

Abstract
Escherichia coli is the leading cause of Gram-negative neonatal bacterial meningitis and also causes meningitis and meningoencephalitis in older and immunocompromised patients. Here, we determined the contribution of granulocytes, monocytes, and TLR signaling cascades in the resistance of adult mice to Escherichia coli K1 brain infection. Deficiency in MyD88 (myd88−/−) but not in TRIF (triflps2) adaptor proteins dramatically reduced the survival of animals. Depletion of CD11b+ Ly-6G+ Ly-6Cint neutrophils by application of the anti-Ly-6G (1A8) monoclonal antibody (MAb) led to higher bacterial loads in cerebellum and spleen tissue and resulted in increased mortality compared to those of isotype-treated controls. Depletion of CD11b+ Ly-6G+ Ly-6Cint neutrophils and CD11b+ Ly-6G− Ly-6Chigh monocytes by administration of the anti-Gr-1 (RB6-8C5) MAb rendered mice even more susceptible to the infection, with higher central nervous system (CNS) and spleen bacterial burdens than anti-Ly-6G-treated animals. Depletion of ∼50% of CD11b+ Ly-6G− Ly-6Chigh monocytes by injection of the anti-CCR2 (MC-21) MAb resulted in a trend toward higher mortality compared to that with isotype treatment. Production of interleukin 1β (IL-1β), IL-6, KC, and MIP-2 in the CNS strongly depended on the bacterial load: increased levels of these cytokines/chemokines were found after depletion of CD11b+ Ly-6G+ Ly-6Cint neutrophils alone or together with CD11b+ Ly-6G− Ly-6Chigh monocytes. These findings identify Toll-like receptor (TLR)-MyD88 signaling and neutrophil and monocyte activity as critical elements in the early host defense against E. coli meningitis.
INTRODUCTION
Escherichia coli K1 is the major cause of Gram-negative meningitis in neonates and young infants. Less frequently, E. coli K1 meningitis can also occur in adults with impaired host defense, head trauma, neurosurgical interventions, or Gram-negative sepsis (1–3). The recent emergence of multidrug-resistant strains of E. coli K1 might further worsen the poor clinical outcome of these patients (4).
Several studies in humans and in the newborn rat model of experimental hematogenous E. coli meningitis suggest that a high degree of bacteremia is required for meningeal invasion (5, 6). The ability of bacteria to escape host defense and achieve the threshold of bacteremia required for invasion of the central nervous system (CNS) is higher in immunocompromised individuals (e.g., neonates) than in immunocompetent adults, thus explaining the differences in the occurrence of E. coli K1 meningitis (1, 3, 7). In addition to bacteremia, invasion of brain microvascular endothelial cells (BMEC) appears to be a prerequisite for E. coli K1 to induce meningitis (7). Some E. coli K1 structures, such as the outer membrane protein A (OmpA), Ibe proteins, and cytotoxic necrotizing factor 1, are necessary for the successful bacterial traversal across the blood-brain barrier or blood-cerebrospinal fluid (CSF) barrier (7). Despite there being an increasing understanding of how E. coli K1 interacts with the host to cause meningitis (reviewed by reference 8), little is known about how the host fights against E. coli once bacteria have entered the CNS. Microglial cells and perivascular and meningeal macrophages represent the first line of defense against microorganisms invading the CNS prior to leukocyte infiltration (9). Microglia express Toll-like receptors (TLRs) that identify pathogen-associated molecular patterns (PAMPs). TLR4 senses lipopolysaccharide (LPS) from Gram-negative bacteria, leading to the recruitment of both adaptor molecules, myeloid differentiation factor 88 (MyD88) and Toll/interleukin 1R (IL-1R) (TIR) domain-containing adaptor protein inducing beta interferon (TRIF), with subsequent downstream signaling consequences (10). Upon such activation, microglia release a wide array of proinflammatory mediators, including KC (rodent homologue of growth-related oncogene α/CXCL1) and macrophage inflammatory protein 2 (MIP-2/CXCL2), which act as potent neutrophil attractants (11, 12). Consistently, MyD88-deficient mice show a markedly reduced CSF leukocyte infiltration associated with decreased brain mRNA levels of KC and MIP-2 in experimental pneumococcal meningitis (13).
Depletion of cell lineage-specific immune cells has been used successfully to elucidate their role in immune responses, including infections. The monoclonal antibody (MAb) RB6-8C5, originally described as binding the granulocyte receptor 1 (Gr-1), has been widely used to induce neutropenia in murine models of disease (14). RB6-8C5, however, does not bind only to the granulocyte surface marker Ly-6G but also to Ly-6C isoforms (15) that are expressed on other leukocyte populations. In contrast, the MAb 1A8 binds specifically to Ly-6Ghigh neutrophils (15), and its administration has no impact on Gr-1+ monocytes (14). Blood monocytes consist of two principal subsets based upon expression of Gr-1, CCR2, and CX3CR1 (16). The MC-21 MAb has been successfully used to study the role of the subset of Gr-1+ inflammatory monocytes (Ly-6Chigh CCR2+ CX3CR1low) during pneumococcal meningitis (17).
Here, we determined individual contributions of the two major TLR4 signaling routes and of circulating granulocytes and monocytes to the host response after intracerebral infection with E. coli K1, using MyD88- and TRIF-deficient (myd88−/− triflps2) mice and performing three antibody-mediated cell depletion strategies: (i) depletion of both granulocytes and monocytes with anti-Gr-1 MAb (RB6-8C5), (ii) specific granulocyte suppression by anti-Ly-6G MAb (1A8), and (iii) specific depletion of inflammatory monocytes by anti-CCR2 MAb (MC-21).
MATERIALS AND METHODS
Bacteria.
The Escherichia coli strain K1 (serotype O18:K1:H7) was originally isolated from the cerebrospinal fluid (CSF) of a child with neonatal meningitis (gift of Gregor Zysk, Institute of Medical Microbiology, Düsseldorf, Germany). Characterization by the Nationales Referenzzentrum für Salmonellen und andere Enteritiserreger at the Robert Koch Institute (Wernigerode, Germany) revealed that this strain expresses sfa (S fimbrial adhesin) and cdt (cytolethal distending toxin) genes. Bacteria were grown overnight on blood agar plates, harvested with 0.9% saline, and stored at −80°C. Frozen aliquots were used for the experiments and adjusted with saline to the required bacterial concentration.
