

Abstract
Candida glabrata is an emerging opportunistic pathogen that is known to develop resistance to azole drugs due to increased drug efflux. The mechanism consists of CgPDR1-mediated upregulation of ATP-binding cassette transporters. A range of gain-of-function (GOF) mutations in CgPDR1 have been found to lead not only to azole resistance but also to enhanced virulence. This implicates CgPDR1 in the regulation of the interaction of C. glabrata with the host. To identify specific CgPDR1-regulated steps of the host-pathogen interaction, we investigated in this work the interaction of selected CgPDR1 GOF mutants with murine bone marrow-derived macrophages and human acute monocytic leukemia cell line (THP-1)-derived macrophages, as well as different epithelial cell lines. GOF mutations in CgPDR1 did not influence survival and replication within macrophages following phagocytosis but led to decreased adherence to and uptake by macrophages. This may allow evasion from the host's innate cellular immune response. The interaction with epithelial cells revealed an opposite trend, suggesting that GOF mutations in CgPDR1 may favor epithelial colonization of the host by C. glabrata through increased adherence to epithelial cell layers. These data reveal that GOF mutations in CgPDR1 modulate the interaction with host cells in ways that may contribute to increased virulence.
INTRODUCTION
Candida glabrata is a commensal yeast that has emerged as an important opportunistic pathogen and has become the second most common cause of candidiasis after Candida albicans (1). Infections caused by C. glabrata have increased steadily in frequency over the last few decades. Previously available epidemiological data showed a proportion of bloodstream infections caused by C. glabrata among all Candida spp. ranging from about 5% in Latin America to 25% in North America (1). The latest data illustrate a continual rise, with C. glabrata now accounting for up to 11.2% and 29% of candidemia episodes in Brazil and the United States, respectively, at the expense of C. albicans (2, 3). Additionally, C. glabrata intrinsically displays reduced susceptibility to azole drugs and shows a high propensity to develop secondary resistance, typically due to increased drug efflux (4). This mechanism is mediated by upregulation of a single or a combination of a few ATP-binding cassette (ABC) transporters, among which are at least CgCDR1, CgCDR2, and CgSNQ2 (5–9). Upregulation of ABC transporters occurs following alterations in their major regulator, the zinc cluster transcription factor CgPDR1 (10, 11). CgPDR1 combines functional attributes of transcription factors PDR1 and PDR3 from the nonpathogenic baker's yeast Saccharomyces cerevisiae (12). We have previously found that gain-of-function (GOF) mutations in CgPDR1 lead not only to azole resistance in vitro and in vivo but also to gain of virulence in murine models of disseminated infection (13). Further work has shown that two CgPDR1-regulated proteins, the ABC transporter CgCdr1 and a hitherto-uncharacterized mitochondrial protein, Pup1, partially contribute to the gain of virulence (14).
C. glabrata is closely related to the nonpathogenic S. cerevisiae, but it appears to have evolved important attributes allowing adaptation to pathogenesis, namely, adherence to host cells, stress resistance, ability to sustain starvation, and antifungal drug resistance (15). Even though it lacks some well-established virulence factors of C. albicans, like true hyphal formation or hydrolase secretion, C. glabrata is able to access the bloodstream and disseminate to internal organs in susceptible patients. These translocation mechanisms are, however, not yet well understood. A remarkable characteristic of C. glabrata infections is that the organism is able to persist over long periods in immunocompetent mice upon systemic challenge without causing disease or high inflammation (16–19). This suggests an ability to subvert the host's first line of defense, which includes cells of the innate immune system. Macrophages, in particular, are essential for both innate and adaptive immunity. They play key roles in microbial phagocytosis and killing, and they are involved in downstream effects such as antigen processing and presentation or cytokine production.
