

Abstract
Helicobacter pylori establishes lifelong infections of the gastric mucosa, a niche considered hostile to most microbes. While responses to gastric acidity and local inflammation are understood, little is known as to how they are integrated into homeostatic control of cell division and growth-stage gene expression. Here we investigate the essential orphan response regulator HP1043, a member of the OmpR/PhoB subfamily of transcriptional regulators that is unique to the Epsilonproteobacteria and that lacks phosphorylation domains. To test the hypothesis that conformational changes in the homodimer might lead to defects in gene expression, we sought mutations that might alter DNA-binding efficiency. Two introduced mutations (C215S, C221S) C terminal to the DNA-binding domain of HP1043 (HP1043CC11) resulted in a 2-fold higher affinity for its own promoter by footprinting. Modeling studies with the crystal structure of HP1043 suggested that C215S might affect the helix-turn-helix domain. Genomic replacement of the hp1043 allele with the hp1043CC11 mutant allele resulted in a 2-fold decrease in protein levels, despite a dramatic increase in mRNA. The mutations did not affect in vitro growth rates or colonization efficiency in a mouse model. Proteomic profiling (CC11 mutant strain versus wild type) identified many expression differences, and quantitative PCR further revealed that 11 out of 12 examined genes had lost growth-stage regulation and that 6 of the genes contained HP1043 binding consensus sequences within the promoter regions (fur, cagA, cag23, flhA, flip, and napA). Our studies show that mutations that affect DNA-binding affinity can be used to identify new members of the HP1043 regulon.
INTRODUCTION
Gastric species of Helicobacter establish lifelong infections of the gastric mucosa in a wide range of mammals, including over 3 billion humans. Aside from the sequelae of chronic infections (chronic gastritis, peptic and duodenal ulcers, mucosa-associated lymphoid tissue lymphoma, and gastric cancer) (1, 2), these organisms resemble normal flora (3, 4). In the absence of much competition from other microbes, competition within the species, through the accumulation of many adaptive mutations in response to an ever-changing gastric environment (often beginning in childhood), is responsible for the remarkable genetic diversity (5). While it has been suggested that genetic diversity might limit fitness for colonization of new hosts, many of the genes required for persistence are also required for new colonization, including those associated with motility, chemotaxis, and acid acclimation (urease) systems (6, 7). In addition, in organisms with small genomes, there are fewer redundant systems, and a greater proportion of genes associated with central metabolism is typically essential (8, 9). Helicobacter pylori also presents a unique opportunity to study the minimum regulatory complement in a small-genome organism residing in a niche with little competition.
While enteric flora such as Escherichia coli must compete fiercely for scarce nutrients, necessitating a plethora of regulatory systems to maximize metabolic efficiency, the gastric helicobacters have relatively few (∼15) transcriptional regulatory factors, more than 80 small noncoding RNAs, and 3 sigma factors (σ80, σ28, and σ54) (10–12). While most of the transcriptional regulatory factors limit cytoplasmic accumulations of metals (iron, copper, and nickel), which tend to concentrate in acid or other environmental insults (HrcA and HspR), they are nonessential in vitro, and their absence manifests as minor colonization defects in animal models of infection (13–15). Of the essential response regulators, ArsR, part of a two-component regulatory system (ArsRS), controls responses to acid, while orphan response regulator HP1043 regulates itself and an acid-sensing chemoreceptor, TlpB (16–18). Both ArsR and HP1043 are members of the OmpR/PhoB family of transcriptional regulators, and while ArsR can be activated by phosphorylation, HP1043 is not phosphorylated (19).
HP1043 was initially described to be a growth-stage regulator (17), but genetic attempts to control expression, including controllable promoters, multiple copies of the gene, or even antisense RNA interference, while affecting gene expression did not change protein levels (17, 20, 21). HP1043 is unique to the epsilonproteobacteria, and the hp1043 allele can be functionally replaced by an orthologue from Campylobacter jejuni (20). Both crystal structure and chemical cross-linking studies indicate that HP1043 forms a dimer (17, 19), but on the basis of footprinting studies, DNA binding appears to be weak compared to that for other H. pylori transcription factors (22, 23). It is therefore possible that posttranslational modifications of HP1043 that are specific to H. pylori might affect DNA-binding efficiency to control gene expression in the absence of phosphorylation.
The inability to manipulate the expression of HP1043 in vivo has proven a major obstacle to elucidation of its biological function. We considered the possibility that subtle mutations to HP1043 that affect DNA-binding efficiency or posttranslational regulatory activity might be permissive and enable elucidation of regulatory functions. We noticed from the crystal structure of HP1043 that just downstream of the C-terminal DNA-binding domains reside two cysteine residues, C215 and C221 (19), one of which (C215) is conserved in all species of Helicobacter and Campylobacter. Cysteine residues are often associated with redox activities or participate in disulfide bond formation with other proteins. We changed the two cysteine residues to serine and replaced the wild-type gene with the mutant allele, designated CC11. Here we show that the recombinant HP1043CC11 protein more efficiently bound its promoter (footprinting) than HP1043. In vivo, the CC11 mutation led to a 40-fold increase in hp1043 mRNA levels and a 2-fold decrease in HP1043CC11 protein levels compared to those for wild-type HP1043. Comparative proteomics and transcriptional profiling identified new genes regulated by HP1043, some of which no longer exhibited growth-phase regulation and many of which contained consensus HP1043 binding sequences within their promoter regions. While gene expression changes did not affect in vitro growth or colonization efficiency in a mouse model, our studies show that mutations to HP1043 can be exploited to identify new members of its regulon.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains and plasmids used in this work are listed in Table 1. All plasmid constructs were verified by sequencing at GeneWIZ. E. coli was routinely grown at 37°C in LB medium (20) containing the appropriate supplements at the following concentrations: ampicillin at 150 μg/ml for plasmids, kanamycin at 20 μg/ml, and chloramphenicol (Cm) at 20 μg/ml. H. pylori strains were grown under humid microaerobic conditions at 37°C on brucella-based medium supplemented with 7.5% newborn calf serum (Gibco Laboratories) and antimicrobials as previously described (24).
Table 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristics | Source |
|---|---|---|
| Strains | ||
| Helicobacter pylori | ||
| G27 | Clinical isolate, wild type | Laboratory collection |
| SS1 | Wild type | Laboratory collection |
| G27 CC11 | hp1043 C215S C221S (CC11) mutant of G27, Cmr | This work |
| G27 HP1043 WT Cmr | Control strain for G27 CC11, G27 Cmr | This work |
| SS1 CC11 | hp1043 C215S C221S (CC11) mutant of SS1, Cmr | This work |
| Escherichia coli | ||
| DH5α | endA1 hsdR17 glnV44 thi-1 recA1 gyrA relA1 Δ(lac-argF)U169 deoR ϕ80dlacZΔM15 | Invitrogen |
| BL21CodonPlus(DE3)-RIPL | ompT hsdS dcm+ Tetr gal (DE3) endA hte [argU ileY proL leuW Cmr Strep/Specr] | |
| Stratagene | ||
| Plasmids | ||
| pET15b | Apr T7 promoter | Novagen |
| pET15b-hp1043 | Apr hp1043, T7 promoter control | This work |
| pET15b-hp1043CC11 | Apr hp1043 CC11, T7 promoter control | This work |
| pCR2.1-TOPO | Apr Kmr | Invitrogen |
| pTOPO-hp1043pr | Derivative of pCR2.1-TOPO containing a 430-bp PCR fragment resulting from amplification of the hp1043 promoter region with oligonucleotides 104-PF and 1043-PF from the G27 genome, Apr Kmr | This work |
Overexpression and purification of HP1043 and HP1043CC11.
