

Abstract
The acute-phase response is characteristic of perhaps all infections, including bacterial pneumonia. In conjunction with the acute-phase response, additional biological pathways are induced in the liver and are dependent on the transcription factors STAT3 and NF-κB, but these responses are poorly understood. Here, we demonstrate that pneumococcal pneumonia and other severe infections increase expression of multiple components of the cellular secretory machinery in the mouse liver, including the endoplasmic reticulum (ER) translocon complex, which mediates protein translation into the ER, and the coat protein complexes (COPI and COPII), which mediate vesicular transport of proteins to and from the ER. Hepatocyte-specific mutation of STAT3 prevented the induction of these secretory pathways during pneumonia, with similar results observed following pharmacological activation of ER stress by using tunicamycin. These findings implicate STAT3 in the unfolded protein response and suggest that STAT3-dependent optimization of secretion may apply broadly. Pneumonia also stimulated the binding of phosphorylated STAT3 to promoter regions of secretion-related genes in the liver, supporting a direct role for STAT3 in their transcription. Altogether, these results identify a novel function of STAT3 during the acute-phase response, namely, the induction of secretory machinery in hepatocytes. This may facilitate the processing and delivery of newly synthesized loads of acute-phase proteins, enhancing innate immunity and preventing liver injury during infection.
INTRODUCTION
Acute lower respiratory tract infections lead to a staggering disease burden worldwide, greater than cancer, HIV/AIDS, ischemic heart disease, or malaria (1, 2). Identification of the biological pathways involved in the innate immune response during such infections is critical to improving outcome (3). The hepatic acute-phase response is characteristic of many diseases, including pneumonia. During bacterial pneumonia, cells in the lung are stimulated to secrete an array of proinflammatory cytokines, including interleukin-6 (IL-6), IL-1α, and IL-1β and tumor necrosis factor alpha (TNF-α) (4–6). A major role of these cytokines is to trigger a systemic immune response, including changes in circulating acute-phase proteins due to transcriptional remodeling in hepatocytes (7, 8). Acute-phase proteins have multiple roles during severe infection that are essential both for innate immune antibacterial defenses and for preventing acute inflammatory tissue injury (9–13).
During pneumonia, mutation of both STAT3 and RelA transcription factors in hepatocytes completely abrogates the hepatic acute-phase response (8). This led to the discovery that blood proteome remodeling by hepatocytes was critical for preventing bacteremia during pneumonia, thereby serving as a systemic innate immune response to compartmentalize local infection. These studies also revealed the pneumonia-induced transcriptional response of the liver to include significant changes in >1,000 genes, far more than the 50 or so known acute-phase proteins. While unknown acute-phase proteins were almost certainly included in these gene sets, other biological pathways were also implicated. Most notably, Gene Ontology categories related to protein secretion, such as protein transport, Golgi vesicle transport, and endoplasmic reticulum (ER)-to-Golgi vesicle-mediated transport, were induced in wild-type (WT) but not mutant mice (8), suggesting for the first time that these cellular pathways are under transcriptional control during the hepatic acute-phase response. These pathways can help prevent the accumulation of excessive protein loads that would otherwise result in ER stress. ER stress activates the unfolded protein response (UPR) to form a negative feedback loop that enhances protein folding and processing while limiting protein translation, thereby mitigating ER stress (14). While ER stress in the liver can elicit an acute-phase response (15, 16), it is unknown whether and how ER stress and the UPR may be involved in naturally occurring acute-phase responses, such as during pneumonia. We hypothesized that these adaptive cellular processes are components of the acute-phase response and are mediated by transcription factors that are themselves central to acute-phase protein induction, STAT3 and NF-κB RelA (7, 8).
MATERIALS AND METHODS
Mice.
Conditional mutant mice lacking STAT3 (STAT3hepΔ), RelA (RelAhepΔ), or both STAT3 and RelA (STAT3/RelAhepΔ) specifically in hepatocytes were generated by crossing mice with LoxP sites flanking STAT3 (17) or RelA (18) with mice expressing Cre-recombinase driven by an albumin promoter (Jackson Laboratories), as previously described (8). The Cre+ mutant mice were compared to Cre− littermates as controls. In addition, C57BL/6 mice, C57BL/6 × 129/Sv random hybrid mice, and outbred ICR mice (Harlan Laboratories, Indianapolis, IN) were used for characterization of responses to ER stress, bacterial pneumonia, and cecal ligation and puncture, respectively. Mice were maintained under pathogen-free conditions, and all studies were approved by the Institutional Animal Care and Use Committee at Boston University.
Mouse challenge studies in vivo.
