

Abstract
Filarial infection is initiated by mosquito-derived third-stage larvae (L3) deposited on the skin that transit through the epidermis, which contains Langerhans cells (LC) and keratinocytes (KC), among other cells. This earliest interaction between L3 and the LC likely conditions the priming of the immune system to the parasite. To determine the nature of this interaction, human LC (langerin+ E-cadherin+ CD1a+) were generated in vitro and exposed to live L3. LC exposed to live L3 for 48 h showed no alterations in the cell surface markers CD14, CD86, CD83, CD207, E-cadherin, CD80, CD40, and HLA-DR or in mRNA expression of inflammation-associated genes, such as those for interleukin 18 (IL-18), IL-18BP, and caspase 1. In contrast to L3, live tachyzoites of Toxoplasma gondii, an intracellular parasite, induced production of CXCL9, IP-10, and IL-6 in LC. Furthermore, preexposure of LC to L3 did not alter Toll-like receptor 3 (TLR3)- or TLR4-mediated expression of the proinflammatory cytokines IL-1β, gamma interferon (IFN-γ), IL-6, or IL-10. Interestingly, cocultures of KC and LC produced significantly more IL-18, IL-1α, and IL-8 than did cultures of LC alone, although exposure of the cocultures to live L3 did not result in altered cytokine production. Microarray examination of ex vivo LC from skin blisters that were exposed to live L3 also showed few significant changes in gene expression compared with unexposed blisters, further underscoring the relatively muted response of LC to L3. Our data suggest that failure by LC to initiate an inflammatory response to the invasive stage of filarial parasites may be a strategy for immune evasion by the filarial parasite.
INTRODUCTION
Lymphatic filariasis (LF) caused by the parasitic nematodes Wuchereria bancrofti, Brugia malayi, and Brugia timori infects approximately 120 million people worldwide in 72 countries. The disfiguring clinical manifestations, such as lymphedema and hydrocele, result in substantial morbidity and social stigma (1). Infection in LF is initiated when mosquito-derived third-stage larvae (L3) are deposited in the skin, an organ containing innate cells, primarily epidermal-resident Langerhans cells (LC) and keratinocytes (KC). Given the parasites' route through the skin, it is likely that the early interaction between L3 and innate cells in the skin—LC in particular—conditions the initiation of the L3-specific immune response, as LC are known to migrate to the draining lymph nodes (LN) soon after antigen internalization.
LC are a subset of antigen-presenting dendritic cells (DC) that are located in the epidermal layer of the skin. LC become activated after antigen internalization and mature phenotypically and functionally (reviewed in references 2 and 3). Upon activation, adhesion molecules (such as E-cadherin) are downregulated to allow migration from the skin to the LN, where mature LC can activate naïve T cells and initiate T cell development (reviewed in references 2 and 3).
Until recently, LC were considered the major antigen-presenting cells (APC) responsible for initiating skin-based immune responses, although other skin-dwelling DC subsets have also been shown to be capable of presenting antigen to T cells (4, 5). While LC mediate a variety of processes, it is clear that the LC function depends greatly on both the nature of the antigens with which they come into contact and the environment in which the LC encounter these antigens. For example, KC, integrally associated with LC anatomically in the epidermis, are critical for optimal LC activation (6).
To date, there are few studies addressing the role of LC in skin-invasive helminth infections. We have shown previously that live infective-stage L3 of B. malayi suppress activation of human epidermal cells (7). In fact, when ex vivo blisters from healthy volunteers were exposed to L3, there was a markedly diminished expression of genes associated with antigen processing and presentation that translated into a diminished ability to activate autologous CD4+ T cells (7). Interestingly, in the same study, live L3 resulted in the upregulation of IL-18 (an inflammatory cytokine important in LC migration) (7). While the ex vivo model of LC approximates the physiological environment, very few LC can be obtained from the model for analysis. Therefore, to better elucidate the consequences of the LC-L3 interaction, in the current study, we generated LC in vitro and assessed their response to live L3. Our data suggest that, compared with a known activator, lipopolysaccharide (LPS), or the intracellular-parasite pathogen Toxoplasma gondii, L3 failed to activate LC. This relatively muted response by the LC to the invading L3 appears to represent a strategy used by the parasite to evade the early host innate response.
MATERIALS AND METHODS
Parasite preparation.
Live L3 of B. malayi isolated from whole infected mosquitoes (Aedes aegypti) were provided by the University of Georgia, Athens, GA. L3 were washed three times with minimal essential medium alpha (MEM-α) containing nucleosides (GlutaMax; Invitrogen, Grand Island, NY) supplemented with penicillin/streptomycin/amphotericin (Invitrogen), as well as ciprofloxacin and ceftazidime (both at 2 μg/ml), and incubated for 2 h at 37°C in culture dishes. The fittest L3 were then picked and added to cell or explant cultures. Purified tachyzoites of the RH strain of T. gondii were provided from the laboratory of A. Sher (National Institute of Allergy and Infectious Disease, National Institutes of Health, Bethesda, MD).
In vitro culture of LC.
Elutriated monocytes from healthy human donors were cultured in RPMI (Gibco, Grand Island, NY) supplemented with penicillin/streptomycin (Invitrogen) and 10% fetal calf serum (Gemini Bio-Products, West Sacramento, CA). Granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin 4 (IL-4) (Peprotech, Rocky Hill, NJ; both at 62.5 ng/ml), and transforming growth factor beta (TGF-β) (Peprotech) at 10 ng/ml were added every 2 days for 6 days. On day 6, the cells were exposed to medium alone, 10 live L3, 10 L3 in a transwell, T. gondii tachyzoites at a ratio of 1:5 (tachyzoite/LC), or LPS at 1 μg/ml (Invitrogen) for 48 h. After stimulation, the cells and supernatants were harvested for RNA expression analysis or cytokine production analysis, respectively.
KC generation.
Primary human KC were isolated as previously described from neonatal foreskins by dispase (Becton, Dickinson, Franklin Lakes, NJ) treatment to separate the epidermis from the dermis (8). The epidermis was then incubated in 0.05% trypsin (Invitrogen). The trypsinized tissue was incubated in F12 medium (Invitrogen) supplemented with Dulbecco's modified Eagle's medium (DMEM) (Invitrogen), 5% fetal bovine serum (FBS) (Gemini Bio-Products), 0.4 μg/ml hydrocortisone (Sigma-Aldrich, St. Louis, MO), 5 μg/ml insulin (Sigma-Aldrich), 8.4 ng/ml cholera toxin (Calbiochem, Rockland, MA), 10 ng/ml endothelial growth factor (Invitrogen), 24 μg/ml adenine (Sigma-Aldrich), l-glutamate (Invitrogen), and penicillin/streptomycin (Invitrogen) to allow the cells to establish, and the medium was changed the following day and every 2 to 3 days afterward. Isolated KC were grown on 3T3 mouse cells (a gift from the laboratory of Alison McBride, NIAID, NIH) in the same medium and split at 80% confluence. To immortalize the KC, 1 μg/ml of Y-27632 dihydrochloride (Tocris Bioscience, Minneapolis, MN) was added to the culture medium.
LC-KC coculture.
In vitro-derived LC were harvested using Versene (Gibco), and KC were harvested using 0.25% trypsin (Gibco). The cells were cocultured at a ratio of 1:5 (LC/KC) or 1:50 (LC/KC) in 24-well plates, with 0.5 × 106 KC in each well. The cells were cultured in 0.5 ml RPMI (Gibco) supplemented with 10% fetal calf serum (FCS) (Gemini Bio-Products) for 48 h in either medium alone or LPS at 1 μg/ml (Invitrogen) or with 10 L3 in contact. The cells were then harvested, and the supernatants were used for cytokine production analysis.