The hp1043 gene and C215S C221S (CC11) double mutant were amplified from strain G27 genomic DNA by PCR using primer pairs HP1043FNdeI/HP1043RBamHI for wild-type (WT) HP1043 and HP1043FNdeI/HP1043SerRBamHI for CC11 (Table 2). Following restriction digestion, fragments were individually cloned in frame with a 6× N-terminal His tag in pET15b and introduced into E. coli BL21CodonPlus(DE3)-RIPL by transformation. Cells were then grown in LB broth supplemented with ampicillin (500 μg/ml) at 37°C, and 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added in early log phase. The temperature was lowered to 20°C and the cells were allowed to grow overnight. The N-terminal His6-tagged HP1043 or HP1043CC11 proteins were purified from cell extracts by Ni2+ affinity chromatography as described in the Novagen standard protocol (Novagen). All buffers contained 10% glycerol and 1 mM dithiothreitol (DTT). Yields of HP1043 and HP1043CC11 proteins were in the range of 2 to 10 mg per liter of culture, and a purity of 90% was estimated by Coomassie brilliant blue staining following sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
Table 2.
Oligonucleotides used in the study
| Oligonucleotide | Sequencea | Positionb |
|---|---|---|
| HP1043FNdeI | gaagtcccatatgATGCGCGTTCTACTGATTG | 1105416 |
| HP1043RBamHI | cagggatccTTACTCTTCACACGCCGGTT | 1104745 |
| HP1043SerRBamHI | cagggatccTTACTCTTCACTCGCCGGTTTTGGGTAGCTAAAACGATA | 1104745 |
| BR | AGGAGTCATACACCATGCGCGT | 1104714 |
| P1-HP1043F | TTCTGTTTTGGGTGGAGAAATTG | 1105390 |
| P2-HP1043R-Cat | atccacttttcaatctatatCACGGAATTTACTCTTCACTCGCCGG | 1104739 |
| P3-HP1044F-Cat | cccagtttgtcgcactgaTAAACTCTTTTCAGTTTTTGTTAGTT | 1104247 |
| P4-HP1042R | TCTGAATTGAATCGTTTCATGTTTG | 1103705 |
| C1c | GATATAGATTGAAAAGTGGAT | |
| C2c | TTATCAGTGCGACAAACTGGG | |
| 1043-PRd (BamHI) | attcagggatccCTCTCTGTTACATCAGCC | 1105322 |
| 1043-PFd (PstI) | agcttctgcagAGCTTAATCGTAATCAAGCGG | 1105755 |
| qPCR primers | ||
| F-HP0597 | TTAGCGGTAGAAGACACCCTC | 632578 |
| R-HP0597 | GCCATAATACCCATGCCCAAA | 632300 |
| F-HP0824 | TTAGTGGATTTTTGGGCGCCA | 876955 |
| R-HP0824 | AACTTTAGTTTGCACGCCCAC | 877177 |
| F-HP0103 | CGTATCATGCTGGTCGTGTTA | 110691 |
| R-HP0103 | TACCACCCCGTTTTTATCCAC | 110392 |
| F-HP1043RT | GAGAGTTTAGAGGATGGGGAA | 1105332 |
| R-HP1043RT | GACTAAAGCCTTAATGCTACG | 1105106 |
| F-HP1573 | TAGACACGCATTGCCATTTGG | 1654614 |
| R-HP1573 | TTCGCCTATCGCCACGCATTT | 1654355 |
| F-HP1456 | ATGAGTGTGGTAGCAGCGATG | 1526716 |
| R-HP1456 | ACCAGAAGCGTCTACCGTTCT | 1526397 |
| F-CsrA | GCTCATACTCAGCCGCAAAGT | 1514560 |
| R-CsrA | CAAGCTTTCATCTACGCACAC | 1514751 |
| F-FlhA | ATGGCAAACGAACGCTCC | 1100926 |
| R-FlhA | AGCGCTAAAATCAGTCGGCTT | 1101156 |
| RT-fur-F | GCATTCTATCCGCCAAAAGG | 1090357 |
| RT-fur-R | TCATGGTGTTCTTTAGCCGC | 1090486 |
| F-Sigma80 | AGCGCGAAAGTCCCCACT | 94715 |
| R-Sigma80 | ATCGCTTCTGCTCCACAC | 94515 |
DNase I footprinting.
To generate template plasmids, the promoter regions were amplified from H. pylori G27 chromosomal DNA by PCR using HotStar Taq DNA polymerase (Qiagen) and cloned into the pCR2.1-TOPO vector (Invitrogen) as specified by the manufacturers (Tables 1 and 2). The pTOPO derivatives containing promoter fragments were first digested with KpnI and XbaI to generate a DNA fragment containing the promoter, which was purified from a 1% agarose gel using a QIAquick gel extraction kit (Qiagen) and then subjected to dephosphorylation by using calf intestinal alkaline phosphatase (CIP; New England BioLabs). Approximately 1.8 μg of each fragment was then 5′ end labeled by [γ-32P]ATP (6,000 Ci/mmol; PerkinElmer) with T4 polynucleotide kinase (New England BioLabs). The labeled DNA fragments were separated from unincorporated [γ-32P]ATP by using a QIAquick nucleotide removal kit (Qiagen), digested with SpeI, and purified using a QIAquick PCR purification kit (Qiagen). For each footprinting experiment, 50 μl of HP1043 or HP1043CC11 protein (5 to 10 mg/ml) was dialyzed for 45 min against FTP buffer (10 mM Tris-HCl at pH 7.5, 50 mM KCl, 10 mM MgCl2, 14 mM 2-mercaptoethanol, 10% glycerol) at 4°C using a Slide-A-Lyzer minidialysis unit (Thermo Scientific Inc., Rockford, IL). The one end-labeled fragment (50,000 cpm) was incubated with specified amounts of purified WT or mutant HP1043 in 50 μl of FTP buffer. After 15 min at 25°C, DNase I digestion was started by addition of 5 μl of the reaction buffer containing 25 mM CaCl2, 25 mM MgCl2, and 80 units/ml DNase I (New England BioLabs). After 1 min at room temperature, 140 μl of stop solution (192 mM sodium acetate, 32 mM Na2-EDTA, 0.14% SDS, 64 mg of sonicated salmon sperm DNA/ml) was added. Samples were then extracted once with an equal volume of phenol-chloroform (1:1), and DNA was precipitated with 95% (vol/vol) ethanol, dried under vacuum, dissolved in loading buffer, and resolved by electrophoresis on 5% sequencing gels (17). Size markers were generated by Maxam-Gilbert sequencing reactions on the same DNA fragment. The gels were dried, and the positions of radioactive DNA fragments were detected with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA).
Generation of polyclonal antibody.
Polyclonal antisera against recombinant HP1043 was produced by immunizing mice with 50 μg of purified recombinant protein in Freund's incomplete adjuvant. Four weeks later, the mice were boosted with 50 μg of the protein in Freund's incomplete adjuvant. At week 6, the animals were terminally bled and the sera were stored at −20°C. Antibody titers were determined, and specificity was evaluated by SDS-PAGE, immunoblotting, and enzyme-linked immunosorbent assay. Immunoblots were prepared and developed as previously described (25).
