

Abstract
Brucella abortus is recognized by several Toll-like receptor (TLR)-associated pathways triggering proinflammatory responses that affect both the nature and intensity of the immune response. Previously, we demonstrated that B. abortus-mediated dendritic cell (DC) maturation and control of infection are dependent on the adaptor molecule MyD88. However, the involvement of all TLRs in response to B. abortus infection is not completely understood. Therefore, we decided to evaluate the requirement for TLR6 in host resistance to B. abortus. Here, we demonstrated that TLR6 is an important component for triggering an innate immune response against B. abortus. An in vitro luciferase assay indicated that TLR6 cooperates with TLR2 to sense Brucella and further activates NF-κB signaling. However, in vivo analysis showed that TLR6, not TLR2, is required for the efficient control of B. abortus infection. Additionally, B. abortus-infected dendritic cells require TLR6 to induce tumor necrosis factor alpha (TNF-α) and interleukin-12 (IL-12). Furthermore, our findings demonstrated that the mitogen-activated protein kinase (MAPK) signaling pathway is impaired in TLR2, TLR6, and TLR2/6 knockout (KO) DCs when infected with B. abortus, which may account for the lower proinflammatory cytokine production observed in TLR6 KO mouse dendritic cells. In summary, the results presented here indicate that TLR6 is required to trigger innate immune responses against B. abortus in vivo and is required for the full activation of DCs to induce robust proinflammatory cytokine production.
INTRODUCTION
The innate immune system is the first line of the defensive mechanisms that protect hosts from invading pathogens. The innate response begins with the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs), triggering intracellular signal transduction that culminates with the transcription of multiple genes that can facilitate efficient pathogen control (1). Among the functional distinct classes of PRRs, Toll-like receptors (TLRs) are the best characterized and are intensively studied due to their ability to recognize a broad range of PAMPs from a variety of pathogens (2). PAMP recognition by its cognate TLR activates intracellular signaling pathways that are mediated by adaptor molecules, such as myeloid differentiation factor 88 (MyD88), resulting in the induction of inflammatory cytokines, chemokines, and interferons (IFNs) and the upregulation of costimulatory molecules (3).
The Gram-negative bacterium Brucella abortus is an intracellular pathogen that not only is able to replicate and survive in macrophages but is capable of infecting nonprofessional phagocyte cells, causing brucellosis in humans and cattle (4). In humans, B. abortus causes undulant fever, endocarditis, arthritis, and osteomyelitis; in animals, it leads to abortion and infertility, resulting in serious economic losses (5, 6). Protection against B. abortus infection requires cell-mediated immunity, which includes CD4+ and CD8+ T lymphocytes, Th1-type cytokines such as IFN-γ and tumor necrosis factor alpha (TNF-α), and activated macrophages and dendritic cells (DCs) (7–9). Therefore, host control of infection requires a set of cells and factors, and together these promote a complex response against Brucella (10).
B. abortus is recognized by several TLR-associated pathways, triggering proinflammatory responses that affect both the nature and the intensity of the immune response (11, 12). Previously, we demonstrated that B. abortus-mediated DC maturation is dependent on the adaptor molecule MyD88 and that MyD88 knockout (KO) mice are highly susceptible to B. abortus infection in vivo compared to wild-type mice (13). However, the involvement of the various TLRs that trigger an effective response is not completely understood. Our group has demonstrated that TLR9 appears to have the most prominent role in host resistance to B. abortus infection among the TLRs, because TLR9 KO infected mice had enhanced susceptibility to B. abortus compared to wild-type mice (13). Nevertheless, the more susceptible phenotype observed in MyD88 KO mice indicates the involvement of other TLRs in host resistance to Brucella infection. Therefore, the concept of a combinatorial repertoire of TLRs to increase the range of PAMPs that are recognized is emerging (14).
TLR6 and TLR2 both are recruited to the macrophage phagosome, where they recognize bacterial peptidoglycan and lipoproteins (15). TLR6 plays a role in the recognition of bacterial diacylated lipopeptides such as MALP2, but it is not essential for cytokine production in response to triacylated lipopeptides. In contrast, TLR2 is essential to signaling in response to both of the aforementioned PAMPs (16). TLR2 clearly does not play a role in controlling B. abortus infection in vivo; however, signaling through this receptor apparently contributes to the full production of proinflammatory cytokines by innate immune cells (17, 18). The main goal of this study was to evaluate the requirement of TLR6 for host resistance to B. abortus and the role of this receptor in the induction of an effective innate immune response against this bacterium. Here, we demonstrated for the first time that TLR6 is an important component for triggering an innate immune response against B. abortus and inducing full resistance against infection. However, it is not necessary for production of IFN-γ and TNF-α by splenic cells in vitro. Nevertheless, bone marrow-derived dendritic cells (BMDCs) require TLR6 to induce interleukin-12 (IL-12) and TNF-α responses mediated by Brucella. Furthermore, our findings demonstrate that the mitogen-activated protein kinase (MAPK) signaling pathway is reduced in TLR2, TLR6, and TLR2/6 KO BMDCs when infected with B. abortus. Taken together, the results presented in this study indicate that TLR6 is critical for initiating innate immune responses against Brucella abortus in dendritic cells, which are related to proinflammatory cytokine production and resistance to infection.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with Brazilian laws 6638 and 9605 regarding animal experimentation. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Federal University of Minas Gerais (permit number CETEA89/2008).
Mice.
TLR2, TLR6, or MyD88 KO mice were kindly provided by Shizuo Akira, Osaka University, Japan. The double knockout mice (TLR2/6 KO) were generated by crossing TLR2 and TLR6 KO mice. Mice from the F2 generation were genotyped using a PCR-based method to identify the double knockout mice. Wild-type strain C57BL/6 mice were purchased from the Federal University of Minas Gerais animal facility (UFMG, Belo Horizonte, Brazil). Genetically deficient and control mice were maintained at UFMG and used at 6 to 8 weeks of age.
Bacteria.
Brucella abortus strain S2308 was obtained from our laboratory collection. The strain S2308 was grown in brucella broth liquid medium (BB) (Difco) at 37°C under constant agitation. After 3 days of growth, the bacterial culture was centrifuged and the pellet was resuspended in 0.15 M phosphate-buffered saline (PBS) (pH 7.4) (2.8 mM Na2PO4, 7.2 mM Na2HPO4, 0.14 M NaCl). Aliquots of these cultures were serially diluted and plated on BB containing 1.5% bacteriological agar. After incubation for 72 h at 37°C, bacterial numbers were determined by counting CFU.