RNA preparation and RT-PCR.
RNA was extracted from harvested LC using an RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. cDNA generation was performed using random hexamers and reverse transcriptase (ABI, Grand Island, NY). Expression of IL-18, IL-18BP, casp1, CD207 (langerin), E-cadherin, NLRP1 (NLR family, pyrin domain containing 1), NLPR3, NLRC4, AIM2, and ASC was determined by quantitative real-time PCR using predeveloped TaqMan reagents (ABI) on an ABI 7900HT. The threshold cycles (CT) for the 18S control and the genes of interest were calculated and used to determine the relative transcript levels. The formula 1/ΔCT was used to determine the relative transcript levels, where ΔCT is the difference between the CT of the target gene and the CT of the corresponding endogenous 18S reference.
Cytokine analysis.
The concentrations of cytokines in supernatants of the LC culture and the LC-KC culture were determined by Duoset enzyme-linked immunosorbent assay (ELISA) for IL-18, IL-18BP, IL-6, and IL-10 according to the manufacturer's instructions (R&D Systems, Minneapolis, MN). The sensitivity of the assays is as follows: 1.56 pg/ml for IL-18, 93.75 pg/ml for IL-18BP, 9.38 pg/ml for IL-6, and 31.25 pg/ml for IL-10.
The remaining cytokines (gamma interferon [IFN-γ], IL-8, IL-6, IL-10, IP-10, CXCL9, IL-12p70, IL-1β, and tumor necrosis factor alpha [TNF-α]) were assessed using Milliplex Max human cytokine panel Luminex kits (Millipore, Billerica, MA). As with the ELISAs, the minimum detectable concentration for the Luminex kits varied by cytokine: 0.8 pg/ml for IFN-γ, 0.4 pg/ml for IL-8, 0.9 pg/ml for IL-6, 1.1 pg/ml for IL-10, 8.6 pg/ml for IP-10, 19.2 pg/ml for CXCL9, 0.8 pg/ml for IL-12p70, 0.4 pg/ml for IL-1β, and 0.7 pg/ml for TNF-α. For samples below the minimum detectable concentration of the assay, the value of 0.001 was assigned for analysis.
Flow cytometry.
Staining of cells was performed according to standard protocols. LC were harvested and washed in fluorescence-activated cell sorter (FACS) buffer (phosphate-buffered saline [PBS] containing 0.5% bovine serum albumin [BSA] [Sigma-Aldrich] and 0.1% sodium azide [Sigma-Aldrich]). The cells were incubated with FACS buffer containing 1% (vol/vol) FcR blocking reagent (Miltenyi, Auburn, CA) for 30 min at 4°C to inhibit nonspecific binding and then washed once with FACS buffer and incubated with mouse or rat anti-human monoclonal antibodies for 30 min at 4°C. The cells were washed again twice with FACS buffer before analysis using an LSRII flow cytometer (BD Biosciences, Franklin Lakes, NJ). Cells were not permeabilized prior to staining for any of the surface markers, including langerin. Antibodies for characterizing LC consisted of the following: fluorescein isothiocyanate (FITC)-conjugated mouse anti-human E-cadherin (Biolegend, San Diego, CA), APC-conjugated mouse anti-human CD207 (langerin) clone 310F7.02/HD36 (Imgenex, San Diego, CA), phycoerythrin (PE)-Cy7-conjugated mouse anti-human CD14 (eBioscience, San Diego, CA), and Pacific Blue-conjugated rat anti-human cutaneous lymphocyte antigen (CLA) (Biolegend). Activation of LC was assessed using the following antibodies: FITC-conjugated mouse anti-human CD83 (eBioscience), PE-Cy7-conjugated mouse anti-human CD80 (Biolegend), PE-conjugated mouse anti-human CD86 (eBioscience), APC-conjugated mouse anti-human CD40 (eBioscience), and eFluor 450-conjugated mouse anti-human HLA-DR (eBioscience).
Microarray.
Skin blisters were cultured in medium alone or exposed to live L3 in a transwell for 48 h, after which crawl-out cells were harvested and placed in RNA lysis buffer. RNA of 12 samples was extracted by using Ambion's RNAqueous-Micro Kit (Life Technologies, Grand Island, NY). After RNA extraction, the concentration of RNA was measured with NanoDrop ND-1000, and the integrity was assessed using an RNA 6000 Nanochip on a Bioanalyzer 2100 (Agilent, Santa Clara, CA). The RNA was amplified using a WT-Ovation pico RNA amplification system (NuGen, San Carlos, CA), and ∼750 ng of the amplified antisense cDNA from each of the 12 biologic replicates was used. cDNA was purified by using a QIAquick PCR purification kit (Qiagen) and then hybridized on a HumanHT-12 Expression BeadChip (Illumina, San Diego, CA) and scanned by the Illumina Beadstation 500 bead array reader. Signal intensity was captured for each probe on the array using BeadArray Reader software from Illumina.
Statistical analysis.
Unless stated otherwise, geometric means were used as measures of central tendency. Statistical analysis of all real-time PCR, Luminex, and ELISA data was done using a Wilcoxon matched-pairs signed-rank test and performed using Graph Pad Prism version 5 (GraphPad Software, Inc., San Diego, CA).
For microarray studies, raw data from the microarray were background subtracted using GenomeStudio Software 2011.1 (Illumina, San Diego, CA). Using GeneSpring GX version 11.5 (Agilent Technologies), the data were further analyzed by setting the threshold of raw signal to 10 and were filtered to select transcripts called present in at least one sample (PALO) using significance of P < 0.01 for the signal detection, eliminating transcripts absent across all samples. There were 847 transcripts that had a 1.5-fold difference, comparing the two group means, and were also statistically significant when using a paired t test with an alpha of 0.05 between LC-L3 and LC. For stringency, a fold change greater than 1.5 in all donors was required. Ingenuity pathway analysis (Ingenuity Systems, Inc., Redwood, CA) was used for the functional analysis.
Microarray accession number.
The genes identified in this study have been deposited in the GEO database under accession number GSE42694.
RESULTS
Live L3 do not alter cell surface molecule expression by human LC.
Human LC are distinguished from other DC subsets in part by the surface expression of langerin (CD207), E-cadherin, CD14, CD1a, and CLA, some of which also perform key functions in antigen capture (CD1a) and migration (E-cadherin) (9, 10). In addition, activated LC are known to express CD80, CD86, CD40, CD83, and HLA-DR following activation (11). To assess the effect of live L3 of B. malayi on human LC, human monocytes were differentiated in vitro using GM-CSF, IL-4, and TGF-β for 7 days; exposed to medium alone, live L3, or LPS; and assessed for surface expression of LC-specific surface molecules and for known markers of LC activation. As expected, in vitro-generated LC express both langerin and E-cadherin at the mRNA level (Fig. 1A); however, exposure to live L3 does not significantly change the expression of either of the genes (langerin, P = 0.7334; E-cadherin, P = 0.1677), Furthermore, stimulation with LPS did not change mRNA expression of these genes compared with unexposed LC (langerin, P = 0.83; E-cadherin, P = 0.0771) (Fig. 1B). When assessed by flow cytometry (Fig. 1C), in vitro-derived LC that expressed surface langerin ranged from 1.23 to 72.2% (data not shown). In response to LPS, the LC demonstrated a predictable increase in the surface expression of CLA and a decrease in langerin, E-cadherin, CD14, and CD1a. In contrast, L3 exposure failed to alter the expression of any of these LC-associated surface molecules when gated on all cells (Fig. 1C). However, when langerin+ cells were gated on, L3 significantly increased the surface expression of E-cadherin (data not shown). Parallel findings were seen when markers of activation were assessed, with LPS clearly upregulating the cell surface expression of CD40, CD86, CD83, and HLA-DR (Fig. 1C), whereas L3 failed to do so.