For bacterial pneumonia studies, mice at 6 to 12 weeks of age were anesthetized by intraperitoneal injection of a cocktail containing ketamine (75 mg/kg of body weight) and xylazine (10 mg/kg). The trachea was surgically exposed, and an angiocatheter was placed down the left bronchus to instill the left lobe of the lung with 50 μl of saline containing 106 CFU of Streptococcus pneumoniae (serotype 3; ATCC 6303) or Escherichia coli serotype 06:K2:H1 (ATCC 19138). For sepsis studies, mice were anesthetized by inhalation of 5% isoflurane in 100% oxygen, and the cecum was accessed through a midline incision and ligated. A 16-gauge needle was used to perforate twice between ligatures, the abdomen was sutured closed, and mice received subcutaneous injections of 25 mg/kg imipenem (Merck, West Point, PA) every 12 h as an antibiotic treatment. For studies of the pharmacological induction of ER stress, mice received intraperitoneal injections of 2 mg/kg tunicamycin (Calbiochem) or vehicle (150 μM dextrose).
qRT-PCR.
Total liver RNA was extracted using TRIzol (Invitrogen) and purified using the RNeasy minikit (Qiagen). Ten or 100 ng of isolated RNA was used in quantitative reverse transcription-PCRs (qRT-PCRs) performed with the TaqMan RNA-to-CT one-step kit on the StepOnePlus Realtime PCR system (Applied Biosystems). Table 1 shows the sequences of primers and probes designed using the CLC DNA Workbench software (CLC Bio). Sequences for ERp72 and 18S rRNA have been reported elsewhere (19, 20). All results were normalized to the quantity of 18S rRNA within each sample and are expressed as the fold induction relative to a control group (21), as indicated in each figure.
Table 1.
Primers and probes for qRT-PCR analysisa
| Gene | Forward primer | Reverse primer | Probe |
|---|---|---|---|
| Arfgap3 | ATGCTTCTTCCTTCTTCC | TCTTGATCTTCTCCCTGT | ACCAAGGACACCAACGCCA |
| Copε | CAGTGAGAACCAGAGGGA | GAGCAGGAAAGTGGTATT | ACACTCCTGCTCATCTCCCG |
| GRP78 | CTATTCCTGCGTCGGTGTGTT | GGGTTGGACGTGAGTTGGTT | TCAGGGCAACCGCATCACGCCG |
| SRPRβ | AGCTAGGGCTGTGGTGTT | AGGCTATTAATAAGGACGGTGAGTT | TTCGGCCACATCTTTCACCTCCCGCT |
| Sec24d | AGTCCCACCATTCTATTTCCAACATC | CGTCTCCTCATGCTCCTCCTT | ACCTCCCAGCCCACCAGCCTT |
| Sec61α1 | GCCCAGAAGTTGTTTGGAATGAT | GCCAGCAACAAAGAGCTGAAT | AGGGTCCCCGTACATGCCCGT |
| CHOP | CCACCACACCTGAAAGCAGAA | ATGAGATATAGGTGCCCCAA | ACGTGCAGTCATGGCAGCTG |
| ATF4 | GCCAAGCACTTGAAACCTCAT | AACATCCAATCTGTCCCGGAA | ACACCGGCAAGGAGGATGCC |
Primer and probe sequences, shown in the 5′-to-3′ direction, were designed (using the CLC DNA workbench software) to amplify a region of 200-bp sequences within the open reading frames of the following transcripts: Arfgap3, Copε, GRP78, SRPRβ, Sec24d, Sec61α1, CHOP, and ATF4.
Liver protein analysis by immunoblotting.
Liver fragments from control and treated mice were snap-frozen in liquid nitrogen and stored at −80°C until protein extraction. The frozen liver fractions were homogenized in protein lysis buffer containing 10% glycerol, 1% Triton X-100, 100 mM Tris-HCl (pH 7.4), 100 mM Na2PO7, 100 mM NaF, 10 mM NaVa, 10 mM EDTA (pH 8), protease inhibitor cocktail (Roche Diagnostics), and dithiothreitol. Immunoblot assays for total STAT3, Y705-phosphorylated STAT3, and glyceraldehyde-3-phosphate dehydrogenase were performed as described previously (7) using antibodies from Cell Signaling.
Serum ALT and AST measurements.
Serum was collected from STAT3hepΔ mice and control littermates 24 h after intraperitoneal injection of tunicamycin. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) concentrations were measured using kinetic assays employing serum glutamic oxaloacetic transaminase (SGOT) and serum glutamic pyruvic transaminase (SGPT) reagents, respectively (Pointe Scientific, Inc., Canton, MI).