Being able to evade the control of cellular innate immunity may thus be a major attribute of C. glabrata pathogenesis. Two general strategies are employed by microbes to survive attacks by the host's cellular innate immunity: on one hand, prevention of phagocytosis, and on the other hand, intracellular survival and escape from phagocytes (20). The former strategy relies mainly on concealing pathogen-associated molecular patterns (PAMPs), which in yeasts consist of cell wall components β-1,3-glucan, mannoproteins, and chitin, from the host's pattern recognition receptors (PRRs). An additional mechanism to avoid phagocytosis consists of preventing opsonization, as described, for example, for C. albicans and Cryptococcus neoformans (20). Survival and escape following phagocytosis depend on additional adaptations. Yeasts undergo massive transcriptional adjustments allowing them to adapt to the hostile conditions within the phagolysosome, and they are able to inhibit phagosome maturation and the oxidative burst mounted by phagocytes (20). In C. glabrata, the ability to escape immune control and persist in the host for long periods of time is not fully understood. To our knowledge, no mechanism of evasion from phagocytosis has so far been identified. However, C. glabrata is clearly able to survive phagocytosis by cells of the innate immune system and even replicate within phagocytes (21–23). The transcriptional adaptations of C. glabrata in the phagolysosome appear to be similar to those of C. albicans (22). Recycling of endogenous cellular components through autophagy (21) and oxidative stress resistance (24) also seem to play important roles. Additionally, recent work by Seider et al. showed that C. glabrata counteracts phagosome maturation and cytokine production to persist within macrophages (23). Interestingly, a family of glycosylphosphatidylinositol (GPI)-linked aspartyl proteases called yapsins have also been shown to contribute to C. glabrata virulence and impact their survival within macrophages (22). By remodeling the cell wall through removal of GPI-anchored cell wall proteins, yapsins are implicated in cell wall integrity and, ultimately, virulence in C. glabrata (22). Another interesting activity may be the control of adherence to mammalian cells, through removal of GPI-anchored cell wall adhesins (22). A dynamic control of the presentation of such adhesins at the cell surface appears to be important, rather than their constitutive expression.
In the present work, we addressed the impact of CgPDR1 GOF mutations on the interaction of C. glabrata with host cells. We hypothesize that the modulation of the interaction with host cells may contribute to the previously found CgPDR1-mediated gain of virulence in C. glabrata. To address this problem, we performed competitive coculture experiments of C. glabrata strains harboring different CgPDR1 alleles with primary murine macrophages and human cell line-derived macrophages as well as with different epithelial cells. We investigated adherence and uptake of C. glabrata by host cells, as well as survival and replication following phagocytosis. Our results reveal that CgPDR1 modulates the interaction of C. glabrata with mammalian cells.
MATERIALS AND METHODS
Strains and growth media.
The C. glabrata strains used in this study are listed in Table 1. Clinical isolates and strains with the prefix SFY are from previously published collections (13, 14). Strains were stored in 20% glycerol stocks at −80°C and cultured on either YPD (1% yeast extract, 2% peptone, 2% d-glucose) or appropriate selective media at 30°C. Selective media for growth of transformed strains were either YPD containing 200 μg/ml of nourseothricin (clonNAT; Werner BioAgents, Germany) or 600 μg/ml of hygromycin B (PAA Laboratories, Austria); additionally, YNB minimal medium (0.67% yeast nitrogen base plus 2% glucose) with appropriate amino acids and bases and without uracil was used for uracil prototrophs. For solid media, 2% agar was added. YPD containing 30 μg/ml of fluconazole (Pfizer) was used when appropriate. Escherichia coli DH5α was used as a host for plasmid construction and propagation. E. coli DH5α was grown in Luria-Bertani broth or on Luria-Bertani agar plates, supplemented with ampicillin (0.1 mg/ml) when required.
Table 1.