Fig 1.

In vitro-generated LC display characteristic LC surface and activation markers that are not altered by L3. In vitro-derived LC were exposed either to medium alone or to LPS (1 μg/ml) or L3 for 48 h. The cells were harvested and assessed for mRNA expression of langerin and E-cadherin (A and B) or for expression of the LC-associated molecules langerin, CD14, CD1a, E-cadherin, and CLA and the activation markers CD40, CD80, CD86, CD83, and HLA-DR (C). (A and B) Genes were measured by TaqMan real-time PCR and normalized to levels of 18S rRNA. Each symbol represents an individual donor (n = 12). (A) Baseline mRNA expression of E-cadherin and langerin, shown as 1/average ΔCT. The horizontal dotted line indicates no mRNA detection below 0.027. (B) Fold change in gene expression in either L3-exposed (solid squares) or LPS-activated (open squares) cells over unexposed cells. A fold change of 1 (dotted horizontal line) indicates no change in expression between stimulated and unexposed cells. (C) Representative set (3 to 7 donors, depending on the marker) of flow histograms in which the given marker is depicted in response to medium alone, LPS, or L3. The histograms were generated by gating on all singlet cells.
Exposure to live L3 does not alter mRNA expression of inflammatory genes or the production of proinflammatory cytokines in human LC.
We have previously shown that exposure of epidermal explants derived from skin blisters of normal volunteers to live L3 of B. malayi results in an increase in production of IL-18, a cytokine involved in LC migration (7). Given this finding, we sought to determine whether other molecules involved in IL-18-mediated signaling or regulation (e.g., IL-18BP, caspase 1, and the inflammasome-associated genes [NLRP1, NLPR3, NLRC4, AIM2, and ASC genes]) were altered in response to L3 in LC (Fig. 2). Although there is baseline expression of each of these mRNAs (data not shown) in in vitro-derived LC, live L3 failed to alter the expression of any of the genes examined, whereas LPS did result in a 3-fold increase in IL-18 expression (P = 0.034), an 8-fold decrease in IL-18BP (P = 0.009), and a 4-fold decrease in caspase 1 expression (P = 0.001) compared with non-L3-stimulated LC (Fig. 2b). Neither LPS nor L3 significantly altered expression of any of the inflammasome-related genes, such as those for NLRP1, NLRP3, and NLRC4 (Fig. 2A). Furthermore, L3 soluble factors (L3 in a transwell) did not change the mRNA expression of any of the genes tested (data not shown), a result similar to that for L3 in contact (Fig. 2).
Fig 2.

Exposure to live L3 does not alter the mRNA expression of inflammatory genes or the production of proinflammatory cytokines in human LC. (A and B) In vitro-derived LC were cultured in medium alone, with L3, or with LPS (1 μg/ml) for 48 h. The cells were harvested, and mRNA levels were measured by real-time PCR and normalized to the levels of 18S rRNA. The fold change in gene expression in either L3-exposed (solid squares) or LPS-activated (open squares) LC over unexposed LC is shown. *, P < 0.05. A fold change of 1 (dotted horizontal line) indicates no change in expression between stimulated and unexposed cells. (C) Supernatants from LC exposed to L3, LPS, or the tachyzoite stage of T. gondii were collected after 48 h and evaluated for levels of IFN-γ, IL-8, IL-6, IL-10, IP-10, CXCL9, and TNF-α by Luminex and of IL-18BP by ELISA. The data are expressed as the geometric mean of the fold change over unexposed LC (n = 6 to 15). *, P < 0.05; **, P < 0.01.
When we assessed the production of proinflammatory cytokines by LC in response to medium alone, L3, LPS, or T. gondii, we found that LPS significantly increased the production of IFN-γ (P = 0.0001; >1,000-fold), IL-8 (P < 0.0001; 70-fold), IL-10 (P < 0.001; 84-fold), TNF-α (P = 0.0039; 160-fold), IP-10 (P = 0.0313; 168-fold), IL-6 (P < 0.0001; >1,000-fold), and CXCL9 (P = 0.0313; 65-fold), confirming that the in vitro-generated LC are capable of mounting a proinflammatory response (Fig. 2C). Moreover, production of IL-6, IP-10, and CXCL9 was also significantly increased by exposure of LC to T. gondii (P = 0.0313), suggesting that the in vitro-generated LC are capable of mounting an inflammatory response to an intracellular parasite. In marked contrast, L3—either in contact or in transwells (data not shown)—failed to induce alterations in most of the cytokines assessed compared with those produced spontaneously (Fig. 2C). Exposure of LC to L3 led to increased production of IL-6 (P = 0.0244; 12-fold). Furthermore, cytokines associated with IL-18 or its known inflammatory effects (IL-33 and IL-1α) were also not induced by L3 (data not shown).
Live L3 does not alter the mRNA expression or function of Toll-like receptor 3 (TLR3) and TLR4 in human LC.
We have shown previously that live B. malayi parasites (L3 and/or microfilaria [mf]) suppress human LC- and monocyte-derived DC (7, 12) function and—for mf at least—also alter TLR3 and TLR4 expression and subsequent downstream signaling (12). Given the lack of proinflammatory cytokine production from in vitro-generated LC in response to L3 (Fig. 2), we investigated whether the L3 were actively suppressing the LC response through alteration of TLR signaling. When LC were exposed to L3 and TLR expression was assessed in comparison with unexposed L3, our data indicate that there was no alteration of TLR3 or TLR4 mRNA expression by L3 (data not shown). More importantly, the L3 did not alter production of IL-1β, IFN-γ, IL-6, IL-10, or IL-12p70 when L3-exposed LC were stimulated with either TLR3 or TLR4 ligands (Fig. 3). Although in vitro LC in response to a TLR4 agonist (LPS) (Fig. 3A) or a TLR3 agonist [poly(I · C)] (Fig. 3B) can produce all the cytokines measured (Fig. 3, Media), the presence of L3 (or their secreted products) prior to TLR stimulation appeared to have little to no effect on TLR-mediated functional responses in these cells (Fig. 3). Live mf of B. malayi were shown to directly activate TLR2 in fibroblasts (12); however, whether the L3 stage of the parasite can activate any of the TLRs is not known. Our preliminary data suggest that L3 (but not Toxoplasma) upregulate only slightly the mRNA expression of TLR2 in LC (data not shown). We have yet to determine if there is any functional significance to this upregulation.
Fig 3.