Generation of H. pylori allelic replacement with the CC11 mutation.
A vector-free strategy was utilized to replace the chromosomal copy of hp1043 with the CC11 mutant sequence (9). Two pairs of primers, P1-HP1043F/P2-HP1043R-Cat and P3-HP1044F-Cat/P4-HP1042R, were used to amplify the C-terminal part of hp1042 and C-terminal part of hp1043 (∼500 bp each), which were combined by PCR with a Campylobacter coli chloramphenicol resistance gene (cat) in the middle. The resulting 1.5-kb fragment was used for natural transformation (9) of strains G27 and SS1. Transformants were selected on brucella agar medium with Cm (10 μg/ml). Replacement of the 490-bp intergenic region between the hp1042 and hp1043 genes (positions 1104738 to 1104248) with the cat cassette was confirmed by PCR with primers BR/C2. The presence of C215S C221S mutations in the chromosomal copy of the hp1043 gene was verified by sequencing of the PCR products amplified from the chromosomal DNAs of Cmr transformants.
RNA preparation and qRT-PCR.
Total H. pylori RNA was extracted from cells at optical densities at 600 nm (OD600s) of 0.9 and 2.0 by the RiboZol RNA extraction reagent (Amresco, OH). First-strand cDNA was generated using a SuperScript first-strand synthesis system for reverse transcription-PCR (RT-PCR; Invitrogen, CA). Quantitative RT-PCR (qRT-PCR) of the target genes was performed for each of the cDNA reactions in a CFX96 real-time system (Bio-Rad Laboratories, CA) with GoTaq quantitative PCR (qPCR) master mix (Promega, WI). The H. pylori 16S rRNA gene was used as the internal control for qRT-PCR (26). The reaction mixtures were incubated for 10 min at 95°C to activate the GoTaq DNA polymerase, followed by 35 amplification cycles of 95°C for 15 s, 55°C for 15 s (optimized by gradient PCR of genomic DNA), and 60°C for 30 s and then a final 10-min extension at 60°C. Following the PCR, the temperature was increased in 0.5°C increments every 10 s to generate melting curves of the PCR amplicons. All samples and RNA standards were assayed without reverse transcriptase to confirm the absence of DNA contamination. All reactions were performed in triplicate. Bio-Rad software was used to perform statistical analyses of the data acquired from the qRT-PCR. The majority of the primers used in this work have been described previously (27, 28). The rest of the primers are shown in Table 2. In this work, we used strains G27 and SS1. For convenience, the positions of the primers are given using the strain 26695 genome, as most published works refer to this genome and comparisons can readily be made.
2D gel electrophoresis.
For two-dimensional (2D) gel electrophoresis (2D PAGE), bacterial pellets from 8 ml of H. pylori culture were resuspended in a modified Celis extraction/rehydration buffer (7 M urea, 2% NP-40, 50 mM DTT, 1 mM EGTA) with protease inhibitors, vortexed, and kept on ice for 20 min. After centrifugation at 12,000 × g for 5 min, the protein concentration in the supernatant was quantified by a protein assay kit (Pierce). Ampholytes (0.2%), pH 3 to 10, and 0.0002% bromophenol blue were added to 120 μg extracted proteins in a 180-μl total volume. The sample was loaded onto 11-cm immobilized-pH-gradient (IPG) strips (pH 3 to 10) and actively rehydrated at 50 V overnight. Isoelectric focusing (IEF) was performed using a Protean IEF cell apparatus (Bio-Rad, CA). After focusing, IPG strips were incubated in equilibration buffer (6 M urea, 2% SDS, 0.05 M Tris-HCl, 2% DTT, 20% glycerol) at room temperature for 10 min. Two-dimensional gel electrophoresis was performed on 12% SDS-PAGE Criterion gels. Silver staining was used to identify the proteins in the gels. The proteins from the 2D gels were identified at the W. M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia.
Morphology.
H. pylori cultures were grown in brucella-based medium supplemented with 7.5% newborn calf serum (Gibco Laboratories). At different optical densities, cells were observed by phase-contrast microscopy with a Zeiss AX10 microscope equipped with a charge-coupled-device camera and Image-Pro software (Media Cybernetics). Images of at least 10 fields were collected and analyzed, and representative photographs are depicted.
Animal colonization.
C57BL6/J mice were challenged with 3 doses of 5 × 107 CFU of H. pylori SS1 wild-type and SS1 CC11 mutant strains in brucella broth on days 1, 3, and 5. Even though the CC11 mutant in the SS1 background did not show differences in cell shape from the wild type, bacterial plating experiments were performed to ensure the accuracy of the doses used to infect animals. An uninfected control group which received only brucella broth was included (n = 10). To facilitate colonization, 5% urea was added to the drinking water for 7 days starting on the day of the first infection (29). Stomachs were collected at weeks 3 and 6 postinfection. Half of the stomach of each mouse was used for Helicobacter pylori reisolation. Briefly, weighed gastric specimens were homogenized in phosphate-buffered saline (PBS) and plated in triplicate onto Columbia agar plates with selective antibiotics: vancomycin (10 μg/ml), trimethoprim (1 μg/ml), amphotericin B (5 μg/ml), and polymyxin B (5 μg/ml). Plates were incubated for 4 days at 37°C in microaerobic jars. Bacterial numbers are reported as the mean and standard deviation of the number of CFU/ml. Statistical differences were determined using Student's t test, with P values of <0.05 considered significant. Four animals from each group were assessed at each time point in each experiment.
Structural analysis of CC11.
The amino acid sequence of HP1043CC11 using the J99 sequence was threaded onto the crystal structure of HP1043 obtained from the J99 strain (19). The Phyre-2 server was used for alignment and secondary structure predictions (www.sbg.bio.ic.ac.uk/phyre2/html), and the PyMOL system was used to superimpose HP1043CC11 onto HP1043 using the protein with Protein Data Bank accession number 2HQR (19).
RESULTS
Mutations to residues C215 and C221 of HP1043 decrease intracellular level of HP1043.
Sequence alignment of HP1043 and its Epsilonproteobacteria homologues revealed a highly conserved DNA-binding domain located in the C terminus of the protein, with two cysteine residues located just downstream of this region (Fig. 1). We hypothesized that conservative residue C215 and variable residue C221 contribute to structural stability of the DNA-binding domain and that their replacement with serine residues might result in subtle but tolerable changes in protein function. In this regard, in silico modeling of the solved crystal structure of HP1043 first showed that the thiol of C215 is close (∼2.5 Å) to an aspartic acid residue (D160) located within the helix-turn-helix DNA-binding domain and that the serine replacement is predicted to change this distance (∼5 Å). Second, we noticed that the distance between the two C215 residues in the homodimer is ∼13 Å and that they are in the same plane (19). While this distance does not favor disulfide bonding, a conformational change similar to the changes reported for nonphosphorylated OmpR/PhoB regulators (30) might bring these subunits sufficiently close to enable disulfide bonding. To test these possibilities, a vector-free approach was utilized to replace the chromosomal copy of hp1043 with the C215S C221S mutant allele designated CC11. The strategy for allelic exchange is depicted in Fig. 2. As a control, allelic replacement of the 490-bp intergenic region between hp1042 and hp1043 that also contained the Cmr cassette (H. pylori G27 HP1043 WT Cmr) was used. As seen in Fig. 3A, the in vitro growth characteristics of the CC11 mutant strain were indistinguishable from those of the wild-type strain. However, when HP1043 protein levels were compared by Western blotting (Fig. 3B) at defined stages of growth, HP1043CC11 levels of the CC11 mutant strain were consistently 2- to 3-fold lower than those of the wild-type strain (validated by densitometry). It is also noteworthy that the HP1043 protein levels (normalized to total protein) for both the CC11 mutant and WT strains did not vary with stage of growth. The HP1043 levels of the control G27 HP1043 WT Cmr strain were also unchanged from those of the WT G27 strain (data not shown), confirming that the observed decrease in HP1043CC11 levels in the CC11 mutant is due to mutation.