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay protocol was modified from a two-step cross-linking method previously reported (22). Briefly, fresh liver tissues from control and infected mice were minced in Dulbecco's phosphate-buffered saline and fixed with 1 mM disuccinimidyl glutarate for 20 min and then with 1% formaldehyde for 15 min at room temperature to cross-link protein-DNA complexes, before quenching with 0.125 M glycine. Liver cells were lysed and sonicated on ice in a Branson ultrasonic sonicator coupled to a Fisher Scientific Sonic Dismembrator 500 power supply to shear the DNA and obtain fragments ranging from 200 to 600 bp, with the majority with a size of 300 bp. Liver cell extracts were precleared at 4°C with Dynabeads linked to protein A (Invitrogen) for 2 h before incubation with phospho-STAT3 (pSTAT3) antibody (9131S; Cell Signaling) or IgG antibody (sc-2027; Santa Cruz Biotechnology) at 4°C. The next day, the beads were washed serially in low-salt buffer, high-salt buffer, lithium chloride buffer, and then Tris-EDTA buffer. The protein-DNA complexes were eluted twice from the beads in elution buffer at 65°C for 10 min. DNA was analyzed using qPCR. ChIP DNA cycle threshold (CT) values were normalized to the input CT for each primer set, IgG control values were subtracted from the phospho-STAT3 values, and pneumonia sample results were expressed as the mean fold signal from uninfected mice. Primer sequences for GRP78 and Arfgap3 are shown in Table 2.
Table 2.
Primer sequences for quantitative PCR after ChIP
| Gene name | Forward primer | Reverse primer |
|---|---|---|
| Arfgap3 (distal) | CCAAGGCTGTCTCTTCTCTAT | CCTGTATGAAGGAGTTGGAGAT |
| Arfgap3 (proximal) | CGCAAAGCCTGGGGTTCCTAT | GGAAGAGACAAGGAGTAGCGGTT |
| GRP78 (distal) | GTGATGCAACTTCTATAGCAAGGAT | AGGCAGGAAATGACTCCAGTA |
| GRP78 (proximal) | GCGGCGGAGAAAGGGAATAGGTT | GAGCTGGGGCCTGATCTGGATT |
Statistics.
Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software). Comparisons between uninfected and infected groups were performed using a two-way analysis of variance (ANOVA) followed by a Tukey post hoc test. For the time course data analyses, a one-way ANOVA followed by Bonferroni's multiple comparison test was used. Comparisons of PCR results from ChIP analyses were performed using Student's t test. Differences between groups were considered statistically significant if P was <0.05.
RESULTS
Secretory protein pathways are induced in the liver during pneumonia.
Microarray analyses suggested that pathways generally related to protein secretion were induced in the liver during pneumococcal pneumonia (8). To determine whether specific secretion-related proteins were induced in the liver during pneumonia, we measured transcripts encoding representative proteins belonging to 3 important groups: the translocon complex (SRPRβ, Sec61α1, and GRP78), the COPII pathway (Sec24d), and the COPI pathway (COPε and Arfgap3). The translocon complex mediates protein translation into the ER lumen, whereas the COPII and COPI pathways mediate anterograde and retrograde vesicular transport with the ER, respectively. Liver mRNA levels increased in response to lung infection with S. pneumoniae, which was apparent by 24 h after intratracheal instillation of bacteria (Fig. 1).
Fig 1.

Secretory pathway gene induction during pneumococcal pneumonia. Quantitative RT-PCR was performed on liver RNA collected from B6/129 mice 0 to 24 h after intratracheal S. pneumoniae infection to evaluate transcripts involved in protein translation into the ER or vesicular transport to and from the ER. Values are the fold induction compared to uninfected controls and are expressed as means ± standard errors of the means. *, P < 0.05 compared to uninfected mice (results for one experiment, with 3 to 5 mice/group).
The induction of secretory protein genes depends on STAT3 but not RelA.