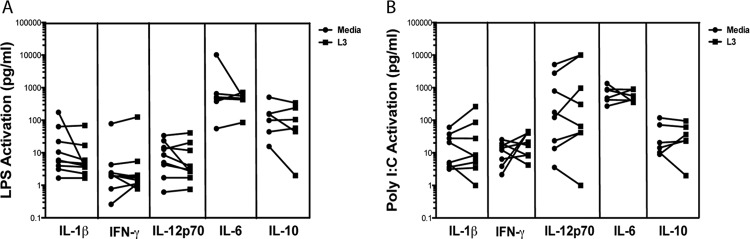
Live L3 do not alter the function of TLR3 or TLR4 in LC. LC were cultured in medium alone or with live L3 for 24 h. The cells were harvested, and viable cells were further activated with medium alone, the TLR4 ligand LPS (A), or the TLR3 ligand poly(I · C) (B) for an additional 48 h. Production of IL-1β, IFN-γ, and IL-12p70 was measured in the culture supernatant with Luminex, and IL-6 and IL-10 levels were measured by ELISA. Each line represents an independent donor (n = 6 to 9).
The presence of KC allows LC to function optimally, and this function is not altered by the presence of L3.
One main difference between the ex vivo blister model of LC interaction with L3 and our in vitro model is the absence of KC in the latter. Given that KC are crucial for LC activation, we assessed the contribution of KC to our in vitro model by culturing the in vitro-generated LC with primary neonatal, immortalized human KC (Fig. 4). As can be seen, in the presence of KC (5:1 [Fig. 4] or 50:1 [not shown] ratio), there was a significant increase in the LC production of IL-18 (P = 0.031), IL-1α (P = 0.016), and IL-8 (P = 0.031) compared with the culture of LC alone (Fig. 4A). Our data also show that LC are required for optimum KC activation. Indeed, compared with KC alone, KC-LC cocultures produced significantly more IL-18BP (P = 0.031), TNF-α (P = 0.03), and IL-8 (P = 0.02) and significantly less IL-1α (P = 0.03) than KC cultures alone (Fig. 4B). The differential expression of cytokines seen between the KC-LC coculture and the individual LC or KC cultures suggests the importance of this interaction occurring in the same anatomical niche.
Fig 4.

The presence of KC allows LC to function optimally, and this function is not altered by the presence of L3. (A and B) Immortalized primary KC and in vitro-derived LC were cocultured at a ratio of 5:1 (KC/LC) for 48 h. The supernatants were collected, and IL-1α, TNF-α, and IL-8 production was assessed with Luminex, while IL-18 and IL-18BP production was assessed by ELISA, and compared with culture of LC alone (A) or culture of KC alone (B). Each line represents an independent donor (n = 7). (C) Immortalized primary KC and in vitro-derived LC were cocultured at a ratio of 5:1 (KC/LC) in medium alone, L3, or 1 μg/ml LPS for 48 h. Cytokine production was determined in culture supernatants with Luminex, while IL-18 and IL-18BP production was assessed by ELISA. The data are expressed as the geometric mean (n = 7) of the fold change over medium for each cytokine. *, P < 0.05.
Because cytokine production from a coculture of KC-LC was significantly different from a culture of LC alone, we assessed cytokine production by KC-LC cocultures exposed to live L3 compared with either unexposed KC-LC or LPS-exposed KC-LC (Fig. 4C). Our data demonstrate that L3 failed to induce significant production of IL-18BP, IL-18, IL-1α, TNF-α, or IL-8 in these KC-LC cocultures above the levels produced spontaneously. LPS, in contrast, induced an 8-fold increase in TNF-α production (P = 0.03) and led to a significant decrease in IL-18BP production in the KC-LC coculture compared with medium alone (P = 0.016; 2-fold decrease) (Fig. 4C). The failure of L3 to induce activation of LC or KC in these KC-LC cocultures suggests a mechanism by which L3 manage to bypass the primary line of defense in the skin, thereby facilitating their migration through the skin to the LN.
Gene expression from epidermal LC is minimally altered by exposure to L3 soluble factors.
Because our data suggest a relatively quiescent response from in vitro-generated human LC to live L3 of B. malayi, we assessed the abilities of the parasites' soluble factors to modify gene expression in epidermal LC from human skin blisters using microarray analysis. To mimic the exposure of human skin to live L3, we obtained epithelial tissue explants (n = 6), as described previously (7), that were exposed to 50 live L3 in transwells (5 L3 per explant blister) and compared them to equal numbers of explants left unexposed. The crawl-out LC were harvested, and RNA was extracted for microarray analysis. Using transwells to separate L3 from the cells allowed us to ensure that the microarray data would be free of contaminating RNA from the worms and would reflect RNA expression changes only in the LC. Our data indicate that of the 48,000 genes in the microarray library, 847 genes were statistically differentially expressed (paired t test [see GEO accession number in Materials and Methods]). between unexposed and L3-exposed LC. To increase the stringency of our analysis further, we required a 1.5-fold change cutoff in L3-exposed LC for all the samples. As can be seen in Table 1, only 51/847 genes met the defined criteria. Among the 13 upregulated genes were heat shock protein 6, fibroblast growth factor 6, and keratin 5 genes, and the 38 genes that were downregulated included IL-32, CCR4-NOT transcription complex subunit 10 (CNOT10), and chromosome 15 open reading frame 44 (C15orf44) genes (Table 1). Functional analysis of significantly altered genes using Ingenuity pathway analysis indicated upregulation of growth factor-related, ion channel, and ligand-dependent nuclear receptor pathways and downregulation of transcription regulators and enzymes in all six donors (data not shown).
Table 1.