Fig 1.

Site-directed mutagenesis strategy. (A) Sequence alignment of the C-terminal sequences of HP1043 homologues from Epsilonproteobacteria species using T-Coffee. Identical amino acid residues are shaded. Mutated residues are shown in red. (B) Locations of the C215S and C221S mutations on the structural model of HP1043 (19).
Fig 2.
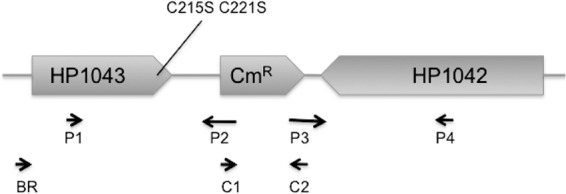
PCR-based approach for identification of allelic exchange performed to introduce the hp1043 CC11 mutation into the H. pylori chromosome. A vector-free strategy was utilized to replace the chromosomal copy of hp1043 with the CC11 mutant copy (9). Two pairs of primers, P1-HP1043F/P2-HP1043R-Cat (P1/P2) and P3-HP1044F-Cat/P4-HP1042R (P3/P4), were used to amplify the C-terminal part of hp1042 and the C-terminal part of hp1043 (∼500 bp each), which were combined by PCR with a Campylobacter coli chloramphenicol resistance gene (cat) in the middle amplified with primers C1 and C2. The resulting 1.5-kb fragment was transformed into strains G27 and SS1 as described in the text.
Fig 3.
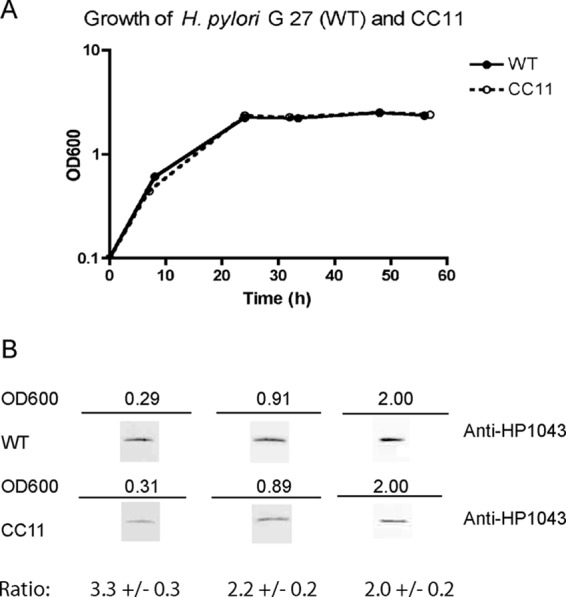
H. pylori G27 WT and CC11 growth and HP1043 protein expression. (A) Cultures of H. pylori G 27 (WT) and CC11 were grown in brucella broth. At the indicated periods of time, OD600s were taken along with samples for the immunoblot. (B) H. pylori cells were harvested, lysed, and standardized on the basis of protein concentration. Cell lysates (4 μg) were subjected to SDS-PAGE and transferred to a nitrocellulose membrane, and the membrane was probed with anti-HP1043 antiserum. Densitometry analysis was performed using ImageJ software (rsbweb.nih.gov/ij/). The experiments were repeated 3 times, and a representative result is presented. Average WT/CC11 ratios and standard deviations are presented in the last lane, showing a 2- to 3-fold decrease of the HP1043 levels in the CC11 mutant.
HP1043CC11 mutations increase DNA-binding efficiency.
To test the functional relevance of the HP1043CC11 mutant protein, we performed in vitro DNA footprinting using the hp1043 promoter (17) as a template and purified His6-HP1043 and His6-HP1043CC11 proteins. To check the possibility that His tags affected DNA binding, they were removed using thrombin protease. No effect of the His6 tags on DNA binding was found (data not presented). For consistency, the promoter fragment used in our studies was similar to that previously described (17). As seen in Fig. 4, both proteins protected an identical area (positions −17 to −47 relative to the start of transcription) described previously for wild-type HP1043 (17). We noted that ∼80% protection from DNase I digestion was achieved at concentrations of 9.6 μg for the wild-type protein and 4.8 μg for the HP1043CC11 mutant protein (Fig. 4, lanes 6 and 9, respectively). The difference in DNA-binding affinity might account for the lower level of protein observed for the CC11 mutant strain, as transcriptional factors often act as autorepressors.
Fig 4.
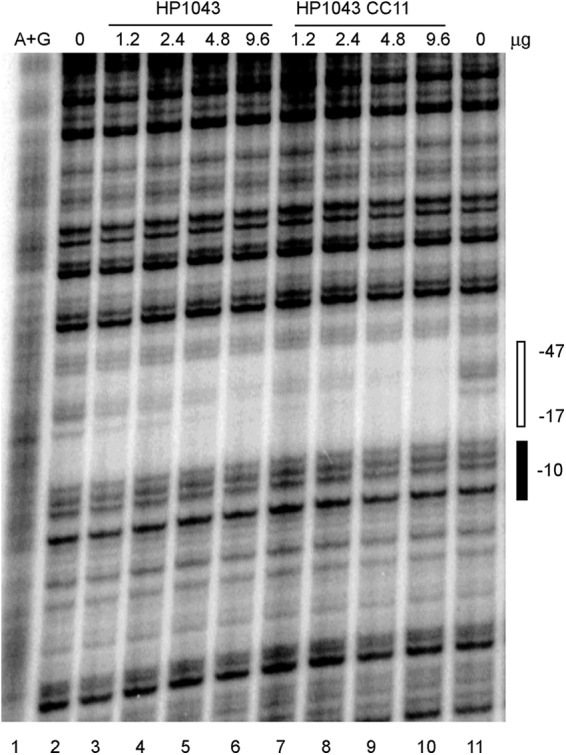
HP1043CC11 and HP1043 binding to the hp1043 promoter. The 430-bp KpnI-XbaI fragment from plasmid pTOPO-hp1043pr (Table 1) labeled at its XbaI site was used as a template. The amounts of the proteins incubated with the DNA template are indicated at the top. Products of A+G sequencing of the same DNA fragment are shown in the left lane. The −10 element and the region protected from DNase I digestion are indicated on the right. Two independent experiments yielded similar results. Representative data from a single experiment are shown.
hp1043 expression is upregulated in the CC11 mutant strain.