Strains used in this study
| Strain | Parental strain | Plasmid used | Description or relevant genotype | Reference |
|---|---|---|---|---|
| DSY562 | Azole-susceptible clinical strain | 13 | ||
| DSY565 | Related to DSY562 | Azole-resistant clinical strain | 13 | |
| SFY114 | SFY93 | DSY562 pdr1Δ::PDR1-SAT1 | 13 | |
| SFY115 | SFY93 | DSY562 pdr1Δ::PDR1L280F-SAT1 | 13 | |
| VSY21 | DSY562 | pVS13 | ura3Δ::hph | This study |
| VSY22 | DSY565 | pVS13 | ura3Δ::hph | This study |
| VSY33 | VSY21 | pCgACU-TDH3p-GFP | DSY562 ura3Δ::hph, ScTDH3p-yEGFP(−3), URA3, CEN-ARS | This study |
| VSY38 | SFY153 | pVS13 | DSY565 cdr1Δ::FRT, ura3Δ::hph | This study |
| VSY40 | SFY155 | pVS13 | DSY565 pup1Δ::FRT, ura3Δ::hph | This study |
| VSY42 | SFY170 | pVS13 | DSY565 cdr1Δ::FRT, pup1Δ::SAT1, ura3Δ::hph | This study |
| VSY43 | SFY114 | pVS13 | DSY562 pdr1Δ::PDR1-SAT1, ura3Δ::hph | This study |
| VSY44 | SFY115 | pVS13 | DSY562 pdr1Δ::PDR1L280F-SAT1, ura3Δ::hph | This study |
| VSY55 | VSY21 | pGRB2.3 | DSY562 ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY56 | VSY22 | pGRB2.3 | DSY565 ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY57 | VSY21 | pVS19 | DSY562 ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY58 | VSY22 | pVS19 | DSY565 ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY59 | VSY21 | pVS20 | DSY562 ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY60 | VSY22 | pVS20 | DSY565 ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY65 | VSY38 | pVS20 | DSY565 cdr1Δ::FRT, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY66 | VSY40 | pVS20 | DSY565 pup1Δ::FRT, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY67 | VSY42 | pVS20 | DSY565 cdr1Δ::FRT, pup1Δ::SAT1, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY68 | VSY38 | pGRB2.3 | DSY565 cdr1Δ::FRT, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY69 | VSY40 | pGRB2.3 | DSY565 pup1Δ::FRT, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY70 | VSY42 | pGRB2.3 | DSY565 cdr1Δ::FRT, pup1Δ::SAT1, ura3Δ::hph, ScPGK1p- yEGFP, URA3, CEN-ARS | This study |
| VSY71 | VSY38 | pVS19 | DSY565 cdr1Δ::FRT, ura3Δ::hph, ScPGK1p- yEmRFP, URA3, CEN-ARS | This study |
| VSY72 | VSY40 | pVS19 | DSY565 pup1Δ::FRT, ura3Δ::hph, ScPGK1p, yEmRFP, URA3-CEN-ARS | This study |
| VSY73 | VSY42 | pVS19 | DSY565 cdr1Δ::FRT, pup1Δ::SAT1, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY101 | VSY43 | pVS20 | DSY562 pdr1Δ::PDR1-SAT1, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY102 | VSY44 | pVS20 | DSY562 pdr1Δ::PDR1L280F-SAT1, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY103 | VSY43 | pGRB2.3 | DSY562 pdr1Δ::PDR1-SAT1, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY104 | VSY44 | pGRB2.3 | DSY562 pdr1Δ::PDR1L280F-SAT1, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY105 | VSY43 | pVS19 | DSY562 pdr1Δ::PDR1-SAT1, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY106 | VSY44 | pVS19 | DSY562 pdr1Δ::PDR1L280F-SAT1, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY119 | SFY93 | pVS13 | DSY562 pdr1Δ::FRT, ura3Δ::hph | This study |
| VSY120 | SFY95 | pVS13 | DSY565 pdr1Δ::FRT, ura3Δ::hph | This study |
| VSY121 | VSY119 | pVS20 | DSY562 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY122 | VSY120 | pVS20 | DSY565 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY123 | VSY119 | pGRB2.3 | DSY562 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY124 | VSY120 | pGRB2.3 | DSY565 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY125 | VSY119 | pVS19 | DSY562 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY126 | VSY120 | pVS19 | DSY565 pdr1Δ::FRT, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY132 | SFY101 | pVS13 | DSY562 pdr1Δ::PDR1R376W-SAT1, ura3Δ::hph | This study |
| VSY133 | SFY105 | pVS13 | DSY562 pdr1Δ::PDR1T588A-SAT1, ura3Δ::hph | This study |
| VSY134 | VSY132 | pVS20 | DSY562 pdr1Δ::PDR1R376W-SAT1, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY135 | VSY133 | pVS20 | DSY562 pdr1Δ::PDR1T588A-SAT1, ura3Δ::hph, ScPGK1p, URA3, CEN-ARS | This study |
| VSY136 | VSY132 | pGRB2.3 | DSY562 pdr1Δ::PDR1R376W-SAT1, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY137 | VSY133 | pGRB2.3 | DSY562 pdr1Δ::PDR1T588A-SAT1, ura3Δ::hph, ScPGK1p-yEGFP, URA3, CEN-ARS | This study |
| VSY138 | VSY132 | pVS19 | DSY562 pdr1Δ::PDR1R376W-SAT1, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
| VSY139 | VSY133 | pVS19 | DSY562 pdr1Δ::PDR1T588A-SAT1, ura3Δ::hph, ScPGK1p-yEmRFP, URA3, CEN-ARS | This study |
Disruption of CgURA3.