Soluble factors from L3 minimally alter gene expression in epidermal LCa
| Probe identifier (ID) | Gene name | Entrez gene name | Fold change (median) |
|---|---|---|---|
| ILMN_1702696 | AKR7L | Aldo-keto reductase family 7-like | 3.35 |
| ILMN_1706418 | EHMT1 | Euchromatic histone-lysine N-methyltransferase 1 | 3.78 |
| ILMN_1728353 | EYA3 | Eyes absent homolog 3 (Drosophila) | 2.52 |
| ILMN_1764605 | FGF6 | Fibroblast growth factor 6 | 2.93 |
| ILMN_1673380 | GNG12 | Guanine nucleotide binding protein (G protein), gamma 12 | 2.84 |
| ILMN_1723486 | HK2 | Hexokinase 2 | 2.98 |
| ILMN_1806165 | HSPA6 | Heat shock 70-kDa protein 6 (HSP70B′) | 4.09 |
| ILMN_1801632 | KRT5 | Keratin 5 | 2.82 |
| ILMN_1807300 | PKD2 (includes EG:18764) | Polycystic kidney disease 2 (autosomal dominant) | 2.17 |
| ILMN_2131103 | PPP1R3B | Protein phosphatase 1, regulatory subunit 3B | 3.93 |
| ILMN_1837297 | Homo sapiens cDNA clone UI-E-EJ1-aje-c-15-0-UI 5 | 1.81 | |
| ILMN_1855493 | Homo sapiens cDNA clone UI-E-EJ1-ajv-h-04-0-UI 3 | 2.79 | |
| ILMN_1796808 | Deleted | 2.41 | |
| ILMN_1713732 | ABL1 | c-abl oncogene 1, nonreceptor tyrosine kinase | −2.45 |
| ILMN_1698369 | AGAP6 (includes others) | ArfGAP with GTPase domain, ankyrin repeat and PH domain 5 | −2.15 |
| ILMN_2395055 | ATPAF1 | ATP synthase mitochondrial F1 complex assembly factor 1 | −2.76 |
| ILMN_1786433 | BCCIP | BRCA2- and CDKN1A-interacting protein | −2.46 |
| ILMN_1771326 | C15orf44 | Chromosome 15 open reading frame 44 | −3.47 |
| ILMN_2078334 | CNOT10 | CCR4-NOT transcription complex, subunit 10 | −3.39 |
| ILMN_1713321 | CYP20A1 | Cytochrome P450, family 20, subfamily A, polypeptide 1 | −3.69 |
| ILMN_1763129 | DCTPP1 | dCTP pyrophosphatase 1 | −2.37 |
| ILMN_1735461 | DDX21 | DEAD (Asp-Glu-Ala-Asp) box helicase 21 | −2.85 |
| ILMN_2145280 | DEF6 | Differentially expressed in FDCP 6 homolog (mouse) | −2.38 |
| ILMN_2112049 | DNLZ | DNL-type zinc finger | −3.61 |
| ILMN_1680130 | DYM | Dymeclin | −4.40 |
| ILMN_1751492 | FAM18B1 | Family with sequence similarity 18, member B1 | −4.50 |
| ILMN_1675401 | GTF2IP1 | General transcription factor IIi, pseudogene 1 | −2.24 |
| ILMN_1773751 | HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog | −1.85 |
| ILMN_1778010 | IL-32 | Interleukin 32 | −4.61 |
| ILMN_2136635 | ISCA2 | Iron-sulfur cluster assembly 2 homolog (Saccharomyces cerevisiae) | −3.40 |
| ILMN_1764861 | ISOC1 | Isochorismatase domain containing 1 | −2.40 |
| ILMN_1741599 | MEMO1 (includes EG:298787) | Mediator of cell motility 1 | −2.72 |
| ILMN_1798288 | MOB3C | MOB kinase activator 3C | −3.61 |
| ILMN_1652246 | NACAD | NAC alpha domain containing | −2.61 |
| ILMN_1758548 | NEK7 | NIMA (never in mitosis gene a)-related kinase 7 | −2.96 |
| ILMN_1806791 | OR4C46 | Olfactory receptor, family 4, subfamily C, member 46 | −4.12 |
| ILMN_1746618 | PAQR7 | Progestin and adipoQ receptor family member VII | −3.87 |
| ILMN_1707548 | RAD18 | RAD18 homolog (S. cerevisiae) | −2.44 |
| ILMN_2186597 | RPP21 | RNase P/MRP 21-kDa subunit | −3.86 |
| ILMN_1719627 | SLC27A3 | Solute carrier family 27 (fatty acid transporter), member 3 | −3.33 |
| ILMN_2400613 | SNX7 | Sorting nexin 7 | −3.33 |
| ILMN_1798827 | SRBD1 | S1 RNA binding domain 1 | −2.17 |
| ILMN_1759549 | SRGAP2 | SLIT-ROBO Rho GTPase-activating protein 2 | −2.37 |
| ILMN_1776333 | TBCEL | Tubulin-folding cofactor E-like | −3.17 |
| ILMN_2118910 | TPRKB | TP53RK binding protein | −2.29 |
| ILMN_1797384 | UROS | Uroporphyrinogen III synthase | −3.53 |
| ILMN_1803744 | VIMP | VCP-interacting membrane protein | −2.31 |
| ILMN_2212354 | WDR46 | WD repeat domain 46 | −3.00 |
| ILMN_1695362 | ZNF32 | Zinc finger protein 32 | −2.41 |
| ILMN_1738793 | ZNF71 | Zinc finger protein 71 | −7.34 |
| ILMN_1720438 | Deleted | −1.99 |
Six ex vivo blisters were cultured for 48 h in a 6-well plate with 50 L3 in transwells. Crawl-out LC were collected, and RNA was extracted for microarray analysis. A PALO filter (see Materials and Methods) and paired t test were used to determine genes differentially expressed in L3-exposed LC compared with unexposed LC. Normalized values from the medium-exposed blisters and values from the L3-exposed blisters were used to calculate the fold change in gene expression. The table represents the 51 genes that were significantly differentially expressed using a cutoff of 1.5-fold change in all donors.
Moreover, similar to our in vitro findings (Fig. 1 to 4), there were no significant differences between unexposed and L3-exposed LC for all the donors in expression of surrogates of LC activation, including IL-18, caspase 1, IL-18BP, TNF-α, and IL-6, or surface markers, such as CD40 and langerin (Table 2). While a few LC activation surrogates, such as langerin, CD40, IL-18, IL-1α, and IL-18BP, have median fold changes over 1.5, in order to ensure that the differences seen were not driven by one or two patients, we used a more stringent criterion of a 1.5-fold change in all six donors for a gene to be considered differentially expressed. Our microarray data, together with our data from in vitro-generated LC, further support our conclusion that L3 induce a relatively unresponsive phenotype in human LC.
Table 2.
Surrogates of LC activation examineda
| Probe ID | Gene name | Entrez gene name | Fold change (median) |
|---|---|---|---|
| ILMN_1718754 | CD207 | CD207 molecule, langerin (CD207), mRNA | 3.54 |
| ILMN_2396444 | CD14 | CD14 molecule (CD14), transcript variant 2, mRNA | 1.00 |
| ILMN_1654210 | CD1C | CD1c molecule (CD1C), mRNA | 1.02 |
| ILMN_2335754 | CD1E | CD1e molecule (CD1E), transcript variant 5, mRNA | 1.74 |
| ILMN_1770940 | CDH1 | Cadherin 1, type 1, E-cadherin (epithelial) (CDH1), mRNA | 1.25 |
| ILMN_2367818 | CD40 | CD40 molecule, TNF receptor superfamily member 5 (CD40), transcript variant 2, mRNA | −2.20 |
| ILMN_1716736 | CD80 | CD80 molecule (CD80), mRNA | −1.58 |
| ILMN_1714602 | CD86 | CD86 molecule (CD86), transcript variant 2, mRNA | −1.44 |
| ILMN_1672097 | CD86 | CD86 antigen (CD28 antigen ligand 2, B7-2 antigen) (CD86), transcript variant 2, mRNA | −1.08 |
| ILMN_1782560 | CD86 | CD86 antigen (CD28 antigen ligand 2, B7-2 antigen) (CD86), transcript variant 1, mRNA | −1.03 |
| ILMN_1780582 | CD83 | CD83 molecule (CD83), transcript variant 2, mRNA | 1.10 |
| ILMN_2328666 | CD83 | CD83 molecule (CD83), transcript variant 1, mRNA | −1.14 |
| ILMN_2157441 | HLA-DRA | Major histocompatibility complex, class II, DR alpha (HLA-DRA), mRNA | 1.17 |
| ILMN_1689655 | HLA-DRA | Major histocompatibility complex, class II, DR alpha (HLA-DRA), mRNA | 1.09 |
| ILMN_1752592 | HLA-DRB4 | Major histocompatibility complex, class II, DR beta 4 (HLA-DRB4), mRNA | −1.05 |
| ILMN_2066060 | HLA-DRB6 | Major histocompatibility complex, class II, DR beta 6 (pseudogene) (HLA-DRB6) on chromosome 6 | 1.47 |
| ILMN_2066066 | HLA-DRB6 | Major histocompatibility complex, class II, DR beta 6 (pseudogene) (HLA-DRB6) on chromosome 6 | 1.19 |
| ILMN_1717261 | HLA-DRB3 | Major histocompatibility complex, class II, DR beta 3 (HLA-DRB3), mRNA | 1.95 |
| ILMN_1715169 | HLA-DRB1 | Major histocompatibility complex, class II, DR beta 1 (HLA-DRB1), mRNA | 1.52 |
| ILMN_2159694 | HLA-DRB4 | Major histocompatibility complex, class II, DR beta 4 (HLA-DRB4), mRNA | 1.62 |