To confirm that the depressed protein level noted in the CC11 mutant strain resulted from increased repression of gene expression, we measured hp1043 transcript levels by qPCR. We found that the mRNA transcript levels were unexpectedly elevated (∼40-fold) in the CC11 strain compared with those in the control strain in both logarithmic and stationary phases of growth (data not presented). These results suggest that mutations influencing DNA-binding affinity by HP1043CC11 enhance gene expression without a concomitant increase in protein levels. Previous studies using gene duplication or controllable promoters have similarly noted up to a 10-fold increase in hp1043 mRNA levels (17, 20). The apparent tight control on HP1043 protein levels throughout the growth cycle suggests a more complex regulatory system that has been disrupted by the CC11 mutations to HP1043. Our studies do not rule out the possibility that the depressed HP1043CC11 mutant protein levels in CC11 might result from overcorrection by an as yet unknown compensatory translational mechanism activated in response to the dramatic increase in mRNA.
CC11 mutation affects cell division.
While routinely checking growing H. pylori cultures by phase-contrast microscopy, we noticed that the G27 CC11 strain formed elongated filaments in early stationary phase, suggesting that the CC11 mutation affects some aspect of cell division (Fig. 5). This effect on cell division might be strain specific, since the phenotype was not observed for the H. pylori SS1 CC11 mutant strain (data not shown). In H. pylori, cell shape determinants include three penicillin-binding proteins (pbp1, pbp2, and pbp3) involved in synthesis of cell wall peptidoglycan (31–33). Deregulation of one or more of these proteins can cause defects in septum formation, resulting in abnormal cell elongation (34).
Fig 5.
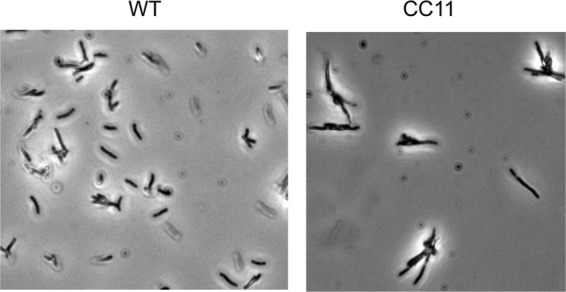
CC11 mutation affects cell morphology. Cultures of H. pylori G27 (WT) and CC11 were grown in brucella broth. Cultures at an OD600 of 1.6 were observed by phase-contrast microscopy with a Zeiss AX10 phase-contrast microscope at a 2,000-fold magnification as described in the text. The depicted micrographs are representative of at least 10 fields recorded for each strain and multiple independent experiments.
Deregulation of genes controlled by growth cycle in CC11 mutant.
The apparent effects of the CC11 mutation on cell division suggested that regulatory defects might extend beyond hp1043 and tlpB. To test this hypothesis, we selected 22 genes associated with virulence and cell growth and assessed their growth-stage regulation by qPCR (Table 3). We calculated growth-phase regulation as a ratio of the mRNA levels at an OD600 of 0.9 (logarithmic phase of growth) to the mRNA levels at an OD600 of 2.0 (stationary phase of growth) (OD 0.9/OD 2.0 ratio). Thus, genes expressed more strongly in the logarithmic phase of growth have OD 0.9/OD 2.0 ratios above 1.0 and genes expressed more strongly in stationary phase of growth have ratios below 1.0. The genes with ratios of 1 ± 0.49 were considered not significantly affected by the growth cycle.
Table 3.
Deregulation of virulence genes controlled by growth cycle in CC11 mutant
| Gene group and gene | Description | OD 0.9/OD 2.0 ratioa |
|
|---|---|---|---|
| WT | CC11 | ||
| Genes induced in logarithmic phase of growth | |||
| HP1027 | fur, ferric uptake regulator | 3.52 | 0.57 |
| HP0527 | cag7, Cag PAIb | 3.26 | 0.70 |
| HP0544 | cag23, Cag PAI | 3.16 | 0.71 |
| HP1041 | flhA, regulator of flagellar biogenesis | 2.82 | 0.76 |
| HP0524 | cag5, Cag PAI | 2.49 | 1.20 |
| HP0597 | pbp1, PBP1 | 2.41 | 0.61 |
| HP1313 | omp24, outer membrane protein HorI | 2.35 | 0.85 |
| HP0685 | fliP, flagellar basal body protein | 1.90 | 0.44 |
| HP0103 | tlpB, chemotaxis | 1.65 | 0.73 |
| HP0887 | vacA, vacuolating cytotoxin | 1.53 | 0.49 |
| Genes induced in stationary phase of growth | |||
| HP0547 | cagA, Cag PAI | 0.29 | 0.40 |
| HP0243 | napA, neutrophil-activating protein | 0.58 | 1.38 |
| Genes less affected by growth cycle (negative controls) | |||
| HPrrnA16S 16S rRNA | 1.00 | 0.99 | |
| HP0875 | katA, catalase | 1.45 | 1.06 |
| HP1573 | tatD, tat translocation system | 1.38 | 1.06 |
| HP0520 | cag1, Cag PAI | 0.93 | 0.70 |
| HP0472 | omp11, outer membrane protein HorE | 0.82 | 0.81 |
| HP0824 | trxA, thioredoxin | 1.44 | 1.30 |
| HP0088 | RNA polymerase σ80 factor | 1.18 | 0.74 |
| HP1456 | lpp20, membrane-associated lipoprotein | 1.03 | 0.66 |
| HP0073 | ureA, subunit of urease enzyme | 0.79 | 0.73 |
| HP1442 | csrA, carbon storage regulator | 1.29 | 0.79 |
RNAs were isolated from H. pylori G27 and G27 CC11 cells in the logarithmic phase of growth (OD600 = 0.9) and stationary phase of growth (OD600 = 2.0). Average changes in gene expression caused by change in growth phase are shown as WT OD 0.9/OD 2.0 and CC11 OD 0.9/OD 2.0 ratios of mRNA levels quantitated by RT-PCR. The studies were repeated three times in triplicate. The errors were less than 15%.
PAI, pathogenicity island.
Of the 22 tested genes, the expression of 10 was increased 1.5- to 3.3-fold in logarithmic phase of growth. The expression of two genes (cagA and napA) was increased 1.7- to 3.3-fold during the switch to the stationary phase of growth, and the rest were only slightly (<50%) affected by the growth stage (Table 3). The known target of HP1043 regulation, chemotaxis gene tlpB (17), was used as a standard to gauge the effects caused by the CC11 mutation. Under our experimental conditions, tlpB was induced in the logarithmic phase of growth (WT OD 0.9/2.0 ratio = 1.65). As seen in Table 3, the effect of the CC11 mutation on the growth-phase regulation of tlpB was reversed from the effect of the WT sequence, being induced in stationary phase of growth. The majority of the genes affected by the growth cycle (fur, cag23, cag7, flhA, omp24, cag5, pbp1, fliP, vacA, and napA) produced a similar pattern: a switch to activation in a different growth stage in the CC11 background, with deregulation occurring over a range of 2- to 6-fold. The effect of the CC11 mutation on cytotoxin-associated gene A (cagA) expression was less significant (∼38%). Importantly, 10 growth-phase-independent genes were unaffected by the CC11 mutation. Thus, our studies reveal a strong correlation between growth-stage-controlled gene expression and the orphan response regulator HP1043.
Proteomic data validate deregulation of growth-phase-dependent genes in the CC11 mutant.
