf mRNAs; ARF transcription, mRNA stability, and protein stability are not altered upon HuR loss. The ARF-p53 checkpoint resides downstream of HuR, as HuR knockdown does not induce cellular senescence of Arf−/− or p53−/− MEFs. Importantly, HuR does not affect Ink4a mRNA or protein levels, demonstrating a clear difference between ARF and INK4a regulation.
Mechanistically, HuR binds to the 5′UTR of Arf mRNA, which prevents its association with actively translating ribosomes. Interestingly, the 5′UTR of Arf mRNA does not contain an ARE sequence, suggesting that the binding of HuR might be indirect or through an unknown HuR binding sequence. Because the localization of Arf mRNA is not altered upon HuR knockdown, the decrease in Arf mRNA translation may be due to HuR-dependent steric hindrance, thereby preventing translation initiation. One possible mechanism of HuR-mediated inhibition of Arf mRNA translation shown is the prevention of nucleolin binding to the 3′UTR of Arf mRNA. Specifically, in the absence of HuR, Arf mRNA is bound to nucleolin, and this correlates with an increase in ARF protein levels. The nucleolin-Arf mRNA complex is predominantly localized to nucleoli, which also correlates with increased Arf mRNA translation. It has previously been shown that nucleolin acts as a bridge between mRNAs and the ribosome, facilitating their interaction in the nucleolus prior to export to the cytoplasm (10). This may provide an efficient mechanism for enhancing the translational efficiency of already-bound ribosome-mRNA complexes. Kawagishi and colleagues demonstrate that a selective deletion of HuR in the adipose tissue of mice results in increased ARF protein expression and slightly increases plasma glucose levels in aged animals, indicating that connections between HuR and ARF protein levels are also manifest in vivo.
The concerted effort of numerous proteins, including HuR, nucleolin, and mTOR (11), to regulate Arf mRNA translation warrants further investigation into its relevance to human cancer (Fig. 1). A general examination of HuR amplification in the cBio Cancer Genomics Portal database of human cancers shows low-grade glioma, cervical, uterine, and ovarian cancer to have an approximately 5% amplification rate (12). In this setting, elevated levels of HuR should decrease ARF protein levels, allowing for deregulation of downstream growth and proliferative pathways. It will be exciting to further examine the role of HuR in tumorigenesis and its potential interplay with the mTOR pathway in determining the overall level of the ARF tumor suppressor.
Footnotes
Published ahead of print 18 March 2013
The views expressed in this Commentary do not necessarily reflect the views of the journal or of ASM.
REFERENCES
- 1. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER. 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486:353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network 2012. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489:519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gebauer F, Hentze MW. 2004. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 5:827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Calin GA, Croce CM. 2006. MicroRNA signatures in human cancers. Nat. Rev. Cancer 6:857–866 [DOI] [PubMed] [Google Scholar]
- 5. Kawagishi H, Hashimoto H, Nakamura H, Tsugawa T, Watanabe A, Kontoyiannis DL, Sugimoto M. 2013. HuR maintains a replicative life span by repressing the ARF tumor suppressor. Mol. Cell. Biol. 33:1886–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brennan CM, Steitz JA. 2001. HuR and mRNA stability. Cell Mol. Life Sci. 58:266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srikantan S, Gorospe M. 2012. HuR function in disease. Front. Biosci. 17:189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, Harokopos V, Aidinis V, Hemberger M, Kontoyiannis DL. 2009. The RNA-binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol. Cell. Biol. 29:2762–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sherr CJ. 2006. Divorcing ARF and p53: an unsettled case. Nat. Rev. Cancer 6:663–673 [DOI] [PubMed] [Google Scholar]
- 10. Abdelmohsen K, Gorospe M. 2012. RNA-binding protein nucleolin in disease. RNA Biol. 9:799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miceli AP, Saporita AJ, Weber JD. 2012. Hypergrowth mTORC1 signals translationally activate the ARF tumor suppressor checkpoint. Mol. Cell. Biol. 32:348–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]