Mice and monitoring.
Animal experiments were approved by the Animal Care Committee of the University Hospital of Göttingen and by the Niedersächsische Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Braunschweig, Lower Saxony, Germany. Meningitis was induced by injection of 3 × 103 to 5.5 × 103 CFU of E. coli K1 into the right frontal neocortex (18, 19) under intraperitoneal anesthesia with ketamine (100 mg/kg of body weight) and xylazine (10 mg/kg of body weight). Mice (2 to 3 months old; weight, 20 to 30 g) were weighed daily and scored clinically (0, no apparent behavioral abnormality; 1, moderate lethargy; 2, severe lethargy; 3, unable to walk; 4, dead) (19).
C57BL/6 mice were used in antibody depletion experiments and as controls to match the genetic background in experiments with MyD88- and TRIF-deficient strains (myd88−/− triflps2) (12). In survival experiments, animals were monitored over a 14-day period after infection. In bacteriological studies, mice were sacrificed 30 h after infection.
Antibodies for depletion.
Anti-Gr-1 MAb (clone RB6-8C5; eBioscience), anti-Ly-6G (clone 1A8; BioXcell, West Lebanon, NH), MC-21 MAb (20), or isotype-matching control antibodies (rat IgG2a, BioXCell; rat IgG2b, eBioscience) were used for cell depletions.
Flow cytometry analysis.
Whole-blood samples (n ≥ 5 mice/group) were diluted at 4°C in buffer solution (phosphate-buffered solution [PBS] containing 2% fetal calf serum [FCS], 1 mM EDTA, and 0.2% NaN3), stained with allophycocyanin (APC)-conjugated anti-mouse CD11b (Integrin αM, Mac-1α; eBioscience), peridinin chlorophyll protein (PerCP)-conjugated rat anti-mouse CD45R/B220 (clone RA3-6B2; BD Pharmingen), fluorescein isothiocyanate (FITC)-conjugated anti-mouse Ly-6C (clone AL-21; BD Pharmingen), and phycoerythrin (PE)-conjugated rat anti-mouse Ly-6G (clone 1A8; BD Pharmingen) antibodies and incubated with fluorescence-activated cell sorter (FACS) lysis solution (BD Biosciences) to lyse erythrocytes. Samples were analyzed with a FACSCanto II flow cytometer (BD Biosciences, Heidelberg, Germany). Data were acquired using WinMDI software.
Sample processing.
Animals were killed by cervical dislocation. The whole brain and spleen were removed, and the cerebellum was dissected from the brain stem. Cerebellum and spleen homogenates were prepared in 0.5 ml 0.9% NaCl solution. One half was serially diluted (1:10) in saline and plated on blood agar plates (detection limit, 200 CFU/ml of homogenate). The other half was frozen at −80°C. The brain was fixed in 4% paraformaldehyde and then embedded in paraffin.
Cytokine and chemokine measurement.
Cytokines and chemokines were measured in cerebellum homogenates of mice sacrificed 30 h after infection. IL-1β, IL-6, KC (CXCL1), and MIP-2 (CXCL2) levels were determined by using DuoSet enzyme-linked immunosorbent assay (ELISA) development kits (R&D Systems, Wiesbaden, Germany). The sensitivity of the assays for these cytokines/chemokines was 7.5 pg/ml. ELISAs were performed according to the manufacturer's instructions (12, 18).
Histological analysis.
Paraffin-embedded, 2-μm coronal brain sections from dead mice from survival experiments as well as from animals sacrificed 30 h after infection were analyzed. Chloroacetate esterase (CAE) stainings were performed to evaluate the degree of inflammation in three superficial meningeal regions and the hippocampal fissure. CAE is a staining method to detect neutrophils. It is, however, not completely specific and can also stain monocytes/macrophages (21). Stained sections were blinded and semiquantitatively scored for the number of CAE-stained leukocytes in one high-power field (×40 objective) per region: no leukocytes, score 0; <10 leukocytes, score 1; 10 to 50 leukocytes, score 2; >50 leukocytes, score 3. For each animal, the scores of the individual fields were added and then divided by the number of scored regions. Ionized calcium-binding adaptor molecule 1 (Iba-1) was used to identify microglial cells. In each animal, Iba-1-positive cells were quantified in at least three different neocortical regions (×20 objective), added, and then divided by the number of scored regions.
Statistical analysis.
Survival rates were compared using the log rank test. The Mann-Whitney U test was used to analyze bacterial loads in cerebellum and spleen, levels of cytokines/chemokines in cerebellar homogenates, leukocyte infiltration, and clinical scores. Granulocyte and monocyte ratios in flow cytometry experiments and quantification of Iba-1+ cells were analyzed by Student's t test. The Bonferroni-Holm method was used to correct for repeated testing. The correlation between bacterial titers and cytokine/chemokine levels was analyzed using Spearman's rank correlation coefficient. For all analyses, GraphPad Prism version 5 (GraphPad Software, San Diego, CA) was used, and a P value of <0.05 was considered statistically significant.
RESULTS
MyD88 but not TRIF signaling has an essential function in the protective host response to E. coli K1 meningitis.
MyD88-deficient mice were unable to control meningitis induced by intracerebral injection of 5.5 × 103 CFU E. coli K1 (Fig. 1A). All myd88−/− mice (n = 8) succumbed to meningitis within 70 h after infection, whereas 43% (6/14) of the wild-type (wt) group survived (P = 0.0006; log rank test). The severity of the disease as assessed by the clinical score was markedly enhanced in myd88−/− mice compared to the wt animals (P = 0.004 at 48 h postinfection). Increased susceptibility to infection was reflected by higher bacterial titers (3 to 4 log CFU/ml) in cerebellar (P = 0.0003; Mann-Whitney U test) and spleen (P = 0.0003; Mann-Whitney U test) homogenates of myd88−/− mice (n = 18) than in wt controls (n = 14) 30 h after infection (Fig. 1B and C).
Fig 1.