Generation and in vitro stimulation of BMDCs.
Bone marrow cells were obtained from femora and tibiae of KO and wild-type mice, and they were differentiated into dendritic cells (BMDCs) as previously described (13). Briefly, bone marrow cells were cultured in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 20 ng/ml murine recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF). Petri dishes containing 1 × 107 cells were incubated at 37°C in an atmosphere of 5% CO2. On day 3 of incubation, a further 5 ml of fresh complete medium containing GM-CSF was added, and on days 5 and 7, 3 ml of medium was replaced with fresh supplemented medium containing GM-CSF. On day 10, nonadherent cells were harvested and seeded in 24-well plates (5 × 105 cells/well). Stimulation of the BMDCs was performed by adding supplemented DMEM (500 μl/well) containing B. abortus (multiplicity of infection [MOI], 100:1), Pam3CSK4 (1 μg/ml; InvivoGen, San Diego, CA), Malp-2 (0.1 μg/ml; Alexis Biochemicals, New York, NY), Escherichia coli lipopolysaccharide (LPS) (1 μg/ml; Sigma-Aldrich), or B. abortus lipidated outer membrane protein-19 (L-Omp19) (500 ng/ml). Culture supernatants of BMDCs were collected after 24 h of stimulation and assayed for the concentrations of IL-12 and TNF-α by enzyme-linked immunosorbent assay (ELISA) (R&D Systems).
Infection and Brucella counts in spleens.
Mice were infected intraperitoneally (i.p.) with 106 CFU of B. abortus strain 2308. To determine residual Brucella CFU in the spleens of mice, five animals from each group were examined at 1, 3, and 6 weeks after infection. Spleens from individual animals were homogenized in PBS, serially diluted 10-fold, and plated on Brucella broth agar (Difco, BD-Pharmingen, San Diego, CA). Plates were incubated at 37°C, and the CFU were counted after 3 days as previously described (19).
Cytokine production by splenocytes and BMDCs.
Splenocyte cultures from KO and wild-type mice were stimulated by the addition of 103 live B. abortus strain S2308 bacteria per cell or with medium as a negative control. Spleen cells were incubated at 37°C with 5% CO2. The supernatant of splenocyte cultures was collected at 48 and 72 h after B. abortus infection to detect TNF-α and IFN-γ, respectively. Additionally, BMDC IL-12 and TNF-α were measured at 24 h after antigen (Ag) stimulation. Levels of cytokines were measured using a commercially available ELISA Duoset kit (R&D Systems, MN).
Real-time RT-PCR.
Bone marrow-derived macrophages (BMDMs) or BMDCs from C57BL/6 mice were infected with B. abortus strain 2308 for 24 h. The infected cells were homogenized in TRIzol reagent (Invitrogen) to isolate total RNA. Reverse transcription (RT) of 1 μg of total RNA was performed using Illustra Ready-To-Go RT-PCR Beads (GE Healthcare, Buckinghamshire, United Kingdom) according to the manufacturer's instructions. Real-time RT-PCR was conducted with a final volume of 10 μl containing SYBR green PCR Master Mix (Applied Biosystems, Carlsbad, CA), oligo(dT) cDNA as the PCR template, and 10 μM primers. Real-time RT-PCR was performed with the ABI 7900 real-time PCR system (Applied Biosystems) using the following cycling parameters: 60°C for 10 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min, and a dissociation stage of 95°C for 15 s, 60°C for 1 min, 95°C for 15 s, and 60°C for 15 s. Primers were used to amplify a specific 100- to 120-bp fragment corresponding to a specific gene. The primers used for the TLR1, TLR2, TLR6, and β-actin genes were as follows: TLR1 forward, 5′-CCGTGATGCACAGCTCCTTGGTTT-3′ ; TLR1 reverse, 5′-TGTCCACAATTGCCTCTGCTCGC-3′ ; TLR2 forward, 5′-GCTGGCGACCGGGAAGTTCG-3′; TLR2 reverse, 5′-TCTCCTGCCAGTGACCGCCT-3′; TLR6 forward, 5′-AGAAAATGGTACCGTCAGTGCTGGA-3′; TLR6 reverse, 5′-AGGCCAGGGCGCAAACAAAG-3′; β-actin forward, 5′-AGGTGTGCACCTTTTATTGGTCTCAA-3′; and β-actin reverse, 5′-TGTATGAAGGTTTGGTCTCCCT-3′. All data are presented as relative expression units after normalization to the β-actin gene, and measurements were conducted in triplicate.
Western blot analysis.
Derived BMDCs were seeded in 24-well plates (5 × 105 cells/well) as described above. On day 10, cells were serum deprived for 16 h and then treated with B. abortus (MOI, 1,000:1), medium, or LPS for 30 min. The appropriate time interval for cell stimulation was determined as the peak of MAPK activation for BMDCs (data not shown). After treatment, cells were washed with Hanks balanced salt solution (HBSS) (Gibco) at room temperature and lysed in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 50 mM NaF, 10 mM β-glycerophosphate, 0.1 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM sodium orthovanadate, and 1:100 protease inhibitor cocktail (Sigma-Aldrich). Protein concentrations were determined by bicinchoninic acid assays. Equal amounts of protein (25 μg) were loaded onto 12% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes (GE Healthcare, Buckinghamshire, United Kingdom) according to standard techniques. Membranes were blocked for 1 h at room temperature with blocking buffer (Tris-buffered saline [TBS] containing 0.1% Tween 20 and 5% nonfat dry milk) before incubation with rabbit anti-mouse phospho-Jun N-terminal protein kinase (phospho-JNK) (clone 81E11), phospho-p38 (clone D3F9), and β-actin (clone 13E5) overnight at 4°C. Subsequently, membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG for 1 h at room temperature, and the protein bands were visualized using the Luminol chemiluminescent HRP substrate (Millipore, Billerica, MA) in a Storm System 860 scanner (GE Healthcare). Monoclonal antibodies were purchased from Cell Signaling Technology Inc. (Danvers, MA). The analysis of densitometry was performed using Kodak software version 1D-3.5.
Luciferase reporter assay.