To test whether the induction of secretory protein genes was dependent upon STAT3 and/or RelA, as inferred from the microarray results (8), we analyzed liver transcripts from STAT3/RelAhepΔ mice and their control littermates 24 h after S. pneumoniae infection. At this time point, there were no significant differences in lung or blood bacterial burdens between the two genotypes (data not shown). The qRT-PCR analyses revealed that the expression levels of genes in the translocon and vesicular transport pathways were significantly diminished in the livers of mutant mice compared to the WT (Fig. 2). Thus, secretory pathway gene induction in the liver during pneumonia requires STAT3 and/or RelA. We next tested whether STAT3 or RelA was individually sufficient to regulate the expression of SRPRβ, Sec61α1, GRP78, Sec24d, COPε, and Arfgap3. STAT3hepΔ mice (with hepatocyte mutation of STAT3 alone) and RelAhepΔ mice (with hepatocyte mutation of RelA alone) were infected with S. pneumoniae 24 h before livers were harvested. The expression pattern of secretory protein genes in STAT3hepΔ mice was similar to that observed in STAT3/RelAhepΔ mice, with pneumonia-induced mRNA induction generally diminished in mutant mice (Fig. 3). In contrast, the mutation of RelA alone did not alter the expression of the secretory protein genes during pneumonia (Fig. 4). These data suggest that STAT3 but not RelA is needed for the hepatic induction of genes mediating protein secretion during pneumonia.
Fig 2.

Secretory protein gene induction is diminished by simultaneous mutation of both STAT3 and RelA. Stat3/RelAhepΔ mice and control (WT) littermates received intratracheal instillations of S. pneumoniae 24 h before the livers were harvested for RNA extraction. Quantitative RT-PCR analysis of secretory protein genes in Stat3/RelAhepΔ mouse liver RNA extracts was performed. Fold induction values for the indicated genes are the means ± standard errors of the means. *, P < 0.05 versus WT (results from 2 independent experiments, with a total of 6 to 10 mice/group).
Fig 3.

Secretory protein gene induction requires STAT3. Stat3hepΔ mice and control (WT) littermates received intratracheal instillations of S. pneumoniae 24 h before the livers were harvested for RNA extraction. Quantitative RT-PCR analysis of secretory protein genes in Stat3hepΔ mouse liver RNA extracts was performed. Fold induction values for the indicated genes are expressed as means ± standard errors of the means. *, P < 0.05 versus WT (results from 2 independent experiments, with a total of 9 to 10 mice/group for pneumonia and 3 mice/group for uninfected controls).
Fig 4.

Secretory protein gene induction is unaffected by mutation of RelA. RelAhepΔ mice and control littermates received intratracheal instillations of S. pneumoniae 24 h before the livers were harvested for RNA extraction. Quantitative RT-PCR analysis of secretory protein genes in RelAhepΔ mouse liver RNA extracts was performed. Fold induction values for the indicated genes are expressed as means ± standard errors of the means. There were no significant effects of genotype for any gene (results from 3 independent experiments, with a total of 5 to 8 mice/group).
ER stress is induced in the liver during pneumonia and dependent on STAT3.
The increased expression of GRP78 suggested that ER stress may occur in the liver during pneumonia, since this protein, also known as binding immunoglobulin protein (BiP) or heat shock protein a5 (Hspa5), is a reliable indicator of that process (23). We therefore measured additional markers of ER stress in livers from pneumonic and nonpneumonic mice, including C/EBP-homologous protein (CHOP; also known as growth arrest and DNA damage-inducible protein 153, or GADD153), ER resident protein 72 (ERp72), and activating transcription factor 4 (ATF4). All 3 trended toward an increase with pneumonia (Fig. 5), suggesting that infection triggers ER stress in hepatocytes. The expression levels of both CHOP and ERp72 during pneumonia were significantly diminished by STAT3 mutation, whereas ATF4 was unaffected (Fig. 5). While revealing complexity to the hepatic ER stress pathways during pneumonia, these data suggest that much of the ER stress response, including the expression of GRP78, CHOP, and ERp72, is downstream of STAT3 in this setting.
Fig 5.

Expression of UPR target genes is diminished by STAT3 mutation. Stat3hepΔ mice and control (WT) littermates received intratracheal instillations of S. pneumoniae 24 h before the livers were harvested for RNA extraction. Quantitative RT-PCR analysis of UPR genes was performed. Fold induction values for the indicated genes are expressed as means ± standard errors of the means. *, P < 0.05 versus WT (results from 2 independent experiments, with a total of 9 or 10 mice/group for pneumonia and 3 mice/group for uninfected).
ER stress-triggered secretory protein pathway gene induction depends on STAT3.
In order to determine whether STAT3 signaling may itself be downstream of ER stress in the induction of secretory pathway genes, we pharmacologically activated ER stress in WT and STAT3hepΔ mice by administering tunicamycin intraperitoneally, to inhibit N-glycosylation of proteins and cause retention of misfolded proteins in the ER. Interestingly, STAT3 became phosphorylated in the liver after tunicamycin administration (Fig. 6A), indicating that this transcription factor was activated during this ER stress in vivo. Although a toxin and strong inducer of ER stress, this tunicamycin treatment did not induce liver injury in WT mice, and there were no significant effects of genotype on circulating liver enzymes (Table 3). As expected, secretory protein genes were strongly induced by tunicamycin stimulation of ER stress (Fig. 6B). Much of this gene induction was blunted by a hepatocyte-specific deficiency of STAT3 (Fig. 6B). Although not universally required, the generally decreased induction of secretory pathway genes due to STAT3 mutation suggests that STAT3 functions downstream of ER stress to drive the expression of secretory machinery.