Uracil auxotrophs of all strains used in this study were constructed through targeted gene disruption of CgURA3. Briefly, the complete CgURA3 open reading frame (ORF) flanked by 500 bp was amplified by PCR from genomic DNA of strain DSY562, using primers CgURA3-KpnI (5′-GTA GGG TAC CTC ATA TCT TGT CAC TAT ATA-3′) and CgURA3-SpeI (5′-GCA AAC TAG TGC AAT AAA GGA TGC AAA AC-3′), and inserted into pBluescript II KS(+) to generate pVS12. This plasmid was amplified by PCR using the primers CgURA3-Inv-SalI (5′-AAA AGT CGA CTG GGA TGC TTA CTT GAA AAG-3′) and CgURA3-Inv-BamHI (5′-TGG AGG ATC CAC TAA TCT ACT GGG ATG ATG-3′). The resulting PCR product was digested by SalI and BamHI and ligated to a 2.1-kb SalI/BamHI fragment from pAP599 containing the hph expression cassette (FRT-ScPGK1p-hph-3′UTR ScHIS3-FRT; conferring resistance to hygromycin B) (25). The resulting plasmid (pVS13) was digested by SpeI and KpnI and used to transform all test strains by an adapted lithium acetate (LiAc) procedure (26). Resulting ura3Δ transformants were selected on hygromycin B-containing YPD plates incubated for 2 to 3 days at 35°C and confirmed by PCR.
Labeling with fluorescent proteins.
A set of plasmids was constructed to label C. glabrata strains with fluorescent proteins. Briefly, pGRB2.3, a CgCEN/ARS URA3 plasmid made by introducing a yeast-enhanced green fluorescence protein (GFP) expression cassette (ScPGK1p-yEGFP-ScHIS3 3′ UTR) in plasmid pGRB2.2 (27), was used for labeling strains with GFP. Yeast-enhanced monomeric red fluorescent protein (RFP) (yEmRFP) was obtained from an available plasmid in our collection, pCgACU-TDH3p-cherry (ScTDH3p-yEmRFP), previously constructed by ligating two KpnI/BamHI fragments consisting of the CgCEN/ARS URA3 backbone from plasmid pCgACU-5 (28) and yEmRFP from plasmid yEpGAP-Cherry (29). yEmRFP was amplified by PCR from pCgACU-TDH3p-cherry using primers TDH3p-GFP (5′-CAC CAA GAA CTT AGT TTC G-3′) and RFP-XhoI (5′-TGA ACT CGA GCT CGG TAC CTT ATT TAT ATA ATT C-3′), digested by EcoRI and XhoI, and ligated to an EcoRI/XhoI fragment from pGRB2.3, thus generating the replacement of yEGFP by yEmRFP (pVS19). An empty control plasmid was constructed by cutting out yEGFP with AvaI, blunting the DNA sticky ends using T4 DNA polymerase, and ligating the blunt ends together (pVS20; ScPGK1p-ScHIS3 3′ UTR). C. glabrata ura3Δ strains were transformed with the three plasmids by an adapted LiAc procedure (26). Transformants were selected based on reversion to uracil prototrophy after growth for 2 to 3 days at 35°C in YNB minimal medium with appropriate amino acids and bases and without uracil.