| ILMN_1697499 | HLA-DRB5 | Major histocompatibility complex, class II, DR beta 5 (HLA-DRB5), mRNA | 1.24 |
| ILMN_1778457 | IL-18 | Interleukin 18 (interferon-gamma-inducing factor) (IL-18), mRNA | −1.04 |
| ILMN_2334296 | IL18BP | Interleukin 18 binding protein (IL18BP), transcript variant A, mRNA | 2.71 |
| ILMN_1727762 | CASP1 | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) (CASP1), transcript variant alpha, mRNA | 1.00 |
| ILMN_2326509 | CASP1 | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) (CASP1), transcript variant delta, mRNA | 2.37 |
| ILMN_2326512 | CASP1 | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) (CASP1), transcript variant delta, mRNA | 2.28 |
| ILMN_1658483 | IL1A | Interleukin 1, alpha (IL1A), mRNA | −3.90 |
| ILMN_1728106 | TNF | Tumor necrosis factor (TNF superfamily, member 2) (TNF), mRNA | 1.14 |
| ILMN_1666733 | IL-8 | IL-8, mRNA | −1.14 |
| ILMN_2184373 | IL-8 | IL-8, mRNA | −1.14 |
| ILMN_1810045 | NLRP1 | NLR family, pyrin domain containing 1 (NLRP1), transcript variant 2, mRNA | −1.04 |
| ILMN_2310896 | NLRP3 | NLR family, pyrin domain containing 3 (NLRP3), transcript variant 1, mRNA | 1.00 |
| ILMN_2398274 | PYCARD | PYD and CARD domain containing (PYCARD), transcript variant 2, mRNA | 1.00 |
| ILMN_1698766 | PYCARD | PYD and CARD domain containing (PYCARD), transcript variant 1, mRNA | 1.26 |
| ILMN_2398274 | PYCARD | PYD and CARD domain containing (PYCARD), transcript variant 2, mRNA | 1.00 |
| ILMN_1699651 | IL-6 | IL-6 (interferon, beta 2), mRNA | −1.32 |
| ILMN_2073307 | IL-10 | IL-10, mRNA | −1.05 |
| ILMN_2334296 | IL18BP | Interleukin 18 binding protein (IL18BP), transcript variant A, mRNA | 2.71 |
| ILMN_1653575 | IL18BP | Interleukin 18 binding protein (IL18BP), transcript variant D, mRNA | 1.68 |
| ILMN_1775501 | IL1B | Interleukin 1, beta (IL1B), mRNA | 1.00 |
| ILMN_1699651 | IL-6 | IL-6 (interferon, beta 2), mRNA | −1.32 |
Normalized values from the medium-exposed blisters and values from the L3-exposed blisters were used to calculate the fold change in gene expression. The table contains all the surrogates of LC activation examined throughout the study. The median fold changes over all 6 donors are given.
DISCUSSION
Knowledge of the role of LC in skin immunity has been evolving since their discovery. Long considered one of the main antigen-presenting cells in the skin and primarily responsible for initiation of adaptive immune responses in the draining LN, alternative explanations of LC function have been recently suggested. With the advent of langerin-specific antibodies, it is quite clear that LC are the primary APC in the epidermis, but it is also clear that other subsets of DC, found in the dermis, may play an equally important role in initiating immune responses at the skin-environment interface (reviewed in reference 2). Additionally, there is increasing evidence that LC—rather than initiating a robust adaptive response—can induce immune tolerance (13).
The role of LC in filarial infection has not been extensively studied. The migratory route of the L3 of filarial worms through the skin, however, implicates the interaction between the L3 stage and epidermal LC as being important for the initial priming of the immune response to the parasite. Whether this interaction between the L3 and the LC leads to robust priming of naïve T cells or alters the function of LC in such a way as to allow the L3 to bypass this innate barrier and successfully enter the host awaits further clarification. Nevertheless, we have shown previously, using skin blister explants, that L3-exposed human LC fail to be activated fully and, to some degree, are functionally suppressed (7). One major drawback of the ex vivo system is that on average, ∼1,000 to 3,000 LC crawl out of each blister. The low yield of LC from these blisters hinders in-depth investigation into the L3-LC interaction. Consequently, we developed an in vitro system by generating LC from monocytes of healthy volunteers cultured with GM-CSF/IL-4 and TGF-β (14); by doing so, we were able to generate large numbers of LC that enabled more in-depth analysis, not only of L3-LC interaction, but also of L3-LC-KC interaction.
There are several surface markers that are generally used to identify human LC, including langerin (CD207), E-cadherin, CLA, CD14, and CD1a (9, 10). Langerin, a C-type lectin receptor, and CD1a, a major histocompatibility complex class I (MHC-I)-type transmembrane glycoprotein, have been shown to be involved in antigen internalization and presentation (15, 16). Both E-cadherin and CLA facilitate LC adherence to KC; their downregulation aids in the migration of LC from the epidermis (17, 18). Therefore, it is of interest to address any changes in these molecules upon pathogen exposure.
In our system of in vitro-generated LC, each of these LC-associated markers could be identified on the surface of the in vitro-generated LC. The percentage of langerin+ cells varied from donor to donor (range, 1.23 to 72.2% [data not shown]). The wide range of langerin expression on our cells could reflect generalized alteration in the surface expression of langerin, which is known to recycle between the plasma membrane and the early endosomes (19). The surface expression of LC-specific molecules did not change in response to L3 when gated on cells (Fig. 1C). However, gating on only the langerin+ cells showed an increase in surface expression of E-cadherin on L3-exposed LC compared to the unexposed cells (data not shown). The downregulation of E-cadherin is associated with activation of LC (2, 3). Thus, this specific upregulation of E-cadherin provides further evidence that the L3 are nonactivating. Additionally, since E-cadherin plays a role in the adherence of the LC to the other cells in the epidermis, the upregulation of this molecule on the surfaces of LC may be an evasion strategy used by the L3 to prevent the LC from migrating out of the epidermis. In contrast, LPS exposure induced the expected decrease in expression of E-cadherin, langerin, and CD14 and the increased expression of CLA (Fig. 1C). We also examined the effect of L3 on the LC activation markers CD40, CD80, CD86, CD83, and HLA-DR. These surface molecules have been shown to be upregulated in activated (i.e., mature) LC (11) and, for CD40, CD80, and CD86, in response to an intracellular parasite, Leishmania major (20). With the exception of CD80, the expression of each of these molecules remained unchanged on LC following exposure to L3 (Fig. 1C). Our data suggest that the LC were not fully activated by L3 but were fully functional in that they maintained the capacity to be activated by other stimuli (e.g., LPS).
LC typically produce cytokines when activated. That the L3 failed to induce cytokine secretion (except IL-6) finds support in previous studies of B. malayi infection in jirds, where a transient neutrophil infiltrate was found surrounding the L3 in the dermis, a process that was preceded by increases in IL-6 and TNF-α mRNAs (21). IL-10 is another cytokine thought to be involved in suppression of the immune response to filarial parasites (22); however, whether IL-10 is produced in the skin following filarial infection is still being defined. Our data indicate that IL-10 was not being produced by LC in response to L3, although it could be induced by TLR ligation (Fig. 2C).