Fig 6.
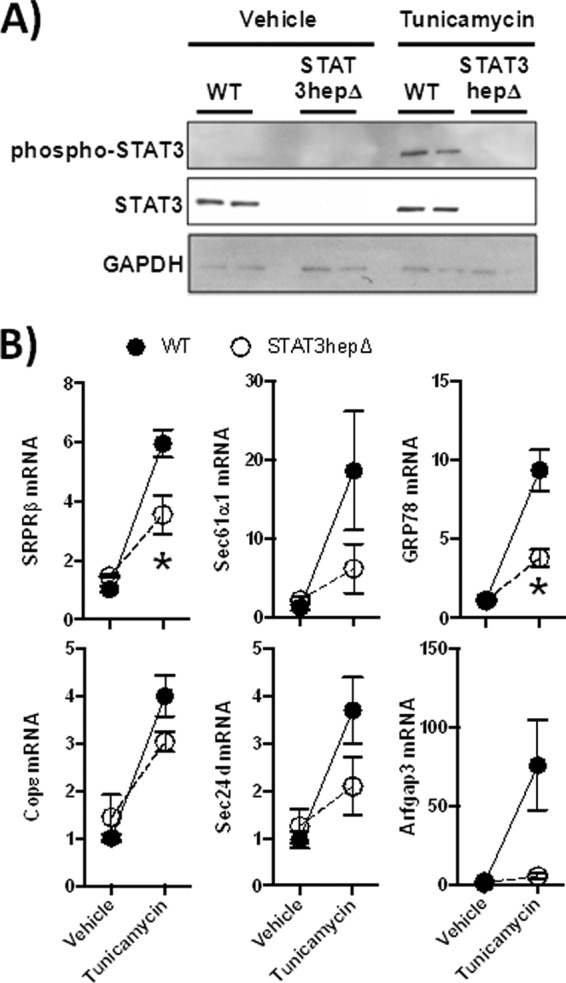
STAT3 mediates secretory protein gene induction elicited by ER stress. Stat3hepΔ mice and control (WT) littermates received intraperitoneal injections of tunicamycin 24 h before the livers were harvested for RNA or protein extraction. (A) Western blot analysis of total and phospho-STAT3, using total liver protein extract. Each lane contains an extract from a different mouse. (B) Quantitative RT-PCR results for secretory protein genes in STAT3hepΔ mice, based on liver RNA extracts. Fold induction values for the indicated genes are means ± standard errors of the mean. *, P < 0.05 compared to WT (results from 2 independent experiments, with a total of 4 to 8 mice/group).
Table 3.
Serum levels of liver enzymes after tunicamycin treatmenta
| Mouse group | ALT (U/liter) |
AST (U/liter) |
||
|---|---|---|---|---|
| Vehicle | Tunicamycin | Vehicle | Tunicamycin | |
| WT | 63.6 ± 50.8 | 50.1 ± 26.7 | 31.8 ± 4.5 | 85.4 ± 16.6 |
| STAT3hepΔ | 49.5 ± 16.0 | 39.8 ± 5.7 | 42.4 ± 7.6 | 154.7 ± 58.0 |
STAT3hepΔ mice and control littermates received i.p. injections of tunicamycin, and serum was collected after 24 h. ALT and AST concentrations were measured in a kinetic spectophotometry assay. Values are means ± standard deviations (results from one experiment; n = 4 to 6 mice/group).
Active STAT3 physically associates with promoters of secretory protein genes.
The observations that STAT3 was essential to hepatic secretory protein transcript induction in response to either pneumococcal pneumonia or tunicamycin-induced ER stress suggested the unexpected possibility that STAT3 directly regulates transcription of secretion-related genes. We analyzed pneumonic and nonpneumonic mouse livers in ChIP assays to test whether and when phosphorylated STAT3 was directly bound to the promoters of secretory protein genes in these cells. We focused on Arfgap3 and GRP78, which were significantly decreased by STAT3 mutation after pneumonia (Fig. 3) and which represent different components of the protein secretion pathway. Figure 7A shows representations of the promoter regions with putative STAT3-binding sites depicted as well as primer pairs (2 sets of opposing arrows) used to differentiate proximal and distal regions. For both the Arfgap3 and GRP78 genes, pneumonia triggered the binding of phosphorylated STAT3 to the proximal but not distal regions of the 5′ promoter (Fig. 7B). Phosphorylated STAT3 binding did not increase during pneumonia in the 3′ region for these genes (data not shown). These data demonstrate that pneumonia induces the association of activated STAT3 with the proximal 5′ promoter region of secretory protein genes in the liver, supporting the conclusion that STAT3 plays a direct role in the regulation of these genes.