HEK 293 cells (ATCC, Manassas, VA) were cultured in DMEM that was supplemented with 10% FBS. Cells were plated in 24-well tissue culture plates (106 cells/well) and were maintained in the above-described medium for 24 h. Cells were transfected using polyethylenimine (PEI) with 1 μg of pcDNA3 that contained cDNA for either TLR1, TLR2, TLR6, TLR2/1, or TLR2/6 or empty vector DNA (pcDNA3; Invitrogen) and 200 ng of NF-κB-Luc plasmid that contained a synthetic firefly luc2 (Photinius pyralis) gene under the control of the NF-κB promoter. All cells were also transfected with 50 ng of the pRL-TK control plasmid, which constitutively expressed the Renilla hRluc (Renilla reniformis) luciferase gene for the normalization of transfection efficiencies. After transfections, the cells were maintained overnight in DMEM with 1% FBS. The following day, cells were either untreated, incubated with Pam3CSK4 (1 μg/ml; InvivoGen), Malp-2 (0.1 μg/ml; Alexis Biochemicals), or L-Omp19 (500 ng/ml), or infected with B. abortus (MOI, 100:1) for 8 h at 37°C with 5% CO2. Next, cells were harvested in lysis buffer and assayed for luciferase activity with the dual-luciferase reporter assay system (Promega, Madison, WI), according to the manufacturer's protocol. Briefly, 5 μl of cell lysate was added to 45 μl of luciferase assay reagent II (Promega), and the luminescence (FLuc) was quantified with a luminometer (LumiCount-Packard). Then, 45 μl of Stop & Glo reagent was added to the mixture, and the luminescence was quantified again with a luminometer (RLuc). The results are expressed as the means of the FLuc/RLuc ratios.
Statistical analysis.
The differences in CFU and cytokine levels between wild-type and KO mice were analyzed using Student's t test with a two-tailed distribution. Luciferase induction or differences in the relative expression of target genes were evaluated by analysis of variance (ANOVA) followed by Tukey's post hoc test. A P value of 0.05 or lower was considered significant.
RESULTS
TLR6 and TLR2 sense Brucella and act synergistically to induce NF-κB activation.
Because TLR2 contributes to the full production of proinflammatory cytokines by innate cells after B. abortus infection (17, 20) and this receptor recruits TLR1 or TLR6 to trigger efficient intracellular signaling, a luciferase assay was used to evaluate the roles of TLR1, TLR2, and TLR6 in NF-κB activation. NF-κB is activated after the recognition of a pathogen by innate immune cells, therefore inducing proinflammatory cytokine expression to control the infection (21). Thus, HEK 293A cells were transfected with a vector containing a firefly luciferase gene under the control of the NF-κB promoter, a constitutive TLR expression vector, and, to normalize the transfection process, a vector with constitutive expression of the Renilla luciferase gene. NF-κB activation was observed in cells expressing TLR2 or TLR6 and stimulated with B. abortus and not in cells transfected with TLR1 alone (Fig. 1). However, when the cells expressed TLR1 and TLR2, the activation of NF-κB increased at the same level as in cells with TLR2 alone. Furthermore, when the TLR2 and TLR6 genes were cotransfected in HEK cells and these cells were infected with Brucella, we observed a synergistic effect, based on increased luciferase activity when the cells were compared to those transfected with each receptor gene separately. These findings demonstrate an in vitro cooperation between TLR2 and TLR6 that results in the activation of NF-κB in cells infected with B. abortus. Additionally, Pam3CSK4 or MALP-2 induced NF-κB activation in TLR2-transfected HEK cells. However, when HEK cells were transfected with TLR2/1 or TLR2/6 and stimulated with Pam3CSK4, we observed higher levels of NF-κB activation than in cells transfected with TLR2 alone. Similarly, when cells were transfected with TLR2/TLR6 and stimulated with MALP-2, we observed a synergistic effect based on increased NF-κB activation compared to cells transfected with TLR2 alone. Furthermore, the results presented here confirmed that L-Omp19 is a TLR2 agonist and not a TLR6 agonist as previously described (20).
Fig 1.
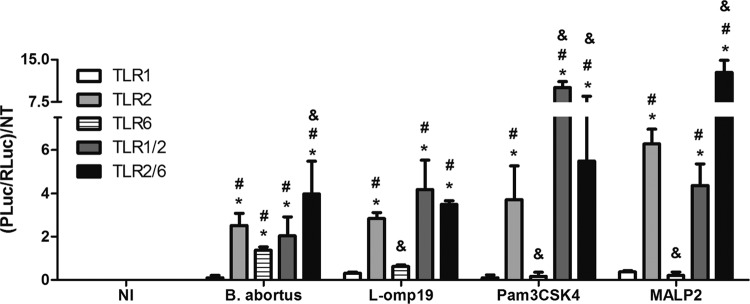
TLR2 and TLR6 cooperate to detect B. abortus and induce NF-κB activation. HEK 293A cells were transiently transfected with plasmids to express TLRs individually (TLR1, TLR2, or TLR6) or in combination (TLR1/TLR2 or TLR2/TLR6). These were stimulated for 24 h with viable B. abortus (MOI, 1:100), Pam3CSK4 (1 μg/ml), MALP2 (100 ng/ml), or L-Omp19 (500 ng/ml). The cells were disrupted with 1× lysis buffer (Promega) and assayed for firefly luciferase normalized with Renilla luciferase. Data are expressed as the mean ± standard deviation (SD). Each experiment was performed three times, and data represent one of the three experiments. Statistically significant differences relative to noninfected cells (NI) are denoted by *, relative to TLR1 by #, and relative to TLR2 by & (P < 0.05).
TLR2 and TLR6 expression is enhanced in Brucella-infected murine macrophages and dendritic cells.
Macrophages and dendritic cells are important cell types in the initial response to B. abortus infection. To investigate whether B. abortus infection activates the expression of the TLR1, TLR2, or TLR6 gene, C57BL/6 mouse BMDMs or BMDCs were infected with B. abortus strain 2308, and the mRNA levels for these receptors were evaluated. The results demonstrated that only TLR1 was not highly upregulated, compared to the augmented levels of TLR2 and TLR6 mRNA expression in both cell types (Fig. 2). In Brucella-infected dendritic cells, TLR6 mRNA levels were greater than TLR2 levels. Nonetheless, all TLRs tested demonstrated statistically significant differences compared to levels in noninfected cells. In conclusion, TLR2 and TLR6 showed higher levels of expression than TLR1 in macrophages and dendritic cells at 24 h following B. abortus infection.