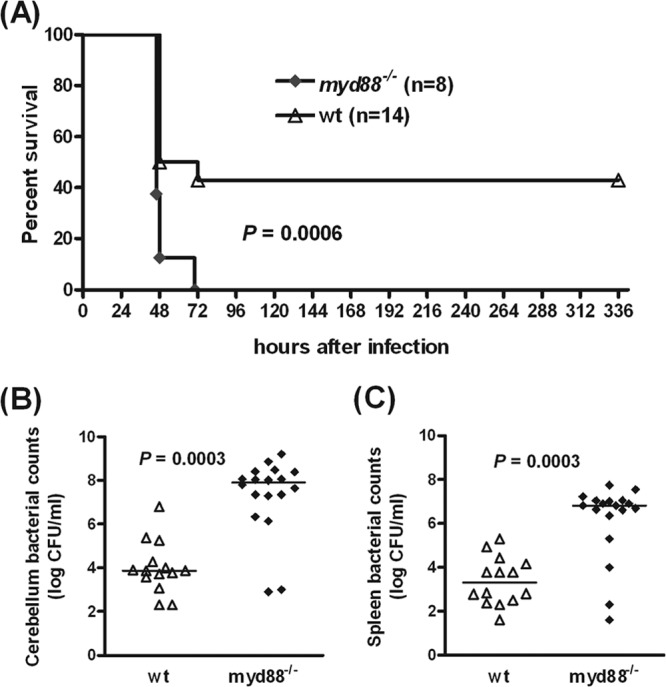
MyD88 signaling plays an essential role in host responses against E. coli K1 meningitis. (A) Survival following E. coli K1 infection (5.5 × 103 CFU/mouse) was significantly lower in myd88−/− mice (0%; P = 0.0006; log rank test) than in C57BL/6 wt mice (43%). After 30 h of infection, bacterial counts in cerebellum (B) and spleen (C) homogenates of myd88−/− mice were significantly higher than those found in wt controls (P = 0.0003; Mann-Whitney U test). Symbols in panels B and C represent individual mice, and bars indicate median values.
In contrast, no differences in survival were observed between triflps2 and wt mice (P = 0.72; log rank test) (Fig. 2), suggesting that the TRIF-dependent pathway was dispensable for the control of intracerebral E. coli infection. Survival rates after 14 days of infection were 58.8% in triflps2 (n = 17) and 53.3% in wt (n = 15) mice. Both triflps2 and wt animals showed no differences in the clinical score throughout the survival experiments.
Fig 2.

TRIF signaling does not contribute to the resistance to E. coli K1 meningitis. Survival following intracerebral E. coli K1 infection (5.5 × 103 CFU/mouse) was not statistically different between triflps2 (58.8%) and C57BL/6 wt (53.3%) mice (P = 0.72; log rank test).
Intracerebral injection of E. coli K1 induces the recruitment of neutrophils and monocytes into the peripheral blood.
During bacterial meningitis, circulating leukocytes, predominantly neutrophilic granulocytes and monocytes, quickly enter the subarachnoid space as key components of the innate immune defense. We first determined the numbers of CD11b+ Ly-6G+ Ly-6Cint granulocytes and CD11b+ Ly-6G− Ly-6Chigh monocytes in the peripheral blood of E. coli K1-infected wt mice (n ≥ 14) over time by flow cytometry (Fig. 3). An increase of granulocytes in the blood was observed beginning at 12 h after E. coli K1 infection. Comparative analysis with B lymphocytes indicated that Ly-6G+ Ly-6Cint granulocytes increased from 0.183 ± 0.102 (mean ± standard deviation [SD]) before infection to 1.112 ± 1.330 at 36 h (P < 0.01; Student's t test) while Ly-6G− Ly-6Chigh monocytes increased from 0.092 ± 0.070 at the time point of 0 h to 0.160 ± 0.137 at the 36-h time point (P > 0.05; Student's t test).
Fig 3.
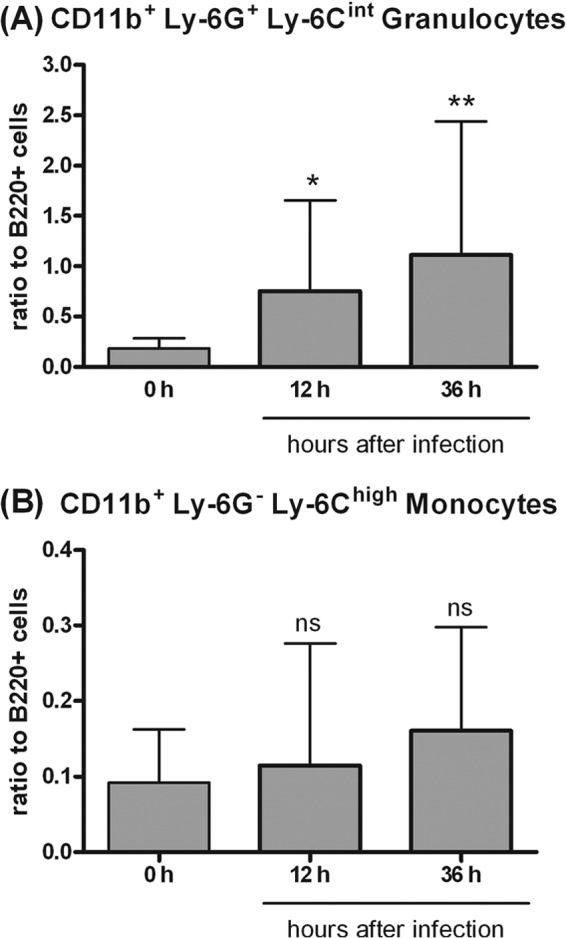
Increases in CD11b+ Ly-6G+ Ly-6Cint granulocytes (A) and in CD11b+ Ly-6G− Ly-6Chigh monocytes (B) in the peripheral blood of E. coli K1-infected C57BL/6 wt mice examined by flow cytometry. Cell counts were normalized to 20,000 B cells (B220+) and expressed as ratios. Data are shown as means ± SD (n = 5 mice per time point). Asterisks indicate statistically significant differences from the noninfected controls by Student's t test, followed by a correction for repeated testing by the Bonferroni-Holm method (*, P < 0.05; **, P < 0.01; ns, not significant).
Characterization of the meningeal inflammation after E. coli K1 challenge.