Fig 7.

Phosphorylated STAT3 binds to secretory protein gene promoters during pneumonia. (A) Schematics of Arfgap3 and Grp78 promoter loci, with putative STAT3-binding sites shown as hash marks on the scaled diagram of the 2,000-bp promoter region and positioned relative to the transcriptional start site. Putative STAT3-binding sites were identified using the TRANSFAC databases, and all sites with a core score greater than 0.99 and matrix score greater than 0.7 (matrix library; TRANSFAC matrix table; release 2012.2) are noted by a hash mark. The locations of the primers used for quantitative PCR are represented by 2 opposing arrows, and positions relative to the transcriptional start site are indicated. The translational start is depicted by a bent arrow. (B) ChIP results with phosphorylated STAT3 revealed a pneumonia-induced association with proximal promoter regions of secretory protein genes. Chromatin was isolated from liver cells collected from C57BL/6 mice 0 or 24 h after S. pneumoniae pneumonia infection, fixed, sheared, and immunoprecipitated with antibodies against phosphorylated STAT3. Precipitated 200- to 600-bp DNA fragments containing the regions amplified were measured using quantitative PCR. Fold induction values for the indicated genes were calculated relative to values observed in uninfected mice (dashed line) and are expressed as means ± standard error of the means. *, P < 0.05 versus distal promoter region (n = 3 mice/group).
Secretory proteins are induced in the liver during diverse infections.
To assess whether the induction of secretory protein genes in the liver is specific to pneumococcal pneumonia, or whether it is instead a more generalizable phenomenon that associates with the acute-phase response elicited by infection, we studied WT mice with pneumonia caused by a Gram-negative bacterium (E. coli) or with sepsis caused by cecal ligation and puncture (CLP). The livers were harvested 24 h after infection, and the expression of secretory protein genes was assessed by qRT-PCR. All of the secretory protein genes examined were elevated in response to both conditions (Fig. 8). The induction of secretory protein genes in the liver following diverse infectious challenges suggests that this pathway may be a fundamental component of the hepatic acute-phase response.
Fig 8.

Secretory protein gene induction in other severe infections. Quantitative RT-PCR was performed on liver RNA collected from ICR mice subjected to CLP-induced sepsis (A) or C57BL/6 mice that received intratracheal instillations of E. coli (B). After 24 h, the livers were harvested for RNA extraction. Fold induction values for the indicated genes (compared to uninfected controls) are means ± standard errors of the means. *, P < 0.05 compared to uninfected mice (each panel represents a different experiment, each with 3 to 5 mice/group).
DISCUSSION
These studies confirmed inferences from microarray analyses (8) and revealed that specific secretory pathways are induced in the liver in association with the acute-phase response during infection. Moreover, they have provided the first evidence to our knowledge that STAT3 is directly involved in the induction of secretory proteins.
Bacterial pneumonia triggers the activation of both STAT3 and RelA in the liver (7, 8). These transcription factors in hepatocytes mediate blood proteome remodeling during pneumonia, which curbs bacterial dissemination (8). The processing of this newly synthesized load of acute-phase proteins would be expected to put significant pressure on the secretory apparatus of hepatocytes. The present studies showed that genes involved in the UPR and in the protein secretion pathway are induced in the liver during severe infections, and this depends on STAT3. After integrating all of these data, we propose that STAT3 is involved in multiple stages of the pathway leading to secretory protein induction during the acute-phase response (Fig. 9).
Fig 9.

Proposed model of STAT3 contributions to the acute-phase response elicited by pneumonia. During pneumonia, hepatocyte STAT3 is activated by IL-6 and also by ER stress resulting from the newly synthesized load of acute-phase proteins. STAT3 induces the expression of secretory pathway-related proteins, such as those in translocon, chaperone, and coat protein complexes, enhancing the extracellular delivery of the newly translated acute-phase proteins to mediate innate immunity in the bloodstream.