Fig 2.
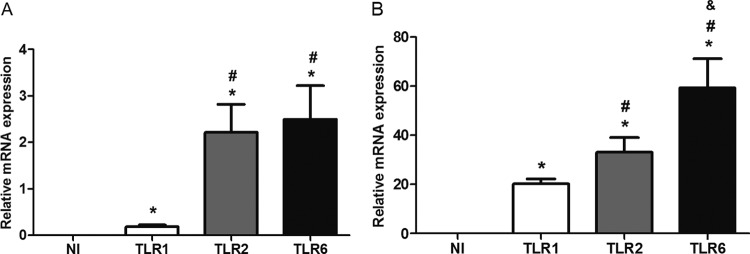
TLR2 and TLR6 mRNAs are expressed in murine macrophages and dendritic cells after B. abortus infection. BMDMs (A) or BMDCs (B) from C57BL/6 mice were infected with B. abortus S2308 strain (MOI, 1:1000) or treated with medium alone (NI). Total RNA was extracted at 24 h postinfection, and cDNA was generated. The primary transcript levels of TLR1, TLR2, and TLR6 were determined using real-time PCR. The data are expressed as the mean ± SD of the relative levels of mRNA after normalization with β-actin mRNA expression. This experiment was performed three times, and the results are representative of one experiment with similar results. Statistically significant differences relative to noninfected cells (NI) are denoted by *, relative to TLR1 by #, and relative to TLR2 by & (P < 0.05).
TLR6 is required to induce proinflammatory cytokine production by dendritic cells.
The recognition of Brucella by innate immune cells, such as macrophages and dendritic cells, results in the activation and the concomitant production of proinflammatory cytokines. In this study, we observed a major reduction in IL-12 (Fig. 3A) and TNF-α (Fig. 3B) synthesis in the supernatants of BMDCs from TLR6 KO mice compared to BMDCs derived from C57BL/6 mice. Furthermore, we observed a greater reduction of IL-12 expression than of TNF-α expression in Brucella-infected TLR6 KO cells. Additionally, a major reduction in IL-12 and TNF-α synthesis was detected in TLR2 KO BMDCs compared to wild-type BMDCs, and a similar profile was observed in TLR2/TLR6 KO cells. Further, when we used Brucella L-Omp19 as an agonist, we confirmed the requirement for TLR2, not TLR6, for BMDC activation. The effect of TLR2 dependency on L-Omp19 activation of BMDCs was more pronounced for TNF-α production than for IL-12 production. In sum, these results demonstrate that TLR6 is required for the full production of IL-12 and TNF-α in Brucella-infected dendritic cells.
Fig 3.

TLR6 is required to induce proinflammatory cytokine production by BMDCs infected with B. abortus. Bone marrow from C57BL/6, TLR2 KO, TLR6 KO, or TLR2/6 KO mice was differentiated in dendritic cells and infected with B. abortus S2308 (MOI, 1:100), Pam3CSK4 (1 μg/ml), MALP2 (100 ng/ml), or L-Omp19 (500 ng/ml) or stimulated with E. coli LPS (1 μg/ml). Supernatants were harvested for measuring IL-12 (A) and TNF-α (B) at 24 h after infection/stimulation by ELISA. Statistically significant differences relative to medium (nonstimulated cells) are denoted by * and those relative to C57BL/6 by # (P < 0.05).
TLR2 and TLR6 recognition of Brucella activates the MAPK intracellular signaling pathway in dendritic cells.
Following TLR stimulation, intracellular signaling events determine the effector functions of activated cells. To investigate the role of TLR2 or TLR6 in the cell signaling pathway induced by B. abortus infection, dendritic cells were stimulated with live bacteria for 30 min, and the components of the MAPK (p38) and JNK pathways were analyzed by examining protein phosphorylation. As observed in Fig. 4, the phosphorylation of c-Jun N-terminal kinases (JNKs) induced by Brucella was abrogated in TLR2, TLR6, TLR2/6, and MyD88 KO dendritic cells. The activation of p38 was reduced in TLR6 KO cells infected with B. abortus, reaching the same level of activation observed in MyD88 KO cells. These results suggest that after binding TLR2 and TLR6, B. abortus activates JNK and p38 signaling pathways to induce proinflammatory cytokines in dendritic cells.
Fig 4.
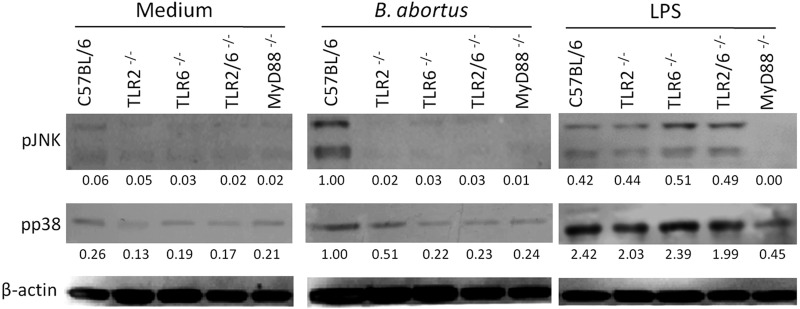
MAPK signaling pathways triggered by B. abortus are dependent on TLR6 in dendritic cells. BMDCs from wild-type C57BL/6, TLR2 KO, TLR6 KO, TLR2/6 KO, and MyD88 KO mice were treated for 30 min with B. abortus strain S2308 (MOI, 1:1,000), LPS (1 μg/ml), or medium alone. Cell lysates were then subjected to Western blot assay using anti-phospho-JNK and anti-phospho-p38 antibodies. β-Actin was used as a loading control. Values below each band indicate the quantification of band intensity relative to the loading control. Data are representative of two independent experiments.
TLR6 is required for the efficient control of Brucella infection in vivo.