Pursuing the hypothesis that the HP1043CC11 mutant protein causes transcriptional changes that can be detected at the protein level, we compared the protein profiles of the WT and CC11 mutant strains collected in the logarithmic phase of growth (OD600 = 0.9) and in the stationary phase of growth (OD600 = 2.0). As seen in Fig. 6 (area 1), about 30 protein spots displayed a decreased intensity between logarithmic-phase cells and stationary-phase cells. These proteins were unchanged by growth stage in the similarly treated CC11 strain. Not all proteins from area 1 showed reproducibly in multiple experiments. Thus, we focused our attention on the three spots inside area 1, designated area 2, that were highly reproducible. Proteins from area 2 were increased in the CC11 mutant under all growth conditions tested (Fig. 6). Individual proteins from three spots in area 2 were identified using mass spectrometry as catalase and penicillin-binding protein 1 (PBP1), which is consistent with the usual presentation of catalase in 2D gels by three spots linked together (a result of posttranslational modifications). Enzymatic assay (35) confirmed an ∼20-fold increase of catalase activity in the CC11 mutant; however, there was no change in ampicillin susceptibility associated with overexpression of PBP1. Thus, proteomic data validate a role of the CC11 mutation in derepression of multiple genes in the stationary phase of growth shown in our transcriptional studies.
Fig 6.

Proteomic profiles of WT and CC11 strains. Cultures of H. pylori G27 (WT) and CC11 were grown in brucella broth and harvested at an OD600 of 0.9 for log phase of cell growth and one of 2.0 for stationary phase of cell growth, lysed, and standardized on the basis of protein concentration. Cell lysates (120 μg of total protein) were subjected to 2D PAGE. Silver staining was used to identify proteins in the gels. Area 1 depicts proteins repressed in stationary phase and derepressed in the CC11 mutant. Individual proteins from area 2 were identified using mass spectrometry as catalase and penicillin-binding protein 1 (PBP1). The study was repeated independently three times. Representative data from a single independent experiment are shown.
Bioinformatic analysis reveals putative new members of the HP1043 regulon.
To distinguish between direct and indirect effects associated with CC11 mutation, we searched for putative HP1043 binding sites in the predicted promoter regions of the 12 selected genes exhibiting growth-stage regulation (Table 3). As seen in Fig. 7, the HP1043 consensus sequence ATTAWDDHnHHnHDnHDnHWWDnHDDDD (where n is any nucleotide) was present in 7 genes. Putative HP1043 DNA-binding sites were located 6 to 21 nucleotides upstream of the identifiable −10 hexamer (TAtaaT, where lowercase indicate variable nucleotides) in fur, cag25, flhA, hp1043, fliP, tlpB, and cagA promoters. No identifiable −10 hexamer was found upstream of the napA gene. A search against H. pylori genomes with the consensus sequence revealed about 70 genes that contained a perfect match (see Table S1 in the supplemental material). Among these are genes encoding proteins associated with cell division, central metabolism, putative membrane proteins, hypothetical proteins, and transposases and proteins of unknown function. A goal of future research will be to directly investigate the HP1043 regulation of these genes.
Fig 7.

Alignment of the HP1043-regulated promoters. Promoters of genes from Table 3 that have sequence similarities to the HP1043 DNA-binding site (17) are shown. Putative HP1043-binding sites are shaded. Sequences similar to −10 promoter regions are shown in bold. Searches against H. pylori genomes using the PyloriGene World Wide Web server (http://genolist.pasteur.fr/PyloriGene/genome.cgi) revealed additional target promoters (see Table S1 in the supplemental material).
Mouse colonization by the CC11 mutant.
To assess the effects of the CC11 mutation on colonization efficiency, we introduced the CC11 mutation into mouse-adapted H. pylori strain SS1 by natural transformation. The wild type and the CC11 mutant were inoculated orgastrically in separated groups of 4 animals, and the colonization efficiency was assessed at 3 and 6 weeks (25). At 3 weeks, there were no differences in colonization efficiency between these strains. At 6 weeks, we noted a slight decrease in the persistence of the CC11 strain that was not significant (P = 0.123; data not shown). Perhaps a larger study over a longer period of time (8 to 10 weeks) might provide results that rise to the level of significance for the CC11 mutant.
DISCUSSION
Orphan or atypical response regulators (ARRs) such as HP1043 are found in many bacterial genera, including Helicobacter, Sinorhizobium, Chlamydia, Myxococcus, Streptomyces, and Synechococcus (19, 36–41). While all ARRs lack phosphorylation domains, they show diversity in growth-stage regulation, DNA-binding affinity, and whether monomer/dimer status controls function. The HP1043 ARR is highly conserved within the Epsilonproteobacteria and is essential for viability in H. pylori and in C. jejuni and even interchangeable between these species (17, 20, 21). Numerous attempts to alter expression of HP1043, including through the use of controllable promoters, gene duplication, and antisense RNA knockdown, have not been successful, thus exhausting molecular strategies proven successful to studies of other regulatory factors (17, 20, 21). We reasoned that mutations that subtly alter HP1043 function might be sufficiently tolerated by H. pylori to enable identification of new members of the regulon by screening for genes with altered expression. In this study, we showed that mutations to HP1043 (C215S C221S) altered in vitro the efficiency of DNA binding to its promoter and that allelic replacement of the CC11 mutant allele in H. pylori resulted in a 40-fold increase in hp1043CC11 gene expression, concomitant with a 2-fold decrease in HP1043CC11 protein levels compared with those in WT G27. The 2- to 3-fold decrease in HP1043CC11 protein level did not manifest in measurable growth defects in vitro or the ability of this mutant to colonize and persist in a mouse infection model. However, the CC11 mutant showed defects in gene expression on the basis of selected transcript profiling and by comparative proteomics. A survey of previously documented growth-phase-regulated genes (42) revealed that 11 out of 12 of them were no longer regulated and that 6 of these genes contained consensus HP1043 binding motifs within their promoters. Some of the regulated genes, such as fur and flhA, are themselves regulators, suggesting that HP1043 participates in a more complex hierarchical regulatory network.
Ferric uptake regulator fur was one of the most strongly affected genes in this study, showing a 3.5-fold increase in expression in exponential phase for WT G27 and the reverse for CC11 (a 1.75-fold decrease in exponential phase). The decreased expression of Fur in the CC11 mutant during exponential phase might alter expression of members of its regulon that might include some of the genes identified in this study that did not contain HP1043 promoter sequences. It is well established that Fur also contributes to the growth-phase-dependent regulation in H. pylori (43).
H. pylori depends on flagellum-driven motility and chemotaxis to both colonize and persist in the gastric mucosa (44, 45). Multiple transcriptional regulators control flagellar biogenesis in H. pylori, including response regulator FlgR, proto channel protein FlhA, sigma factors σ28 and σ54, and anti-sigma factor FlgM (46). We showed that expression of FlhA, the master regulator of flagellar and nonflagellar genes that controls about 10% of the H. pylori genome, is coordinated with the cell cycle through HP1043. Importantly, hp1043 and flhA (HP1041) appear to be evolutionarily linked, a feature common to other members of the Epsilonproteobacteria. Bioinformatic analysis of the fur and flhA promoter regions revealed consensus HP1043 binding sequences. Additional genes containing consensus HP1043 sequences included cag25, fliP, cagA, and napA promoters (Fig. 7; see Table S1 in the supplemental material), thus suggesting that HP1043 likely binds to these promoters. However, additional experimental evidence is required to confirm this possibility and to eliminate indirect regulatory effects. It is likely that more complete transcriptome profiling will be required to complete the analysis of the HP1043 regulon. It should be possible to connect regulons for Fur, FlhA, and many others into a regulatory network that accounts for both direct and indirect regulatory activities.