The transcription of acute-phase proteins driven by STAT3 contributes to the expression of a protein load in the ER that triggers ER stress. Secretory cells such as hepatocytes have a well-developed ER, which facilitates efficient processing of secreted proteins (24). When this secretory capacity becomes overwhelmed, ER stress induces the UPR, which increases expression of protein folding and secretion machinery (14). During bacterial pneumonia, IL-6 stimulates STAT3-driven expression of acute-phase transcripts (7, 8), which are then translated via the translocon into the ER. This initial protein load can likely be handled by the constitutively expressed secretory machinery of hepatocytes, but as the acute-phase response continues to increase through 24 h (7), the folding and vesicular transport capacity of the ER in hepatocytes becomes overwhelmed. As a result, misfolded proteins accumulate and deplete the reserve of chaperones and foldases in the ER, causing ER stress and triggering the UPR (Fig. 9). The UPR then increases expression of multiple proteins involved in translocation and folding in the ER as well as in vesicular transport between the ER and Golgi complex. Consistent with this, we observed multiple markers of ER stress in the liver during pneumonia, including GRP78, CHOP, ATF4, and ERp72, most of which were diminished by the interruption of STAT3. These data suggest that STAT3 can function upstream of ER stress in the liver during bacterial pneumonia.
In addition, STAT3 may directly drive the transcription of secretory protein genes. STAT3 binds to the promoter regions of secretory protein genes during pneumonia. During pneumonia, STAT3 activation in the liver requires IL-6 (7), which may form a direct conduit to secretory protein expression (Fig. 9). Furthermore, tunicamycin administration in vivo led to phosphorylation of STAT3 and the STAT3-dependent expression of secretory proteins in the liver, suggesting that STAT3 can function downstream of ER stress. Autocrine, paracrine, or endocrine signaling from IL-6 or other cytokines may activate STAT3 in this setting. Alternatively, it is conceivable that intracellular pathways triggered by ER stress can directly activate STAT3 in hepatocytes. Prior studies revealed connections between ER resident proteins, such as ERp57, and STAT3 transcriptional activity (25, 26), although the mechanisms and significance are unclear. An intriguing speculation is that protein kinase R-like ER kinase (PERK), an ER membrane-localized tyrosine kinase that becomes activated in the UPR, might phosphorylate STAT3 and thereby directly link ER stress to STAT3 activity (27, 28). STAT3 may be activated by multiple different pathways during the acute-phase response, resulting in its association with proximal promoter regions of secretory protein genes with STAT3-dependent induction of these transcripts.
To our knowledge, secretory protein gene induction has previously been demonstrated to be mediated only by traditional transcription factors of the UPR, including XBP-1, ATF6, and ATF4 (14). As with perhaps any gene, secretory protein gene transcription is certain to be mediated by multiple transcription factors acting cooperatively. The dependence on STAT3 in the present studies does not suggest an independence from other transcription factors previously implicated in the UPR. However, the present data provide the first demonstration, to our knowledge, that STAT3 is involved in either the UPR or secretory protein expression.
Finally, it will be important to determine the functional significance of STAT3-mediated induction of secretory proteins during the acute-phase response. The increased secretory machinery should enhance acute-phase protein translation into the ER, acute-phase protein folding in the ER, and the passage of acute-phase proteins from the ER to the Golgi apparatus. The net effects of these processes should be 2-fold, to promote immunity and to limit injury. Increased acute-phase proteins in the blood enhances humoral and phagocytic defenses against bacteria (8, 9, 13), and so enhancing delivery of this secreted cargo to the blood compartment is an innate immune response that improves systemic defense. Furthermore, sustained or severe ER stress results in cell death (14), and the relief of ER stress due to enhanced secretory machinery during the acute-phase response may help prevent liver injury during severe infection. Altogether, these studies identify STAT3 as a novel mediator of the UPR during the acute-phase response. We propose that, in addition to driving the expression of acute-phase proteins, STAT3 functions in the liver during the acute-phase response to enhance the delivery of blood-borne innate immunity proteins and to limit liver injury.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants R01 HL-068153, R01 HL-079392, R00 HL-092956, and T32 HL-007035.
We thank Shizuo Akira (Osaka, Japan) and Roland Schmid (Munich, Germany) for providing the mice with LoxP sites in the genes for STAT3 and RelA, respectively. We thank Jean-Bosco Tagne (Boston, MA) for technical assistance with the ChIP assays.