In this study, we demonstrate cooperation between TLR2 and TLR6 signaling through NF-κB activation in vitro. Therefore, we generated TLR2/TLR6 double knockout mice to evaluate the contributions of both TLRs in vivo in an innate immune response against B abortus. It was previously demonstrated and confirmed that TLR2 does not play any role in controlling B. abortus infection in vivo (17, 18); however, the participation of TLR6 had not yet been evaluated. Thus, TLR2, TLR6, TLR2/6, or MyD88 KO mice were infected intraperitoneally with 1 × 106 CFU of B. abortus, and the course of infection was observed for 6 weeks. Confirming previous results obtained by Campos et al. (17) and Weiss et al. (18), TLR2 was found to play no role in controlling B. abortus infection in vivo (Fig. 5). On the other hand, it was observed that TLR6 KO mice were more susceptible to B. abortus infection than the wild-type C57BL/6 mice at all time points analyzed (Fig. 5). Furthermore, TLR2/6 KO mice did not demonstrate increased susceptibility to B. abortus infection compared to TLR6 KO animals. These findings suggest that TLR2 is totally dispensable for host innate immune control of this bacterial infection in vivo. However, the enhanced susceptibility of TLR6 KO animals confirms that this receptor participates in cell signaling to activate innate immune responses and resistance against B. abortus infection.
Fig 5.
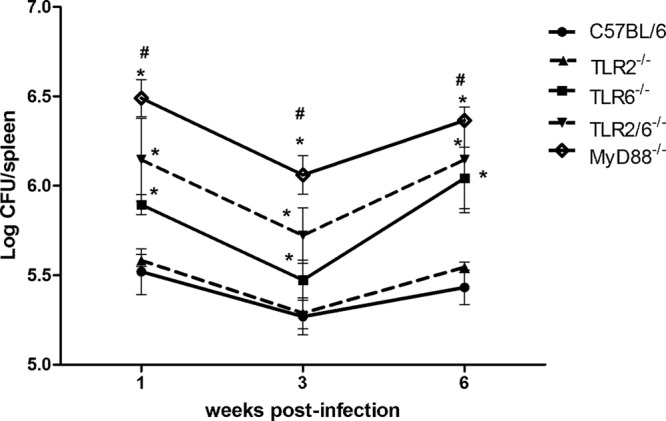
TLR6 is required for full resistance to B. abortus infection in vivo. C57BL/6, TLR2 KO, TLR6 KO, TLR2/6 KO, and MyD88 KO mice were infected, and the number of bacteria in the spleens was analyzed by CFU counting at 1, 3, and 6 weeks after infection. Data are expressed mean ± SD for five animals per group. The results are representative of four independent experiments. Statistically significant differences relative to C57BL/6 and TLR2 KO mice are denoted by * and those relative to TLR6 and TLR2/6 KO mice by # (P < 0.05).
TLR2 but not TLR6 is partially required for production of TNF-α in splenocytes from mice infected with Brucella.
Protective immunity against infection by B. abortus is directly related to the induction of a type 1 pattern of immune response. IFN-γ and TNF-α are key cytokines involved in this type of immunity. Although TLR2 is not required for resistance against B. abortus in vivo, this receptor seems to be important for generating a proinflammatory response against this bacterium (17, 20). Here, we showed that TLR6 KO mice were more susceptible to B. abortus infection than C57BL/6 and TLR2 KO animals. To determine whether this susceptibility is related to a deficiency in proinflammatory cytokine production, splenocytes from Brucella-primed C57BL/6, TLR2 KO, TLR6 KO, and TLR2/6 KO mice were infected, and IFN-γ or TNF-α was measured in cell supernatants. TLR2 KO cells produced lower levels of TNF-α but not IFN-γ than C57BL/6 cells when stimulated with B. abortus at 3 and 6 weeks postinfection (Fig. 6A and B). In contrast, TLR6 KO cells did not demonstrate any difference in IFN-γ (Fig. 6A) or TNF-α (Fig. 6B) production compared to wild-type cells. These results demonstrate that despite the importance of TLR6 in host control of B. abortus infection, this innate immune receptor is not required for inducing IFN-γ or TNF-α production in splenocytes infected with Brucella. According to our results, splenocytes from TLR2/6 KO mice showed the same profile of proinflammatory cytokine production as TLR2 KO cells (Fig. 6A and B). Taken together, these results demonstrated that TLR6 is not required for proinflammatory cytokine production by splenocytes infected with this bacterium.
Fig 6.

TLR2 but not TLR6 is partially required for full production of TNF-α in splenocytes of Brucella-infected mice. Spleen cells were stimulated with B. abortus S2308 (MOI, 1:100) or medium as a negative control. Splenocyte supernatants were harvested for measuring IFN-γ (A) or TNF-α (B) levels by ELISA at 1, 3, or 6 weeks postinfection. Data are expressed the mean ± SD for five animals per group. These results are representative of three independent experiments. Statistically significant differences relative to medium are denoted by * and those relative to C57BL/6 mice by # (P < 0.05).
DISCUSSION
Since the description of TLRs by Medzhitov et al. (22), these innate immune receptors have been extensively studied and implicated in the recognition of different pathogenic bacteria (23). The recognition of a wide range of PAMPS, including lipids, proteins, lipoproteins, glycans, and nucleic acids, by TLRs positions this receptor family as the best characterized among innate immune cell receptors. Furthermore, TLRs are able to recruit adaptor intracellular proteins, triggering signal transduction for an efficient response against pathogens that culminates in the production of proinflammatory cytokines and the expression of costimulatory molecules, ultimately linking innate immunity and adaptive immunity (10–13).
The involvement of TLRs in host resistance to B. abortus infection was investigated using TLR2-, TLR4-, TLR9-, and MyD88-deficient mice. MyD88-dependent signaling was shown to be essential for the activation of IFN-γ-producing cells and dendritic cells during Brucella infection. Copin et al. (24) also demonstrated that MyD88 deficiency strongly reduces the number of IFN-γ-producing cells during Brucella melitensis infection. In a previous study, we demonstrated that B. abortus-mediated DC maturation is dependent on the adaptor molecule MyD88 (13); additionally, we showed that MyD88 KO mice are highly susceptible to B. abortus infection in vivo, and this result was also confirmed by Copin et al. using B. melitensis (24). Here, we confirmed the involvement of MyD88 in protection against B. abortus infection by showing the enhanced susceptibility of MyD88 KO mice at 1, 3, and 6 weeks after infection. Recently, Copin and colleagues characterized Brucella-infected cells by microscopy of the spleen and liver, demonstrating that the majority of these cells were from the macrophage lineage (25). These authors also showed that Brucella infection induces splenic dendritic cell migration to the infection site and the maturation of these cells and that effective immunity against this bacterium is dependent on MyD88 during liver granuloma formation. Furthermore, our group demonstrated that IL-1 receptor-associated kinase 4 (IRAK-4) signaling downstream of MyD88 is critical for triggering innate immune responses and resistance at the initial phase of Brucella infection (26).