Unlike most growth-stage regulators that become activated or repressed postexponentially, our studies show that HP1043 protein levels did not vary with stage of growth in any of the strains examined in this study (Fig. 3). Based on footprinting studies with recombinant HP1043CC11 protein, we initially concluded that loss of growth-stage regulation most likely resulted from an increased affinity for DNA relative to that of WT HP1043. However, in vivo studies seem to suggest the opposite, as autoregulatory transcription factors are generally believed to function as repressors and the location of the HP1043 DNA-binding motif (positions −17 to −35) within the promoter region is consistent with this regulatory mechanism. Moreover, the nearly 40-fold increase in hp1043CC11 transcript levels relative to that of the WT is also consistent with decreased DNA binding and derepression. The apparent posttranscriptional control of translation has been noted in previous studies, and the underlying regulatory mechanisms are not known (17, 20). In other systems, translation can be controlled by riboswitches (47) or through the action of noncoding regulatory RNAs (48). Regulatory noncoding RNAs have been studied in H. pylori (12), but so far, none have been shown to form duplexes with hp1043 mRNA. Similarly, it is not known whether DNA-binding activity is modulated posttranslationally through conformational changes or by interactions with other proteins, such as anti-sigma factors or proteases that maintain steady-state levels across the growth cycle. While our studies do suggest that the CC11 mutation, particularly C215S, most likely affects DNA binding, they do not rule out other mechanisms involving conformational changes or oxidation/reduction involving the cysteine residues. We are intrigued by the possibility that conformational changes to HP1043, particularly under conditions of oxidative stress, might result in disulfide bond cross-linking of the homodimer, thus arresting growth until cellular redox is restored. Such redox switches are common in other bacteria that use OxyR to control gene expression (49). Studies are in progress to explore these possibilities for control of functional activity for HP1043.
The apparent discrepancy between in vitro and in vivo results, together with observations reported by others, suggests that recombinant HP1043 may differ from the native protein. Accordingly, most transcription factors, including others from H. pylori, bind DNA with a 5- to 100-fold higher affinity than HP1043 (17, 22, 23). In the absence of any direct study with native HP1043, we employed a gel shift strategy to enrich for native proteins that bound promoter DNA sequences of pfor, which encodes the pyruvate:ferredoxin oxidoreductase (50). After several purification steps, one fraction with demonstrated activity by gel shift was analyzed by liquid chromatography-mass spectrometry, and the amount of HP1043 in this fraction was enriched ∼5-fold over the amount in crude extracts. When tested with the hp1043 promoter sequences, these fractions were dramatically more active by gel shift than the recombinant protein obtained from E. coli (I. N. Olekhnovich and P. S. Hoffman, unpublished data). Studies are in progress to complete purification of the native HP1043 to address these possibilities.
Based on our studies and those of others, we suggest that HP1043 may serve as a homeostatic marker whose steady-state level is maintained at the transcriptional, posttranscriptional, and posttranslational levels. We speculate that the CC11 mutation perturbs this regulation at the level of transcription and that the 40-fold increase in mRNA levels results in an overcorrection at the posttranscriptional level to control translation. It is possible that this overcorrection can be exploited in future strategies to identify additional regulatory factors. In summary, our studies with the CC11 mutant variant of HP1043 revealed pleiotropic effects on many genes and operons and enabled identification of new members of the regulon. Our studies show that a mutation-based approach to study the regulatory activities of an essential transcription factor can be used to explore regulatory functions.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant R01-DK073823 to P.S.H., NIAID contract no. HHSN272201000056C to J.B.-R., and funds from the Nutritional Immunology and Molecular Medicine Laboratory.
Footnotes
Published ahead of print 19 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01193-12.
REFERENCES
- 1. Marshall BJ, Warren JR. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311–1315 [DOI] [PubMed] [Google Scholar]
- 2. Peek RM, Jr, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2:28–37 [DOI] [PubMed] [Google Scholar]
- 3. Allen LA. 2007. Phagocytosis and persistence of Helicobacter pylori. Cell. Microbiol. 9:817–828 [DOI] [PubMed] [Google Scholar]
- 4. Fischer W, Prassl S, Haas R. 2009. Virulence mechanisms and persistence strategies of the human gastric pathogen Helicobacter pylori. Curr. Top. Microbiol. Immunol. 337:129–171 [DOI] [PubMed] [Google Scholar]
- 5. Blaser MJ. 2012. Heterogeneity of Helicobacter pylori. Eur. J. Gastroenterol. Hepatol. 9(Suppl 1):S3–S6 [DOI] [PubMed] [Google Scholar]
- 6. Yoshiyama H, Nakazawa T. 2000. Unique mechanism of Helicobacter pylori for colonizing the gastric mucus. Microbes Infect. 2:55–60 [DOI] [PubMed] [Google Scholar]
- 7. Croxen MA, Sisson G, Melano R, Hoffman PS. 2006. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 188:2656–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thiele I, Vo TD, Price ND, Palsson BO. 2005. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. J. Bacteriol. 187:5818–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chalker AF, Minehart HW, Hughes NJ, Koretke KK, Lonetto MA, Brinkman KK, Warren PV, Lupas A, Stanhope MJ, Brown JR, Hoffman PS. 2001. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J. Bacteriol. 183:1259–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danielli A, Amore G, Scarlato V. 2010. Built shallow to maintain homeostasis and persistent infection: insight into the transcriptional regulatory network of the gastric human pathogen Helicobacter pylori. PLoS Pathog. 6:e1000938 doi:10.1371/journal.ppat.1000938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barnard FM, Loughlin MF, Fainberg HP, Messenger MP, Ussery DW, Williams P, Jenks PJ. 2004. Global regulation of virulence and the stress response by CsrA in the highly adapted human gastric pathogen Helicobacter pylori. Mol. Microbiol. 51:15–32 [DOI] [PubMed] [Google Scholar]
- 12. Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermuller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 13. Danielli A, Scarlato V. 2010. Regulatory circuits in Helicobacter pylori: network motifs and regulators involved in metal-dependent responses. FEMS Microbiol. Rev. 34:738–752 [DOI] [PubMed] [Google Scholar]
- 14. Miles S, Piazuelo MB, Semino-Mora C, Washington MK, Dubois A, Peek RM, Jr, Correa P, Merrell DS. 2010. Detailed in vivo analysis of the role of Helicobacter pylori Fur in colonization and disease. Infect. Immun. 78:3073–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baldwin DN, Shepherd B, Kraemer P, Hall MK, Sycuro LK, Pinto-Santini DM, Salama NR. 2007. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75:1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loh JT, Gupta SS, Friedman DB, Krezel AM, Cover TL. 2010. Analysis of protein expression regulated by the Helicobacter pylori ArsRS two-component signal transduction system. J. Bacteriol. 192:2034–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delany I, Spohn G, Rappuoli R, Scarlato V. 2002. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184:4800–4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2007. The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J. Bacteriol. 189:2426–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hong E, Lee HM, Ko H, Kim DU, Jeon BY, Jung J, Shin J, Lee SA, Kim Y, Jeon YH, Cheong C, Cho HS, Lee W. 2007. Structure of an atypical orphan response regulator protein supports a new phosphorylation-independent regulatory mechanism. J. Biol. Chem. 282:20667–20675 [DOI] [PubMed] [Google Scholar]