Representative examples of the different degrees of meningeal inflammation (scores 0 to 3) in CAE-stained brain sections are shown in Fig. 4. CAE staining performed in sections from isotype-treated and depleted animals which died during the acute phase of infection (Fig. 5A) confirmed that anti-Gr-1-treated and anti-Ly-6G-treated mice had fewer infiltrating leukocytes than isotype-treated animals (P = 0.0009 and P = 0.11, respectively). Because mortality in the control groups was low, to perform statistical analysis, all IgG2a- and IgG2b-treated animals were pooled in one isotype group. Meningeal inflammation scores from CAE stainings of animals which were sacrificed 30 h after infection are presented in Fig. 5B. Here, the absence of inflammation in isotype-treated animals that had already efficiently cleared the infection gave a false display of a weaker meningeal inflammatory reaction in immunocompetent animals than in immunocompromised animals.
Fig 4.

Illustrative examples of meningeal inflammation scores (0 [A], 1 [B], 2 [C], 3 [D]) in chloroacetate esterase stainings of brain sections of depleted and control animals 30 h after intracerebral challenge with E. coli K1. Arrows in panel B indicate CAE-positive cells. Brain sections were prepared and assessed as described in Materials and Methods. Magnification, ×40.
Fig 5.
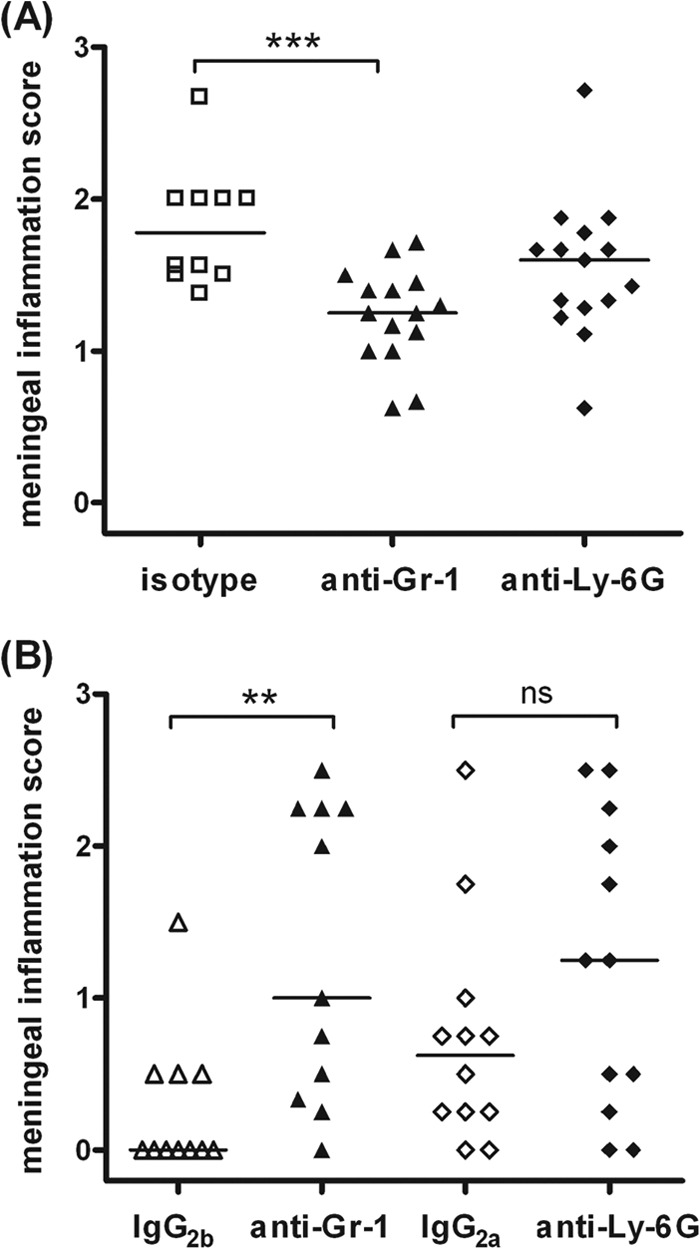
Meningeal infiltration score calculated in chloroacetate esterase-stained brain sections of isotype (IgG2a- and IgG2b-treated) mice and depleted (anti-Gr-1- and anti-Ly-6G-treated) mice which died during the acute phase of E. coli meningitis (A) and nondepleted and depleted animals which were sacrificed after 30 h of E. coli meningitis (B). Each symbol represents an individual mouse. Bars indicate median values. Statistical analysis was performed by Mann-Whitney U test followed by the Bonferroni-Holm method to correct for repeated testing (**, P < 0.01; ***, P < 0.001; ns, not significant). The few nondepleted mice which succumbed to the infection showed the highest degree of meningeal inflammation (A). Conversely, when animals were sacrificed 30 h after infection, depleted mice displayed higher scores of meningeal inflammation than isotype-treated animals because many immunocompetent mice had already overcome infection.
Quantification of microglial cells after E. coli K1 injection.
We evaluated cortical regions of Iba-1-stained brain sections to quantify microglial cells from isotype-treated and depleted animals which died during the acute phase of infection (Fig. 6A) as well as animals that were sacrificed after 30 h of E. coli K1 meningitis (Fig. 6B). Representative examples of Iba-1+ cells are shown in Fig. 6C. The number of Iba-1+ cells in anti-Gr-1-treated animals was significantly decreased compared to that in the isotype-treated mice (P < 0.05) in sections from both groups of mice which either died or were sacrificed at early infection. The number of Iba-1+ cells in anti-Ly-6G-treated animals was higher than that in the isotype-treated mice, with the difference being significant in mice which were sacrificed after 30 h of E. coli K1 infection (P < 0.05).
Fig 6.

Microglial cells were quantified in ionized calcium-binding adaptor molecule 1 (Iba-1)-stained brain sections of isotype (IgG2a- and IgG2b-treated) mice and depleted (anti-Gr-1- and anti-Ly-6G-treated) mice which died during the acute phase of E. coli meningitis (A) and nondepleted and depleted animals which were sacrificed after 30 h of E. coli meningitis (B). Each symbol represents an individual mouse. Bars indicate mean values. Statistical analysis was performed by Student's t test, followed by a correction for repeated testing by the Bonferroni-Holm method (*, P < 0.05). (C) Illustrative example of a neocortical region with Iba-1+ microglial cells (arrows). Brain sections were prepared and assessed as described in Materials and Methods. Magnification, ×20.
CD11b+ Ly-6G+ Ly-6Cint granulocytes and CD11b+ Ly-6G− Ly-6Chigh monocytes are required for the control of E. coli K1 meningitis.