Regarding TLRs, TLR2, TLR4, and TLR9 have been implicated in host responses to Brucella; however, to date, TLR9 appears to have the most important role in controlling this infection in vivo (13). The results observed using MyD88 KO mice strongly suggest that TLRs other than TLR9 may be related to an efficient immune response against B. abortus (10–13). In the context of PAMP recognition by TLRs, TLR1, TLR2, and TLR6 are extensively studied because these receptors establish a combinatorial repertoire to discriminate among the large number of ligands found in nature (15). TLR2 can form functional pairs with TLR6 or TLR1, and this interaction leads to cytokine induction after lipoprotein recognition (15). TLR2 has evolutionarily developed its unique ability to form heterodimers with TLR1 or TLR6 to attain specificity for the diverse lipoprotein repertoire (27). Studies have shown an important role of TLR2 and TLR6 acting in cooperation to recognize diacylated lipopeptides during bacterial infection (16). The phenol-soluble modulin (PSM), a complex of three small secreted polypeptides from Staphylococcus epidermidis, was shown to activate NF-κB and induce cytokine release in macrophages (28), suggesting that this complex signals through TLR2 and also requires TLR6 (29). Additionally, Bulut and colleagues demonstrated the requirement for the cooperation of TLR2 and TLR6 in cellular activation by Mycobacterium tuberculosis-conditioned culture supernatant soluble tuberculosis factor (STF) and Borrelia burgdorferi outer surface protein A lipoprotein (OspA-L) (14). Clearly, TLR2 is not involved in the host control of B. abortus infection, as demonstrated here and by others (17, 18), nor is it essential for the resolution of other Gram-negative bacterial infections such as those with Salmonella, Klebsiella, and Haemophilus (30, 31, 32). Despite the lack of a role for TLR2 in host resistance to B. abortus infection in vivo, signaling through this receptor contributes to the full production of proinflammatory cytokines by innate immune cells. The contribution of TLR2 in Brucella-induced cytokine production has already been shown in studies using live B. abortus (18), HKBA (33), or Brucella lipoproteins (20, 30). Moreover, this TLR2-mediated cytokine production has been shown to contribute to the inflammatory pathogenesis of brucellosis (34, 35). Here, we determined that L-Omp19, a model B. abortus lipoprotein, is recognized by TLR2 and not by TLR6. Further, L-Omp19 induces IL-12 and TNF-α production by BMDCs in a TLR2-dependent fashion. Several studies have reported that the recognition of Brucella L-Omp19 by TLR2 interferes with phagocytosis, major histocompatibility complex class II (MHC-II) expression and Ag processing in human monocyte/macrophages, which leads to reduced Ag-specific T-cell proliferation and IFN-γ production by peripheral blood mononuclear cells (PBMCs) of Brucella-infected patients (36, 37). Additionally, a recently identified Brucella protein, Btp1, is able to control dendritic cell function by interfering with the TLR2 signaling pathway (38). These studies suggest that TLR2 recognition of Brucella can actually downmodulate host innate and adaptive immune responses to this bacterium. These mechanisms provide a potential explanation for the resistance of TLR2 KO mice to infection compared to wild-type animals. Additionally, it is important to mention that the in vitro results represent one example of a specific cell type and that the in vivo responses to these pathogens are complex, involve different cell types and PRRs, and are therefore not easily predicted.
Microbial recognition by DCs constitutes a major step in the activation of immune mechanisms that lead to the development of resistance against many infectious agents. To determine whether TLR2 and TLR6 cooperate during Brucella infection, we generated double TLR2/TLR6 KO mice. In vivo analysis demonstrated that TLR2/6 KO mice were more susceptible to infection with B. abortus than wild-type mice. However, we detected similar Brucella CFU numbers in the spleens of TLR2/6 KO and TLR6 KO animals. The results presented here suggest that TLR6 interacts with TLR2 to induce innate immune responses against Brucella PAMPs in vitro; however, TLR2 plays no role in B. abortus control in vivo. On the other hand, we have shown for the first time that TLR6 is important for host resistance against Brucella infection. Initial human studies with mycobacteria demonstrated that TLR6 polymorphisms were associated with lower IL-6 production, NF-κB signaling, and increased risks for tuberculosis (TB) disease in certain populations (39, 40). Huang et al. have shown that B. abortus can induce mouse DCs to migrate to T-cell areas in the spleen, where they secrete IL-12 (41). Furthermore, in vivo protection against B. abortus was shown to require IL-12 and IFN-γ (42, 43). Here, we did not observe a significant reduction in IFN-γ production in the spleen cells of TLR6 KO mice compared to C57BL/6 mice, although TLR-deficient animals are more susceptible to infection. However, we detected a major reduction in IL-12 production by TLR6 KO dendritic cells compared to that in wild-type animals. It is possible that the reduced levels of IL-12 detected in TLR6 KO BMDCs affects IFN-γ production in tissues other than the spleen or interferes directly with T-cell activation. In addition to their expression in professional antigen-presenting cells (APCs), such as DCs and macrophages, TLRs can be expressed in T cells and serve as costimulatory signals in T-cell activation. It has been demonstrated that activation of TLR6 expressed in T cells blocks the suppressive function of Treg cells, activating effector lymphocytes (44). In contrast, TLR2 agonists can act directly on purified Treg cells, resulting in augmented Treg cell proliferation and, in some cases, a reduction of T-cell effector function. It is possible that the difference observed in TLR2 and TLR6 KO mice in controlling Brucella infection in vivo may be related to differences in the ability to activate T-cell function in these animals. Alternatively, a lack of TLR6 affects other components of the immune system during Brucella infection that require further characterization.