- 20. Muller S, Pflock M, Schar J, Kennard S, Beier D. 2007. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 162:1–14 [DOI] [PubMed] [Google Scholar]
- 21. Croxen MA, Ernst PB, Hoffman PS. 2007. Antisense RNA modulation of alkyl hydroperoxide reductase levels in Helicobacter pylori correlates with organic peroxide toxicity but not infectivity. J. Bacteriol. 189:3359–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Danielli A, Romagnoli S, Roncarati D, Costantino L, Delany I, Scarlato V. 2009. Growth phase and metal-dependent transcriptional regulation of the fecA genes in Helicobacter pylori. J. Bacteriol. 191:3717–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dietz P, Gerlach G, Beier D. 2002. Identification of target genes regulated by the two-component system HP166-HP165 of Helicobacter pylori. J. Bacteriol. 184:350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoffman PS, Goodwin A, Johnsen J, Magee K, Veldhuyzen van Zanten SJ. 1996. Metabolic activities of metronidazole-sensitive and -resistant strains of Helicobacter pylori: repression of pyruvate oxidoreductase and expression of isocitrate lyase activity correlate with resistance. J. Bacteriol. 178:4822–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoffman PS, Vats N, Hutchison D, Butler J, Chisholm K, Sisson G, Raudonikiene A, Marshall JS, Veldhuyzen van Zanten SJ. 2003. Development of an interleukin-12-deficient mouse model that is permissive for colonization by a motile KE26695 strain of Helicobacter pylori. Infect. Immun. 71:2534–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu H, Rahman A, Semino-Mora C, Doi SQ, Dubois A. 2008. Specific and sensitive detection of H. pylori in biological specimens by real-time RT-PCR and in situ hybridization. PLoS One 3:e2689 doi:10.1371/journal.pone.0002689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boonjakuakul JK, Canfield DR, Solnick JV. 2005. Comparison of Helicobacter pylori virulence gene expression in vitro and in the rhesus macaque. Infect. Immun. 73:4895–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruzsovics A, Molnar B, Unger Z, Tulassay Z, Pronai L. 2001. Determination of Helicobacter pylori cagA, vacA genotypes with real-time PCR melting curve analysis. J. Physiol. Paris 95:369–377 [DOI] [PubMed] [Google Scholar]
- 29. Bassaganya-Riera J, Dominguez-Bello MG, Kronsteiner B, Carbo A, Lu P, Viladomiu M, Pedragosa M, Zhang X, Sobral BW, Mane SP, Mohapatra SK, Horne WT, Guri AJ, Groeschl M, Lopez-Velasco G, Hontecillas R. 2012. Helicobacter pylori colonization ameliorates glucose homeostasis in mice through a PPAR gamma-dependent mechanism. PLoS One 7:e50069 doi:10.1371/journal.pone.0050069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu. Rev. Microbiol. 63:133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 32. DeLoney CR, Schiller NL. 1999. Competition of various beta-lactam antibiotics for the major penicillin-binding proteins of Helicobacter pylori: antibacterial activity and effects on bacterial morphology. Antimicrob. Agents Chemother. 43:2702–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rimbara E, Noguchi N, Kawai T, Sasatsu M. 2008. Mutations in penicillin-binding proteins 1, 2 and 3 are responsible for amoxicillin resistance in Helicobacter pylori. J. Antimicrob. Chemother. 61:995–998 [DOI] [PubMed] [Google Scholar]
- 34. Cabeen MT, Jacobs-Wagner C. 2005. Bacterial cell shape. Nat. Rev. Microbiol. 3:601–610 [DOI] [PubMed] [Google Scholar]
- 35. Pine L, Hoffman PS, Malcolm GB, Benson RF, Keen MG. 1984. Determination of catalase, peroxidase, and superoxide dismutase within the genus. Legionella. J. Clin. Microbiol. 20:421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rotter C, Muhlbacher S, Salamon D, Schmitt R, Scharf B. 2006. Rem, a new transcriptional activator of motility and chemotaxis in Sinorhizobium meliloti. J. Bacteriol. 188:6932–6942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hickey JM, Weldon L, Hefty PS. 2011. The atypical OmpR/PhoB response regulator ChxR from Chlamydia trachomatis forms homodimers in vivo and binds a direct repeat of nucleotide sequences. J. Bacteriol. 193:389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fraser JS, Merlie JP, Jr, Echols N, Weisfield SR, Mignot T, Wemmer DE, Zusman DR, Alber T. 2007. An atypical receiver domain controls the dynamic polar localization of the Myxococcus xanthus social motility protein FrzS. Mol. Microbiol. 65:319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kato H, Chibazakura T, Yoshikawa H. 2008. NblR is a novel one-component response regulator in the cyanobacterium Synechococcus elongatus PCC 7942. Biosci. Biotechnol. Biochem. 72:1072–1079 [DOI] [PubMed] [Google Scholar]
- 40. Mittal S, Kroos L. 2009. A combination of unusual transcription factors binds cooperatively to control Myxococcus xanthus developmental gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:1965–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ainsa JA, Parry HD, Chater KF. 1999. A response regulator-like protein that functions at an intermediate stage of sporulation in Streptomyces coelicolor A3(2). Mol. Microbiol. 34:607–619 [DOI] [PubMed] [Google Scholar]
- 42. Thompson LJ, Merrell DS, Neilan BA, Mitchell H, Lee A, Falkow S. 2003. Gene expression profiling of Helicobacter pylori reveals a growth-phase-dependent switch in virulence gene expression. Infect. Immun. 71:2643–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Choi YW, Park SA, Lee HW, Lee NG. 2009. Alteration of growth-phase-dependent protein regulation by a fur mutation in Helicobacter pylori. FEMS Microbiol. Lett. 294:102–110 [DOI] [PubMed] [Google Scholar]
- 44. McGee DJ, Coker C, Testerman TL, Harro JM, Gibson SV, Mobley HL. 2002. The Helicobacter pylori flbA flagellar biosynthesis and regulatory gene is required for motility and virulence and modulates urease of H. pylori and Proteus mirabilis. J. Med. Microbiol. 51:958–970 [DOI] [PubMed] [Google Scholar]
- 45. Kavermann H, Burns BP, Angermuller K, Odenbreit S, Fischer W, Melchers K, Haas R. 2003. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 197:813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niehus E, Gressmann H, Ye F, Schlapbach R, Dehio M, Dehio C, Stack A, Meyer TF, Suerbaum S, Josenhans C. 2004. Genome-wide analysis of transcriptional hierarchy and feedback regulation in the flagellar system of Helicobacter pylori. Mol. Microbiol. 52:947–961 [DOI] [PubMed] [Google Scholar]
- 47. Smith AM, Fuchs RT, Grundy FJ, Henkin TM. 2010. Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol. 7:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gottesman S, Storz G. 2011. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3(12):pii=a003798. doi:10.1101/cshperspect.a003798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Antelmann H, Helmann JD. 2011. Thiol-based redox switches and gene regulation. Antioxid. Redox Signal. 14:1049–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, Berg DE, Hoffman PS. 2002. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob. Agents Chemother. 46:2116–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.