In depletion experiments, animals were treated intraperitoneally (12 h prior to induction of meningitis and thereafter every 24 h with a total of three injections) with anti-Gr-1 (RB6-8C5), anti-Ly-6G (1A8), MC-21 monoclonal antibody (MAb), or respective isotype controls. Blood samples underwent flow cytometric analysis based on CD45R/B220, CD11b, Ly-6C, and Ly-6G staining (Fig. 7). While neutropenia was established upon injection of 1A8, administration of RB6-8C5 resulted in depletion of not only CD11b+ Ly-6G+ Ly-6Cint granulocytes but also CD11b+ Ly-6G− Ly-6Chigh monocytes. Administration of 10 μg of MC-21 led to a depletion of 51.6% ± 30.4% (mean ± SD) of CD11b+ Ly-6G− Ly-6Chigh inflammatory monocytes compared to the isotype-treated animals (10 μg of rat IgG2b).
Fig 7.

Flow cytometric analysis of blood from antibody-treated C57BL/6 mice at 24 and 48 h after intracerebral challenge with E. coli K1. The panel at 0 h represents a nondepleted and noninfected mouse. Administration of 20 μg of anti-Gr-1 MAb (RB6-8C5) led to an efficient depletion of Ly-6G+ Ly-6Cint granulocytes and Ly-6G− Ly-6Chigh inflammatory monocytes compared to the isotype control (20 μg of rat IgG2b). Mice treated with 50 μg of anti-Ly-6G MAb (1A8) showed an absence of Ly-6G+ Ly-6Cint granulocytes compared to the isotype control (50 μg of rat IgG2a). Application of 10 μg of anti-CCR2 MAb (MC-21) resulted in a partial depletion of Ly-6G− Ly-6Chigh inflammatory monocytes compared with the isotype control (10 μg of rat IgG2b). Data from one representative animal out of five are shown.
Survival experiments showed that neutrophils were essential for combating E. coli K1 infection (n ≥ 9). Anti-Gr-1-treated animals died significantly earlier and at a higher rate than mice treated with the isotype control (P = 0.0002; log rank test) (Fig. 8A). Survival in anti-Ly-6G-treated mice (Fig. 8D) was also lower (40.74%) than that in controls (65.39%; P = 0.049). Anti-Gr-1- and anti-Ly-6G-treated animals exhibited more-severe clinical symptoms of infection than isotype-treated control mice (P < 0.01; data not shown). Depletion of ∼50% of the CD11b+ Ly-6G− Ly-6Chigh inflammatory monocytes resulted in a tendency toward higher mortality (25%; 4/16) compared to that of the isotype-treated group (7%; 1/14) (P = 0.18; log rank test). The disease score did not differ between MC-21- and isotype-treated mice.
Fig 8.

CD11b+ Ly-6G+ Ly-6Cint granulocytes and CD11b+ Ly-6G− Ly-6Chigh monocytes were essential for the control of E. coli K1 meningitis. Kaplan-Meier survival curves from C57BL/6 mice treated with 20 μg of anti-Gr-1 MAb (RB6-8C5) or matching isotype (IgG2b) (A) and 50 μg of anti-Ly-6G MAb (1A8 clone) and matching isotype (IgG2a) (D) following intracerebral E. coli K1 infection (3 × 103 CFU/mouse). Bacterial counts were measured 30 h after infection in cerebellum (B and E) and spleen (C and F) homogenates (CFU/ml) from anti-Gr-1-, anti-Ly-6G-, and isotype-treated mice. Each symbol represents an individual mouse. Bars indicate median values. Statistical analysis was performed by log rank tests (A and D) and Mann-Whitney U tests (B, C, E, and F).
All animals that survived the infection were sacrificed after 14 days, and bacterial titers in cerebellar and spleen homogenates were determined. Three out of 11 anti-Ly-6G-treated animals showed positive cerebellum cultures, while in all isotype- and MC-21-treated animals, no viable bacteria were detected.
The impaired ability to overcome infection was illustrated by increased bacterial loads in cerebellum and spleen 30 h after infection in anti-Gr-1-treated mice (P < 0.0001; Mann-Whitney U test) (Fig. 8B and C) and in anti-Ly-6G-treated mice (P ≤ 0.004; Mann-Whitney U test) (Fig. 8E and F) compared to those found in the respective controls (n = 11 to 12/group). At 30 h, anti-Gr-1-depleted mice also showed higher concentrations of E. coli K1 in the cerebellum and spleen than did anti-Ly-6G-depleted mice (P ≤ 0.003; Mann-Whitney U test).
Absence of CD11b+ Ly-6G+ Ly-6Cint granulocytes and CD11b+ Ly-6G− Ly-6Chigh monocytes results in an increased production of inflammatory cytokines and chemokines 30 h after induction of E. coli K1 meningitis.
Based on the importance of neutrophils and monocytes in ensuring survival and controlling bacterial burdens during early E. coli K1 meningitis, we next investigated whether anti-Gr-1- and anti-Ly-6G-treated mice showed alterations in the levels of proinflammatory mediators. We measured IL-1β and IL-6 as key cytokines of innate defense during infection and the chemokines KC and MIP-2 as key players in the recruitment of neutrophils into the CNS (22, 23). Interestingly, anti-Gr-1- and anti-Ly-6G-treated mice showed a significantly enhanced production of inflammatory cytokines/chemokines in the cerebellar homogenates compared with that of their respective isotype-treated counterparts (Fig. 9). Anti-Gr-1 pretreatment resulted in a 60-fold increase of IL-1β, a 200-fold increase of IL-6, a 70-fold increase of KC, and a 20-fold increase of MIP-2 levels (P < 0.01 versus the isotype-treated mice). Anti-Ly-6G pretreatment led to an increase of the production of IL-1β by 30-fold, IL-6 by 17-fold, KC by 45-fold, and MIP-2 by 4-fold (P < 0.05 versus the isotype-treated mice). IL-6, KC, and MIP-2 levels were higher in anti-Gr-1-treated mice than in anti-Ly-6G-treated mice (P < 0.05). The two groups of depleted mice showed similar amounts of IL-1β (P = 0.08). The low levels of proinflammatory mediators found in the cerebellum homogenate of the majority of isotype-treated mice were most probably due to an effective bacterial clearance immediately after infection, which prevented the development of a strong inflammatory reaction in the whole brain. We hypothesized that the high concentrations of proinflammatory mediators were the consequence of the high bacterial load. Therefore, we examined the correlation between bacterial burdens and cytokine/chemokine production (Table 1). Bacterial titers in cerebellum homogenates strongly correlated with the levels of IL-1β, IL-6, KC, and MIP-2 (P < 0.0001) in all mice studied (n = 45). Thus, high concentrations of these proinflammatory mediators in the anti-Gr-1-treated and anti-Ly-6G-treated mice represented an ineffective inflammatory response in the brain after rapid bacterial clearance had failed.