We have shown previously that 72-h exposure of ex vivo skin blisters to live L3 of B. malayi upregulates both the mRNA expression and the production of IL-18 (7). Therefore, we examined IL-18 production from LC exposed to L3, as well as production of IFN-γ and TNF-α, all of which are crucial for LC migration (23–25). To our surprise, we were unable to demonstrate IL-18 production from the in vitro-derived LC regardless of the stimuli used (data not shown). Moreover, mRNA expression of genes involved in IL-18 regulation and signaling was also not changed by L3 exposure (Fig. 2B), nor was mRNA expression of the NLRP3, NLRP1, NLRC4, AIM2, or ASC inflammasome gene (Fig. 2A).
The lack of IL-18 production from the in vitro LC was in stark contrast to the ex vivo blister LC used previously (7). Because LC function is optimal in the presence of KC-produced cytokines, such as IL-1 and TNF-α (6), and the ex vivo blisters contained both LC and KC, we examined the LC requirement for KC for full activation using LC-KC coculture (Fig. 4). Not surprisingly, LC activation was indeed significantly better in the presence of KC; however, even when conditions were optimized for LC-KC interactions, L3 remained relatively inert in their ability to activate LC, in contrast to what was seen in response to T. gondii, an intracellular protozoan parasite known to induce both IP-10 and CXCL9 in mice (26, 27). Both IP-10 and CXCL9 are T cell chemokines regulated by IFN-γ (27). The lack of induction of these chemokines by L3 in LC suggests that the IFN-γ response—and consequently, the recruitment of T cells—is diminished in this model; however, LC exposed to T. gondii do upregulate IP-10 and CXCL9, so the in vitro cells are capable of activating this pathway of T cell recruitment. While the specific contribution of each cell type to the overall levels of cytokine production was not determined in the cocultures, the differences in cytokine levels between the monocultures and the cocultures highlight the interrelationship of the two cell types. Additionally, the observation that L3 did not alter cytokine production from the KC-LC cocultures suggests that L3 elicit a relatively quiescent response from both cell types.
Human LC express a full complement of TLRs (1 to 10), which are activated by specific TLR ligands (28). TLRs on LC are important for the cutaneous immune response to microbial pathogens, such as bacteria and viruses (29). As TLRs are one of the main signaling pathways following pathogen recognition and because L3 fail to activate in vitro-generated LC, we examined the effect of L3 on expression of and signaling through two of the most highly expressed TLRs by LC. Both TLR3 and TLR4 have been shown previously to be functionally repressed by the mf stage of B. malayi (12), a stage that shares some (but not all) proteins with the L3 (30), while TLR2 was activated by mf (12). The finding that L3 exposure failed to alter TLR expression or responses to TLR ligands in LC points to the concept of parasite stage specificity (30) and the anatomical location and/or specificity of LC (12).
While there are intrinsic differences between LC generated in vitro from monocytes or CD34+ cells and ex vivo LC, a microarray analysis of ex vivo blisters exposed to L3 soluble factors through transwells showed that none of the genes used as surrogates for LC activation, such as those for IL-18, caspase 1, IL-18BP, NLRPs, IL-6, and IL-8, were significantly up- or downregulated by the parasite (Table 2). Also, of the over 48,000 genes present on the array, only 51 were significantly differentially expressed in all the donors between unexposed LC and LC exposed to L3 (Table 1). Of those 51 differentially expressed genes, the majority (38/51) were downregulated by exposure to L3. While the downregulated genes were found to be related to antigen processing and presentation, as found in the previous study (7), the pattern of downregulation of LC gene expression by L3 remains the same. Differences between the two studies may be related to the length of exposure to the parasites (72 h in the previous study [7] compared with 48 h in the present study). While the difference in exposure time could account for the lack of response by the LC to L3, it is unlikely that L3 remain in the skin during migration for an extended period. Studies in mice show that the majority of L3 injected into the skin are present in the draining LN within 24 h (31). Other differences between the two studies may involve using L3 in contact or its effect on the entire blister (7) compared with L3 in transwells and their influence on crawl-out LC (present study).
In general, LC may respond differently to live parasites than to the parasite-secreted soluble factors. The paucity of changes seen in the mRNA expression of LC exposed to soluble factors of L3 was slightly surprising given that epidermal LC do internalize L3 antigens (7). However, the possibility that additional changes in LC function are the result of direct cell-parasite contact cannot be completely excluded. To date, there are no data on the receptors required for L3 antigen internalization or recognition. A homologue of Acanthocheilonema viteae-derived ES-62, is expressed in molting L3 (30) of B. malayi and is recognized by TLR4 (32), a TLR known to be expressed on LC that may represent a target for antigen internalization by LC. Using proteomics to identify the L3 secretome, only relatively few secreted proteins have been identified: several thioredoxin peroxidases known to neutralize host reactive oxygen (33). Among the nonsecreted proteins that may modulate the host immune response is a TGF-β homologue (TGH-2) that was found to be expressed primarily in the L3 stage of the parasite and that may mimic human TGF-β (30). Studies are ongoing in our laboratory to identify additional immunomodulatory proteins that are parasite derived.
The concept that LC may not be the only innate cells in the skin that respond to invading tissue-dwelling parasites is supported by data from a mouse model of schistosomiasis in which LC do migrate to draining LN in response to larval exposure, but they fail to prime protective CD4+ cells in the LN (34). In L. major infection in mice, LC have been shown to reduce regulatory T cell immigration to the site of infection, which leads to and enhances effector T cell function and attenuation of disease (35).
In conclusion, our studies of LC exposed to the L3 stage of B. malayi have shown a relatively quiescent response of the LC to this infective stage of the parasite. This quiescent response adds to the increasing evidence that LC have many different functions in the skin other than priming the adaptive immune response, that these functions depend on the type and nature of the stimulus involved, and that skin-transiting helminths have evolved methods for bypassing the hosts' first line of immune defense by failing to fully activate LC.
ACKNOWLEDGMENTS
We thank Dragana Jankovic, Alan Sher, and Kevin Tosh for providing us with T. gondii. We thank Allison McBride for supplying feeder cells and primary neonatal keratinocytes. We thank Brenda Rae Marshall, DPSS, NIAID, for editing.