Footnotes
Published ahead of print 4 March 2013
REFERENCES
- 1. Mizgerd JP. 2006. Lung infection: a public health priority. PLoS Med. 3:e76 doi:10.1371/journal.pmed.0030076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizgerd JP. 2012. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am. J. Respir. Crit. Care Med. 186:824–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizgerd JP. 2008. Acute lower respiratory tract infection. N. Engl. J. Med. 358:716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quinton LJ, Jones MR, Simms BT, Kogan MS, Robson BE, Skerrett SJ, Mizgerd JP. 2007. Functions and regulation of NF-κB RelA during pneumococcal pneumonia. J. Immunol. 178:1896–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung cxc chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. 2005. Lung NF-κB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J. Immunol. 175:7530–7535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quinton LJ, Jones MR, Robson BE, Mizgerd JP. 2009. Mechanisms of the hepatic acute-phase response during bacterial pneumonia. Infect. Immun. 77:2417–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinton LJ, Blahna MT, Jones MR, Allen E, Ferrari JD, Hilliard KL, Zhang X, Sabharwal V, Algul H, Akira S, Schmid RM, Pelton SI, Spira A, Mizgerd JP. 2012. Hepatocyte-specific mutation of both NF-κB RelA and STAT3 abrogates the acute phase response in mice. J. Clin. Invest. 122:1758–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD. 2008. Structural recognition and functional activation of FcγR by innate pentraxins. Nature 456:989–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Renckens R, Roelofs JJ, Knapp S, de Vos AF, Florquin S, van der Poll T. 2006. The acute-phase response and serum amyloid a inhibit the inflammatory response to Acinetobacter baumannii pneumonia. J. Infect. Dis. 193:187–195 [DOI] [PubMed] [Google Scholar]
- 11. Sakamori R, Takehara T, Ohnishi C, Tatsumi T, Ohkawa K, Takeda K, Akira S, Hayashi N. 2007. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology 46:1564–1573 [DOI] [PubMed] [Google Scholar]
- 12. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, Blander JM, Tacke F, Trautwein C. 2010. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207:1453–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuste J, Botto M, Bottoms SE, Brown JS. 2007. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog. 3:1208–1219 doi:10.1371/journal.ppat.0030120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hetz C. 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13:89–102 [DOI] [PubMed] [Google Scholar]
- 15. Luebke-Wheeler J, Zhang K, Battle M, Si-Tayeb K, Garrison W, Chhinder S, Li J, Kaufman RJ, Duncan SA. 2008. Hepatocyte nuclear factor 4α is implicated in endoplasmic reticulum stress-induced acute phase response by regulating expression of cyclic adenosine monophosphate responsive element binding protein H. Hepatology 48:1242–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, Back SH, Kaufman RJ. 2006. Endoplasmic reticulum stress activates cleavage of crebh to induce a systemic inflammatory response. Cell 124:587–599 [DOI] [PubMed] [Google Scholar]
- 17. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. 1998. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 161:4652–4660 [PubMed] [Google Scholar]
- 18. Algul H, Treiber M, Lesina M, Nakhai H, Saur D, Geisler F, Pfeifer A, Paxian S, Schmid RM. 2007. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J. Clin. Invest. 117:1490–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Terao A, Steininger TL, Hyder K, Apte-Deshpande A, Ding J, Rishipathak D, Davis RW, Heller HC, Kilduff TS. 2003. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience 116:187–200 [DOI] [PubMed] [Google Scholar]
- 20. Jones MR, Quinton LJ, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. 2006. Roles of interleukin-6 in activation of Stat proteins and recruitment of neutrophils during Escherichia coli pneumonia. J. Infect. Dis. 193:360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. 2000. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal. Biochem. 285:194–204 [DOI] [PubMed] [Google Scholar]
- 22. Ahyi AN, Chang HC, Dent Nutt ALSL, Kaplan MH. 2009. IFN regulatory factor 4 regulates the expression of a subset of Th2 cytokines. J. Immunol. 183:1598–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee AS. 2005. The ER chaperone and signaling regulator GRP78/Bip as a monitor of endoplasmic reticulum stress. Methods 35:373–381 [DOI] [PubMed] [Google Scholar]
- 24. Nunnari J, Walter P. 1996. Regulation of organelle biogenesis. Cell 84:389–394 [DOI] [PubMed] [Google Scholar]
- 25. Coe H, Jung J, Groenendyk J, Prins D, Michalak M. 2010. Erp57 modulates Stat3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 285:6725–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Eufemi M, Coppari S, Altieri F, Grillo C, Ferraro A, Turano C. 2004. Erp57 is present in Stat3-DNA complexes. Biochem. Biophys. Res. Commun. 323:1306–1312 [DOI] [PubMed] [Google Scholar]
- 27. Malhi H, Kaufman RJ. 2011. Endoplasmic reticulum stress in liver disease. J. Hepatol. 54:795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, Koromilas AE. 2008. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J. Biol. Chem. 283:469–475 [DOI] [PubMed] [Google Scholar]