Fig 9.

Inflammatory cytokines and chemokines in cerebellar homogenates from anti-Gr-1-, anti-Ly-6G-, and isotype-treated mice. Levels (in pg/ml) of IL-1β (A), IL-6 (B), KC (C), and MIP-2 (D) 30 h after intracerebral E. coli K1 infection (3 × 103 CFU/mouse) were measured by an ELISA. Each symbol represents an individual mouse. Bars indicate median values. Statistical analysis was performed by Mann-Whitney U test followed by the Bonferroni-Holm method to correct for repeated testing (*, P < 0.05; **, P < 0.01; ***, P < 0.001). The concentrations of the proinflammatory mediators were higher in depleted animals, because at this time point, the majority of immunocompetent mice had already cleared the bacteria and overcome infection (Fig. 8B and E).
Table 1.
Spearman's rank correlation coefficients between bacterial burdens and cytokine/chemokine levels in cerebellar homogenates of anti-Gr-1-, anti-Ly-6G-, and isotype-treated micea
| Mouse group | Correlation coefficient for each cytokine/chemokine |
|||
|---|---|---|---|---|
| IL-1β | IL-6 | KC | MIP-2 | |
| Anti-Gr-1 | 0.54 | 0.86*** | 0.36 | 0.92*** |
| IgG2b | 0.17 | 0.19 | 0.42 | 0.24 |
| Anti-Ly-6G | 0.52 | 0.71** | 0.70* | 0.77** |
| IgG2a | 0.89*** | 0.47 | 0.82*** | 0.45 |
| All mice | 0.73*** | 0.84*** | 0.93*** | 0.90*** |
After 30 h of intracerebral infection with E. coli K1. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
The immune defense of the CNS has been compared with a two-walled castle moat (24). The blood-brain barrier (BBB) consists of the endothelium of cerebral vessels and capillaries linked by tight junctions and surrounded by tight astrocytes and pericytes. It regulates the controlled entry or exclusion of soluble molecules, pathogens, and host cells into the brain. Together with the BBB, the blood-CSF barrier, i.e., the cylindric epithelium of the choroid plexus, serves as the outer wall. The CSF-drained fluid spaces represent the moat. The glia limitans and perivascular and meningeal macrophages serve as the inner wall. Inside the wall, microglial cells continually survey their environment with extremely motile processes (25). Microglia can phagocytose and kill many pathogens which have passed the two-walled castle moat, and stimulation of TLRs increases both phagocytosis and intracellular killing (26). Upon pathogen encounter, the function of microglia in the vicinity decides whether the infection remains limited and pathogens can be eliminated rapidly or whether the infection spreads and eventually causes the death of the host subsequent to meningitis or accompanying sepsis.
Microglia express molecules for the recognition of diverse PAMPs which are critical to mount a response to infection. Intact TLR→MyD88 signaling appears to be necessary to protect the brain tissue against invading microorganisms. Indeed, myd88−/− mice rapidly succumbed to pneumococcal meningitis within 24 h of intracerebral infection and developed higher bacterial counts in the CNS and more-severe bacteremia than wt controls (13). The importance of MyD88 signaling in the host defense against E. coli was demonstrated in an experimental sepsis model (27). More recently, Krishnan et al. reported that infection with E. coli K1 OmpA+ selectively upregulates TLR2 but not TLR4 expression in BMEC (28). Accordingly, upon intranasal inoculation of E. coli K1 OmpA+, newborn TLR2−/− mice were resistant to meningitis while newborn TLR4−/− animals succumbed to the infection within 72 h (28). Here, we show that absence of MyD88 decreased the resistance of the brain of adult mice to E. coli K1 meningitis. Even with very low doses of bacteria (250 CFU/mouse), mice lacking MyD88 were clearly more susceptible to intracerebral infection (60% mortality) than were their wt counterparts (0% mortality; data not shown). The higher bacterial titers in target organs of myd88−/− mice indicate that this signaling adaptor molecule has a critical role in the control of E. coli K1 meningitis and sepsis. Conversely, we observed that TRIF signaling is not (essentially) required for protection against E. coli K1 meningitis, since triflps2 mice showed mortality rates similar to those of wt mice upon bacterial challenge. Similar to our results, TRIF-deficient mice had a significant survival advantage over myd88−/− mice in a model of E. coli sepsis (27).
Microglia and other CNS macrophages communicate with circulating blood cells by multiple mechanisms (for a review, see reference 29). Neutrophils are produced in great numbers in bone marrow and circulate in blood for a few hours. Upon infection, circulating neutrophils are attracted by endothelial cells. The neutrophil-endothelium interactions comprise an initial attachment (rolling), followed by firm adhesion and ultimately transendothelial migration (for a review, see reference 30). Direct recognition of bacterial products (e.g., LPS) by endothelial cells can cause adhesion of neutrophils to the walls of CNS vessels (31). Additionally, endothelial cells can be activated by proinflammatory mediators (e.g., KC and MIP-2) released by resident brain cells.