TLRs signal through MyD88 and IRAK-4 to mediate many cellular responses, including proinflammatory cytokine production (26). To investigate whether Brucella requires TLR6 to modulate intracellular signaling, we examined the phosphorylation of JNK and p38 in dendritic cells. We observed that Brucella-mediated activation of MAPKs in dendritic cells is TLR6 and MyD88 dependent. Therefore, the lack of Brucella-induced signaling in BMDCs may account for the lower IL-12 and TNF-α production observed in TLR6 KO mouse cells. Recently, Zhang et al. (2012) demonstrated that heat-killed Brucella activates IL-12 and TNF-α secretion in DCs through a p38-dependent pathway (45). These authors claimed that TLR2 mediates the activation of p38 MAPK, which is a critical step for phagocytosis of heat-killed Brucella, consequently inducing IL-12 production via TLR9 activation. The intracellular cross talk scenarios are essential for understanding host-microbe interactions, because most pathogens have multiple molecules that are recognized by multiple innate receptors. Therefore, it is possible that TLR6 can interact with another innate receptor, such as C-type lectin receptors (CLRs), NOD-like receptors (NLRs), or RIG-I like receptors (RLRs) to sense Brucella and enhance host resistance to infection. A complete understanding of TLR detection of PAMPs and the subsequent signaling pathway is crucial for deciphering the pathogenesis of brucellosis.
Collectively, we have demonstrated that TLR6 is important for initiating innate immune responses against B. abortus in dendritic cells. Furthermore, reduced p38 and JNK phosphorylation in the absence of TLR6 culminates in the reduced production of proinflammatory cytokines. Finally, a lack of TLR6, not TLR2, enhanced the susceptibility of mice to B. abortus infection.
ACKNOWLEDGMENTS
This work was supported by grants from CNPq, CNPq/CONICET, FAPEMIG, FAPEMIG/CNPq (PRONEX), CAPES/PNPD, CNPq/MAPA, CNPq/REPENSA, and INCT-Vacinas.
Footnotes
Published ahead of print 4 March 2013
REFERENCES
- 1. Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826 [DOI] [PubMed] [Google Scholar]
- 2. Kawai T, Akira S. 2007. TLR signaling. Semin. Immunol. 19:24–32 [DOI] [PubMed] [Google Scholar]
- 3. Kawai T, Akira S. 2007. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 13:460–469 [DOI] [PubMed] [Google Scholar]
- 4. Corbel MJ. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franco MP, Mulder M, Gilman RH, Smits HL. 2007. Human brucellosis. Lancet Infect. Dis. 7:775–786 [DOI] [PubMed] [Google Scholar]
- 6. Boschiroli M-L, Foulongne V, O'Callaghan D. 2001. Brucellosis: a worldwide zoonosis. Curr. Opin. Microbiol. 4:58–64 [DOI] [PubMed] [Google Scholar]
- 7. Golding B, Scott DE, Scharf O, Huang Zaitseva L-YM, Lapham C, Eller N, Golding H. 2001. Immunity and protection against Brucella abortus. Microbes Infect. 3:43–48 [DOI] [PubMed] [Google Scholar]
- 8. Murphy EA, Sathiyaseelan J, Parent MA, Zou B, Baldwin CL. 2001. Interferon-gamma is crucial for surviving a Brucella abortus infection in both resistant C57BL/6 and susceptible BALB/c mice. Immunology 103:511–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oliveira SC, Splitter GA. 1995. CD8+ type 1 CD44hi CD45 RBlo T lymphocytes control intracellular Brucella abortus infection as demonstrated in major histocompatibility complex class I- and class II-deficient mice. Eur. J. Immunol. 25:2551–2557 [DOI] [PubMed] [Google Scholar]
- 10. Oliveira SC, de Almeida LA, Carvalho NB, Oliveira FS, Lacerda TLS. 2012. Update on the role of innate immune receptors during Brucella abortus infection. Vet. Immunol. Immunopathol. 148:129–135 [DOI] [PubMed] [Google Scholar]
- 11. Oliveira SC, de Oliveira FS, Macedo GC, de Almeida LA, Carvalho NB. 2008. The role of innate immune receptors in the control of Brucella abortus infection: Toll-like receptors and beyond. Microbes Infect. 10:1005–1009 [DOI] [PubMed] [Google Scholar]
- 12. Gomes MTR, Campos PC, de Almeida LA, Oliveira FS, Costa MMS, Marim FM, Pereira GSM, Oliveira SC. 2012. The role of innate immune signals in immunity to Brucella abortus. Front. Cell. Infect. Microbiol. 2:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Macedo GC, Magnani DM, Carvalho NB, Bruna-Romero O, Gazzinelli RT, Oliveira SC. 2008. Central role of MyD88-dependent dendritic cell maturation and proinflammatory cytokine production to control Brucella abortus infection. J. Immunol. 180:1080–1087 [DOI] [PubMed] [Google Scholar]
- 14. Bulut Y, Faure E, Thomas L, Equils O, Arditi M. 2001. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2. J. Immunol. 167:987–994 [DOI] [PubMed] [Google Scholar]
- 15. Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. 2000. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. U. S. A. 97:13766–13771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Mühlradt PF, Akira S. 2000. Preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164:554–557 [DOI] [PubMed] [Google Scholar]
- 17. Campos MA, Rosinha GMS, Almeida IC, Salgueiro XS, Jarvis BW, Splitter GA, Qureshi N, Bruna-Romero O, Gazzinelli RT, Oliveira SC. 2004. Role of Toll-like receptor 4 in induction of cell-mediated immunity and resistance to Brucella abortus infection in mice. Infect. Immun. 72:176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiss DS, Takeda K, Akira S, Zychlinsky A, Moreno E. 2005. MyD88, but not Toll-like receptors 4 and 2, is required for efficient clearance of Brucella abortus. Infect. Immun. 73:5137–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trant CGMC, Lacerda TLS, Carvalho NB, Azevedo V, Rosinha GMS, Salcedo SP, Gorvel Oliveira J-PSC. 2010. The Brucella abortus phosphoglycerate kinase mutant is highly attenuated and induces protection superior to that of vaccine strain 19 in immunocompromised and immunocompetent mice. Infect. Immun. 78:2283–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giambartolomei GH, Zwerdling A, Cassataro J, Bruno L, Fossati CA, Philipp MT. 2004. Lipoproteins, not lipopolysaccharide, are the key mediators of the proinflammatory response elicited by heat-killed Brucella abortus. J. Immunol. 173:4635–4642 [DOI] [PubMed] [Google Scholar]
- 21. Akira S. 2006. TLR signaling. Curr. Top. Microbiol. Immunol. 311:1–16 [DOI] [PubMed] [Google Scholar]