This work was supported by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print 19 February 2013
REFERENCES
- 1. Lustigman S, Prichard RK, Gazzinelli A, Grant WN, Boatin BA, McCarthy JS, Basanez MG. 2012. A research agenda for helminth diseases of humans: the problem of helminthiases. PLoS Negl. Trop. Dis. 6:e1582 doi:10.1371/journal.pntd.0001582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Romani N, Brunner PM, Stingl G. 2012. Changing views of the role of Langerhans cells. J. Investig. Dermatol. 132:872–881 [DOI] [PubMed] [Google Scholar]
- 3. Mathers AR, Larregina AT. 2006. Professional antigen-presenting cells of the skin. Immunol. Res. 36:127–136 [DOI] [PubMed] [Google Scholar]
- 4. Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR. 2003. Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301:1925–1928 [DOI] [PubMed] [Google Scholar]
- 5. Ritter U, Meissner A, Scheidig C, Korner H. 2004. CD8 alpha- and Langerin-negative dendritic cells, but not Langerhans cells, act as principal antigen-presenting cells in leishmaniasis. Eur. J. Immunol. 34:1542–1550 [DOI] [PubMed] [Google Scholar]
- 6. Sugita K, Kabashima K, Atarashi K, Shimauchi T, Kobayashi M, Tokura Y. 2007. Innate immunity mediated by epidermal keratinocytes promotes acquired immunity involving Langerhans cells and T cells in the skin. Clin. Exp. Immunol. 147:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Semnani RT, Law M, Kubofcik J, Nutman TB. 2004. Filaria-induced immune evasion: suppression by the infective stage of Brugia malayi at the earliest host-parasite interface. J. Immunol. 172:6229–6238 [DOI] [PubMed] [Google Scholar]
- 8. Meyers C, Mayer TJ, Ozbun MA. 1997. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 71:7381–7386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Musso T, Scutera S, Vermi W, Daniele R, Fornaro M, Castagnoli C, Alotto D, Ravanini M, Cambieri I, Salogni L, Elia AR, Giovarelli M, Facchetti F, Girolomoni G, Sozzani S. 2008. Activin A induces Langerhans cell differentiation in vitro and in human skin explants. PLoS One 3:e3271 doi:10.1371/journal.pone.0003271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elder JT, Reynolds NJ, Cooper KD, Griffiths CE, Hardas BD, Bleicher PA. 1993. CD1 gene expression in human skin. J. Dermatol. Sci. 6:206–213 [DOI] [PubMed] [Google Scholar]
- 11. Nguyen VA, Dubrac S, Forstner M, Huter O, Del Frari B, Romani N, Ebner S. 2011. CD34+-derived Langerhans cell-like cells are different from epidermal Langerhans cells in their response to thymic stromal lymphopoietin. J. Cell. Mol. Med. 15:1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Semnani RT, Venugopal PG, Leifer CA, Mostbock S, Sabzevari H, Nutman TB. 2008. Inhibition of TLR3 and TLR4 function and expression in human dendritic cells by helminth parasites. Blood 112:1290–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shklovskaya E, O'Sullivan BJ, Ng LG, Roediger B, Thomas R, Weninger W, Fazekas de St Groth B. 2011. Langerhans cells are precommitted to immune tolerance induction. Proc. Natl. Acad. Sci. U. S. A. 108:18049–18054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geissmann F, Prost C, Monnet JP, Dy M, Brousse N, Hermine O. 1998. Transforming growth factor beta1, in the presence of granulocyte/macrophage colony-stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J. Exp. Med. 187:961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stoitzner P, Romani N. 2011. Langerin, the “Catcher in the Rye”: an important receptor for pathogens on Langerhans cells. Eur. J. Immunol. 41:2526–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peiser M, Grutzkau A, Wanner R, Kolde G. 2003. CD1a and CD1c cell sorting yields a homogeneous population of immature human Langerhans cells. J. Immunol. Methods 279:41–53 [DOI] [PubMed] [Google Scholar]
- 17. Yasaka N, Furue M, Tamaki K. 1996. Expression of cutaneous lymphocyte-associated antigen defined by monoclonal antibody HECA-452 on human Langerhans cells. J. Dermatol. Sci. 11:19–27 [DOI] [PubMed] [Google Scholar]
- 18. Blauvelt A, Katz SI, Udey MC. 1995. Human Langerhans cells express E-cadherin. J. Investig. Dermatol. 104:293–296 [DOI] [PubMed] [Google Scholar]
- 19. Stambach NS, Taylor ME. 2003. Characterization of carbohydrate recognition by langerin, a C-type lectin of Langerhans cells. Glycobiology 13:401–410 [DOI] [PubMed] [Google Scholar]
- 20. von Stebut E, Belkaid Y, Nguyen BV, Cushing M, Sacks DL, Udey MC. 2000. Leishmania major-infected murine Langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur. J. Immunol. 30:3498–3506 [DOI] [PubMed] [Google Scholar]
- 21. Porthouse KH, Chirgwin SR, Coleman SU, Taylor HW, Klei TR. 2006. Inflammatory responses to migrating Brugia pahangi third-stage larvae. Infect. Immun. 74:2366–2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitre E, Chien D, Nutman TB. 2008. CD4(+) (and not CD25+) T cells are the predominant interleukin-10-producing cells in the circulation of filaria-infected patients. J. Infect. Dis. 197:94–101 [DOI] [PubMed] [Google Scholar]
- 23. Cumberbatch M, Dearman RJ, Antonopoulos C, Groves RW, Kimber I. 2001. Interleukin (IL)-18 induces Langerhans cell migration by a tumour necrosis factor-alpha- and IL-1beta-dependent mechanism. Immunology 102:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caceres-Dittmar G, Sanchez MA, Oriol O, Kraal G, Tapia FJ. 1992. Epidermal compromise in American cutaneous leishmaniasis. J. Investig. Dermatol. 99:95S–98S [DOI] [PubMed] [Google Scholar]
- 25. von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. 1998. Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J. Exp. Med. 188:1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, Luster AD. 2000. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity 12:483–494 [DOI] [PubMed] [Google Scholar]
- 27. Wen X, Kudo T, Payne L, Wang X, Rodgers L, Suzuki Y. 2010. Predominant interferon-gamma-mediated expression of CXCL9, CXCL10, and CCL5 proteins in the brain during chronic infection with Toxoplasma gondii in BALB/c mice resistant to development of toxoplasmic encephalitis. J. Interferon Cytokine Res. 30:653–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Renn CN, Sanchez DJ, Ochoa MT, Legaspi AJ, Oh CK, Liu PT, Krutzik SR, Sieling PA, Cheng G, Modlin RL. 2006. TLR activation of Langerhans cell-like dendritic cells triggers an antiviral immune response. J. Immunol. 177:298–305 [DOI] [PubMed] [Google Scholar]
- 29. Miller LS. 2008. Toll-like receptors in skin. Adv. Dermatol. 24:71–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bennuru S, Meng Z, Ribeiro JM, Semnani RT, Ghedin E, Chan K, Lucas DA, Veenstra TD, Nutman TB. 2011. Stage-specific proteomic expression patterns of the human filarial parasite Brugia malayi and its endosymbiont Wolbachia. Proc. Natl. Acad. Sci. U. S. A. 108:9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chirgwin SR, Coleman SU, Porthouse KH, Klei TR. 2006. Tissue migration capability of larval and adult Brugia pahangi. J. Parasitol. 92:46–51 [DOI] [PubMed] [Google Scholar]
- 32. Goodridge HS, Marshall FA, Else KJ, Houston KM, Egan C, Al-Riyami L, Liew FY, Harnett W, Harnett MM. 2005. Immunomodulation via novel use of TLR4 by the filarial nematode phosphorylcholine-containing secreted product, ES-62. J. Immunol. 174:284–293 [DOI] [PubMed] [Google Scholar]
- 33. Bennuru S, Semnani R, Meng Z, Ribeiro JM, Veenstra TD, Nutman TB. 2009. Brugia malayi excreted/secreted proteins at the host/parasite interface: stage- and gender-specific proteomic profiling. PLoS Negl. Trop. Dis. 3:e410 doi:10.1371/journal.pntd.0000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumkate S, Jenkins GR, Paveley RA, Hogg KG, Mountford AP. 2007. CD207+ Langerhans cells constitute a minor population of skin-derived antigen-presenting cells in the draining lymph node following exposure to Schistosoma mansoni. Int. J. Parasitol. 37:209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kautz-Neu K, Noordegraaf M, Dinges S, Bennett CL, John D, Clausen BE, von Stebut E. 2011. Langerhans cells are negative regulators of the anti-Leishmania response. J. Exp. Med. 208:885–891 [DOI] [PMC free article] [PubMed] [Google Scholar]