Several studies suggest that defective neutrophil recruitment during microbial infection increases host susceptibility. Mice lacking expression of CD11b+ Ly-6G+ CCR2− granulocytes showed elevated brain bacterial titers, an aggravated clinical course, and higher mortality during Streptococcus pneumoniae meningitis (17). To determine whether granulocytes also contribute to the protection of mice early in the course of E. coli meningitis, we used two different approaches for cell depletion, based on the antibody clones RB6-8C5 and 1A8. Our findings support the use of anti-Ly-6G MAb (1A8) to specifically study the role of neutrophils in the resistance to infection (14), since administration of RB6-8C5 led to the depletion of not only CD11b+ Ly-6G+ Ly-6Cint granulocytes but also CD11b+ Ly-6G− Ly-6Chigh monocytes. In our experimental setting, induction of specific neutropenia significantly impaired the outcome of adult mice following intracerebral infection with E. coli K1. These mice showed higher bacterial loads than controls in both cerebellum and spleen homogenates 30 h after infection. Even more, in 27% of the depleted animals, E. coli was still detected in target organs 14 days after infection. Absence of CD11b+ Ly-6G+ Ly-6Cint granulocytes and CD11b+ Ly-6G− Ly-6Chigh monocytes after anti-Gr-1 treatment rendered mice even more susceptible. In our model, the contribution of inflammatory monocytes to controlling the infection was evident, since the effect of immune cell depletion was much more pronounced with the Gr-1 antibody than with the Ly-6G antibody. Moreover, animals with a reduction of ∼50% of the subset of circulating monocytes after administration of MC-21 MAb showed a trend toward a higher mortality than the isotype-treated group. Unlike in an autoimmune model (20), we were unable to completely eliminate Gr-1+ monocytes from the circulation, even with high doses of MC-21 MAb (data not shown). This is probably a consequence of the strong stimulation of the systemic immune response by bacteria entering the bloodstream. In contrast to E. coli meningitis, depletion of inflammatory monocytes did not aggravate the course of pneumococcal meningitis after intralumbar infection (17).
An exacerbated proinflammatory phenotype has been reported in animals receiving a systemic administration of RB6-8C5 (14). In the present study, depleted mice sacrificed 30 h after infection were subjects for the study of proinflammatory mediator levels. Interestingly, IL-1β, IL-6, KC, and MIP-2 levels were significantly elevated in the cerebellum of anti-Gr-1- and anti-Ly-6G-depleted mice compared with those in isotype-treated animals. The positive correlation between the bacterial burdens and these cytokine/chemokine levels in cerebellum homogenates of all mice suggests that the cytokine/chemokine levels depend mainly on the bacterial load and that high cytokine/chemokine concentrations do not reflect resistance to infection. Microglia, perivascular macrophages, astrocytes, and brain endothelial cells produce IL-1β, IL-6, KC, and MIP-2 in response to experimental stimulation by bacterial agents (9, 26, 32–34). Exposure to viable E. coli induced the release of high levels of tumor necrosis factor alpha (TNF-α) and KC by primary mouse microglia in a time-of-exposure-dependent manner (S. Ribes, unpublished data). Since important effector cells were lacking in depleted mice, in our model, high cytokine/chemokine levels were paradoxically associated with low resistance to infection.
The inoculum size was selected to induce a mortality rate of 50% in depleted mice but was insufficient to induce high bacterial titers and, subsequently, high levels of cytokines/chemokines in cerebellum homogenates of most of the isotype-treated animals. Since they had already overcome infection, the degree of meningeal inflammation 30 h after infection in most isotype animals was lower than that in depleted animals. The highest degree of meningeal inflammation was observed in the few isotype-treated mice that died during the acute phase of the infection.
The strong detrimental effect of neutropenia on the resistance of the brain to intracerebral infection was unexpected since microglia as the primary defense line appeared to be unaffected. The present data suggest that the protective functions of microglia and other CNS-resident macrophages critically depend on a cross talk with and the activities of circulating granulocytes and monocytes—especially in the early course of an intracerebral challenge and probably before circulating leukocytes enter the brain and CSF.
In conclusion, our study demonstrates the essential function of MyD88 signaling and the crucial role of neutrophils and monocytes in the recognition and subsequent elimination of E. coli K1 in mice after intracerebral infection. It also identifies them as putative therapeutic targets to improve the resistance to E. coli K1 CNS infections in the immunocompromised host. We recently reported enhanced phagocytosis and intracellular killing of E. coli strains by primary murine microglia upon stimulation with ligands of TLR1/2, -4, and -9 (26) that are all using MyD88 for signaling. Neutropenic mice upon administration of MAb 1A8 may thus serve as a suitable model of the immunocompromised host for investigating the effects of TLR agonists on the resistance of the brain to infections.
ACKNOWLEDGMENTS
This work was supported by grants from the Else Kröner-Fresenius-Stiftung (to R.N.), CAREPNEUMO (to R.N.), and the German Research Council (DFG), SFB/TRR43, The brain as a target of inflammatory processes, as well as FOR1336, From monocytes to brain macrophages—conditions influencing the fate of myeloid cells in the brain (both to U.K.H.).
We thank Stephanie Bunkowski, Elke Pralle, and Susanne Kieke for the technical assistance. We also thank Angelika Fruth from the Nationales Referenzzentrum für Salmonellen und anderen Enteritiserreger at the Robert Koch Institute, Wernigerode, Germany, for the characterization of the E. coli strain. This work is dedicated to Nora Papiol.