- 22. Medzhitov R, Preston-Hurlburt P, Janeway CA. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397 [DOI] [PubMed] [Google Scholar]
- 23. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 24. Copin R, De Baetselier P, Carlier Y, Letesson J-J, Muraille E. 2007. MyD88-dependent activation of B220-CD11b+LY-6C+ dendritic cells during Brucella melitensis infection. J. Immunol. 178:5182–5191 [DOI] [PubMed] [Google Scholar]
- 25. Copin R, Vitry Hanot Mambres M-AD, Machelart A, De Trez C, Vanderwinden Magez J-MS, Akira S, Ryffel B, Carlier Y, Letesson J-J, Muraille E. 2012. In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog. 8:e1002575 doi:10.1371/journal.ppat.1002575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliveira FS, Carvalho NB, Brandão APMS, Gomes MTR, de Almeida LA, Oliveira SC. 2011. Interleukin-1 receptor-associated kinase 4 is essential for initial host control of Brucella abortus infection. Infect. Immun. 79:4688–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820 [DOI] [PubMed] [Google Scholar]
- 28. Mehlin C, Headley CM, Klebanoff SJ. 1999. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J. Exp. Med. 189:907–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hajjar AM, O'Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. 2001. Functional interactions between Toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J. Immunol. 166:15–19 [DOI] [PubMed] [Google Scholar]
- 30. Seibert SA, Mex P, Köhler A, Kaufmann SHE, Mittrücker H-W. 2010. TLR2-, TLR4- and Myd88-independent acquired humoral and cellular immunity against Salmonella enterica serovar Typhimurium. Immunol. Lett. 127:126–134 [DOI] [PubMed] [Google Scholar]
- 31. Moranta D, Regueiro V, March C, Llobet E, Margareto J, Larrarte E, Larrate E, Garmendia J, Bengoechea JA. 2010. Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect. Immun. 78:1135–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lorenz E, Chemotti DC, Jiang AL, McDougal LD. 2005. Differential involvement of Toll-like receptors 2 and 4 in the host response to acute respiratory infections with wild-type and mutant Haemophilus influenzae strains. Infect. Immun. 73:2075–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang L-Y, Aliberti J, Leifer CA, Segal DM, Sher A, Golenbock DT, Golding B. 2003. Heat-killed Brucella abortus induces TNF and IL-12p40 by distinct MyD88-dependent pathways: TNF, unlike IL-12p40 secretion, is Toll-like receptor 2 dependent. J. Immunol. 171:1441–1446 [DOI] [PubMed] [Google Scholar]
- 34. Delpino MV, Barrionuevo P, Macedo GC, Oliveira SC, Genaro Di S, Scian R, Miraglia MC, Fossati CA, Baldi PC, Giambartolomei GH. 2012. Macrophage-elicited osteoclastogenesis in response to Brucella abortus infection requires TLR2/MyD88-dependent TNF-α production. J. Leukoc. Biol. 91:285–298 [DOI] [PubMed] [Google Scholar]
- 35. Scian R, Barrionuevo P, Giambartolomei GH, Fossati CA, Baldi PC, Delpino MV. 2011. Granulocyte-macrophage colony-stimulating factor- and tumor necrosis factor alpha-mediated matrix metalloproteinase production by human osteoblasts and monocytes after infection with Brucella abortus. Infect. Immun. 79:192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barrionuevo P, Cassataro J, Delpino MV, Zwerdling A, Pasquevich KA, García Samartino C, Wallach JC, Fossati CA, Giambartolomei GH. 2008. Brucella abortus inhibits major histocompatibility complex class II expression and antigen processing through interleukin-6 secretion via Toll-like receptor 2. Infect. Immun. 76:250–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barrionuevo P, Delpino MV, Velásquez LN, García Samartino C, Coria LM, Ibañez AE, Rodríguez ME, Cassataro J, Giambartolomei GH. 2011. Brucella abortus inhibits IFN-γ-induced FcγRI expression and FcγRI-restricted phagocytosis via Toll-like receptor 2 on human monocytes/macrophages. Microbes Infect. 13:239–250 [DOI] [PubMed] [Google Scholar]
- 38. Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, Muller A, Lapaque N, Demaria O, Alexopoulou L, Comerci DJ, Ugalde RA, Pierre P, Gorvel J-P. 2008. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4:e21 doi:10.1371/journal.ppat.0040021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma X, Liu Y, Gowen BB, Graviss EA, Clark AG, Musser JM. 2007. Full-exon resequencing reveals Toll-like receptor variants contribute to human susceptibility to tuberculosis disease. PLoS One 2:e1318 doi:10.1371/journal.pone.0001318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shey MS, Randhawa AK, Bowmaker M, Smith E, Scriba TJ, de Kock M, Mahomed H, Hussey G, Hawn TR, Hanekom WA. 2010. Single nucleotide polymorphisms in Toll-like receptor 6 are associated with altered lipopeptide- and mycobacteria-induced interleukin-6 secretion. Genes Immun. 11:561–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang LY, Reis e Sousa C, Itoh Y, Inman J, Scott DE. 2001. IL-12 induction by a TH1-inducing adjuvant in vivo: dendritic cell subsets and regulation by IL-10. J. Immunol. 167:1423–1430 [DOI] [PubMed] [Google Scholar]
- 42. Brandão APMS, Oliveira FS, Carvalho NB, Vieira LQ, Azevedo V, Macedo GC, Oliveira SC. 2012. Host susceptibility to Brucella abortus infection is more pronounced in IFN-γ knockout than IL-12/β2-microglobulin double-deficient mice. Clin. Dev. Immunol. 2012:589494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vitry M-A, De Trez C, Goriely S, Dumoutier L, Akira S, Ryffel B, Carlier Y, Letesson J-J, Muraille E. 2012. Crucial role of gamma interferon-producing CD4+ Th1 cells but dispensable function of CD8+ T Cell, B cell, Th2, and Th17 responses in the control of Brucella melitensis infection in mice. Infect. Immun. 80:4271–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin B, Sun T, Yu X-H, Yang Y-X, Yeo AET. 2012. The effects of TLR activation on T-cell development and differentiation. Clin. Dev. Immunol. 2012:836485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang C-Y, Bai N, Zhang Z-H, Liang N, Dong L, Xiang R, Liu C-H. 2012. TLR2 signaling subpathways regulate TLR9 signaling for the effective induction of IL-12 upon stimulation by heat-killed Brucella abortus. Cell. Mol. Immunol. 9:324–333 [DOI] [PMC free article] [PubMed] [Google Scholar]