Footnotes
Published ahead of print 11 March 2013
REFERENCES
- 1. Briongos-Figuero LS, Morchón-Simón D, Aparicio-Blanco M, Garea García-Malvar MJ. 2008. Spontaneous meningitis due to Escherichia coli in the adult: a case report. Rev. Clin. Esp. 208:262. [DOI] [PubMed] [Google Scholar]
- 2. Gilmore RL, Lebow R, Berk SL. 1983. Spontaneous Escherichia coli K1 meningitis in an adult. South. Med. J. 76:1202–1203 [DOI] [PubMed] [Google Scholar]
- 3. Pitt J. 1978. K-1 antigen of Escherichia coli: epidemiology and serum sensitivity of pathogenic strains. Infect. Immun. 22:219–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyer-Mariotte S, Duboc P, Bonacorsi S, Lemeland JF, Bingen E, Pinquier D. 2008. CTX-M-15-producing Escherichia coli in fatal neonatal meningitis: failure of empirical chemotherapy. J. Antimicrob. Chemother. 62:1472–1474 [DOI] [PubMed] [Google Scholar]
- 5. Kim KS, Itabashi H, Gemski P, Sadoff J, Warren RL, Cross AS. 1992. The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J. Clin. Invest. 90:897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dietzman DE, Fischer GW, Schoenknecht FD. 1974. Neonatal Escherichia coli septicemia—bacterial counts in blood. J. Pediatr. 85:128–130 [DOI] [PubMed] [Google Scholar]
- 7. Kim KS. 2002. Strategy of Escherichia coli for crossing the blood-brain barrier. J. Infect. Dis. 186(Suppl 2):S220–S224 [DOI] [PubMed] [Google Scholar]
- 8. Kim KS. 2012. Current concepts on the pathogenesis of Escherichia coli meningitis: implications for therapy and prevention. Curr. Opin. Infect. Dis. 25:273–278 [DOI] [PubMed] [Google Scholar]
- 9. Hanisch UK, Kettenmann H. 2007. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10:1387–1394 [DOI] [PubMed] [Google Scholar]
- 10. Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11:373–384 [DOI] [PubMed] [Google Scholar]
- 11. Häusler KG, Prinz M, Weber JR, Schumann RR, Kettenmann H, Hanisch UK. 2002. Interferon-γ differentially modulates the release of cytokines and chemokines in lipopolysaccharide- and pneumococcal cell wall-stimulated mouse microglia and macrophages. Eur. J. Neurosci. 16:2113–2122 [DOI] [PubMed] [Google Scholar]
- 12. Regen T, van Rossum D, Scheffel J, Kastriti ME, Revelo NH, Prinz M, Brück W, Hanisch UK. 2011. CD14 and TRIF govern distinct responsiveness and responses in mouse microglial TLR4 challenges by structural variants of LPS. Brain Behav. Immun. 25:957–970 [DOI] [PubMed] [Google Scholar]
- 13. Koedel U, Rupprecht T, Angele B, Heesemann J, Wagner H, Pfister HW, Kirschning CJ. 2004. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain 127:1437–1445 [DOI] [PubMed] [Google Scholar]
- 14. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70 [DOI] [PubMed] [Google Scholar]
- 15. Fleming TJ, Fleming ML, Malek TR. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J. Immunol. 151:2399–2408 [PubMed] [Google Scholar]
- 16. Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82 [DOI] [PubMed] [Google Scholar]
- 17. Mildner A, Djukic M, Garbe D, Wellmer A, Kuziel WA, Mack M, Nau R, Prinz M. 2008. Ly-6G+ CCR2− myeloid cells rather than Ly-6Chigh CCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J. Immunol. 181:2713–2722 [DOI] [PubMed] [Google Scholar]
- 18. Scheffel J, Regen T, Van Rossum D, Seifert S, Ribes S, Nau R, Parsa R, Harris RA, Boddeke HW, Chuang HN, Pukrop T, Wessels JT, Jürgens T, Merkler D, Brück W, Schnaars M, Simons M, Kettenmann H, Hanisch UK. 2012. Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia 60:1930–1943 [DOI] [PubMed] [Google Scholar]
- 19. Gerber J, Raivich G, Wellmer A, Noeske C, Kunst T, Werner A, Brück W, Nau R. 2001. A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol. 101:499–508 [DOI] [PubMed] [Google Scholar]
- 20. Bruhl H, Cihak J, Plachy J, Kunz-Schughart L, Niedermeier M, Denzel A, Rodriguez GM, Talke Y, Luckow B, Stangassinger M, Mack M. 2007. Targeting of Gr-1+, CCR2+ monocytes in collagen-induced arthritis. Arthritis Rheum. 56:2975–2985 [DOI] [PubMed] [Google Scholar]
- 21. Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. 2000. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol. Dial. Transplant. 15:1562–1574 [DOI] [PubMed] [Google Scholar]
- 22. Spanaus KS, Nadal D, Pfister HW, Seebach J, Widmer U, Frei K, Gloor S, Fontana A. 1997. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. J. Immunol. 158:1956–1964 [PubMed] [Google Scholar]
- 23. Kielian T, Barry B, Hickey WF. 2001. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J. Immunol. 166:4634–4643 [DOI] [PubMed] [Google Scholar]
- 24. Engelhardt B, Coisne C. 2011. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nimmerjahn A, Kirchhoff F, Helmchen F. 2005. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318 [DOI] [PubMed] [Google Scholar]
- 26. Ribes S, Ebert S, Czesnik D, Regen T, Zeug A, Bukowski S, Mildner A, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R. 2009. Toll-like receptor prestimulation increases phagocytosis of Escherichia coli DH5α and Escherichia coli K1 strains by murine microglial cells. Infect. Immun. 77:557–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van 't Veer C, van den Pangaart PS, Kruijswijk D, Florquin S, de Vos AF, van der Poll T. 2011. Delineation of the role of Toll-like receptor signaling during peritonitis by a gradually growing pathogenic Escherichia coli. J. Biol. Chem. 286:36603–36618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Krishnan S, Chen S, Turcatel G, Arditi M, Prasadarao NV. 2013. Regulation of Toll-like receptor 2 interaction with Ecgp96 controls Escherichia coli K1 invasion of brain endothelial cells. Cell. Microbiol. 15:63–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perry VH, Cunningham C, Holmes C. 2007. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7:161–167 [DOI] [PubMed] [Google Scholar]
- 30. Borregaard N. 2010. Neutrophils, from marrow to microbes. Immunity 33:657–670 [DOI] [PubMed] [Google Scholar]
- 31. Zhou H, Andonegui G, Wong CH, Kubes P. 2009. Role of endothelial TLR4 for neutrophil recruitment into central nervous system microvessels in systemic inflammation. J. Immunol. 183:5244–5250 [DOI] [PubMed] [Google Scholar]
- 32. Luo Y, Fischer FR, Hancock WW, Dorf ME. 2000. Macrophage inflammatory protein-2 and KC induce chemokine production by mouse astrocytes. J. Immunol. 165:4015–4023 [DOI] [PubMed] [Google Scholar]
- 33. Diab A, Abdalla H, Li HL, Shi FD, Zhu J, Höjberg B, Lindquist L, Wretlind B, Bakhiet M, Link H. 1999. Neutralization of macrophage inflammatory protein 2 (MIP-2) and MIP-1α attenuates neutrophil recruitment in the central nervous system during experimental bacterial meningitis. Infect. Immun. 67:2590–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fabry Z, Fitzsimmons KM, Herlein JA, Moninger TO, Dobbs MB, Hart MN. 1993. Production of the cytokines interleukin 1 and 6 by murine brain microvessel endothelium and smooth muscle pericytes. J. Neuroimmunol. 47:23–34 [DOI] [PubMed] [Google Scholar]