Macrophage culture and infection for yeast replication assays.
Bone marrow was extracted from both femurs and tibiae of 8- to 10-week-old female BALB/c mice (approximately 20 g; the mice were obtained from Charles River Laboratories, France, and housed in filter top cages with free access to food and water, under the surveillance of the local governmental veterinarian offices, Affaires Vétérinaires du Canton de Vaud, Switzerland; authorization number 2240). A bone marrow cell suspension was filtered through a 40-μm cell strainer filter and suspended in culture medium (high-glucose Iscove's modified Dulbecco's medium with GlutaMAX [IMDM]; Life Technologies), supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies), 10% fetal bovine serum (FBS; Life Technologies), and 20% L-cell-conditioned medium (as a source of macrophage colony-stimulating factor [M-CSF]). Cells were seeded into 150- by 20-mm petri dishes (Sarstedt) at 4 × 106 cells/plate and incubated at 37°C in the presence of 5% CO2. Cultures were fed by adding fresh medium after 2 and 4 days of incubation. After 6 days of incubation, bone marrow-derived macrophages (BMDMs) that selectively adhered to the dishes were harvested with the nonenzymatic solution Versene (Life Technologies) and transferred to 24-well plates at a density of 3.0 × 105 cells/well in 1 ml of medium (IMDM with antibiotics and 10% FBS). Yeast strains were ura3Δ strains carrying either GFP/RFP-expressing plasmids or empty control plasmids (reconstituting the ura3 deletion), and inoculum concentrations were confirmed by plating serial dilutions in YPD agar plates. After an additional 20- to 24-h incubation, preconfluent BMDM monolayers were used to perform fungal competition assays. To prepare C. glabrata suspensions for infection, overnight cultures of fluorescent protein-labeled or empty plasmid control strains in selective medium were diluted in fresh medium and grown for a minimum of 2 generations to mid-log phase. Log-phase cells were washed and resuspended in phosphate-buffered saline solution (PBS). For staining with superficial stains, yeasts were washed and resuspended in carbonate buffer (0.1 M Na2CO3, 0.15 M NaCl, pH 9.0), stained with fluorescein isothiocyanate (FITC; Thermo Scientific) or tetramethylrhodamine isothiocyanate (TRITC; Thermo Scientific) for 30 min at 37°C at final concentrations of 100 μg/ml and 10 μg/ml, respectively, and then washed and resuspended in PBS. Macrophage cultures were infected with either single or 1:1 mixed yeast strain suspensions at a multiplicity of infection (MOI) of 1 (according to the macrophage seeding density), and the plates were centrifuged at 200 × g for 1 min. Cocultures were incubated at 37°C in 5% CO2, and at selected time points non-macrophage-associated yeasts were removed by washing. Yeasts were recovered by lysis of the macrophages in 0.1% Triton X-100 and plated onto YPD plates for quantification of CFU. YPD agar and YPD agar plates containing 30 μg/ml of fluconazole were used to distinguish between azole-susceptible and azole-resistant yeast strains. Alternatively, cocultures established on top of round cover slides were mounted onto microscopy slides and observed using a Zeiss Axioplan 2 epifluorescence microscope (images were recorded using a Visitron Systems HistoScope Camera and VisiView Imaging Software).
Macrophage culture and infection for yeast adherence and phagocytosis assays.
BMDM cultures established in 24-well plates on top of round cover slides as described above were infected with 1:1 mixed yeast suspensions at an MOI of 0.6, 2, or 6, and the plates were centrifuged at 200 × g for 1 min. For adherence experiments, BMDM cultures were treated with 1.0 μM cytochalasin D at 37°C in 5% CO2 starting 30 min before infection and then throughout coincubation. To test the influence of galactose, the culture medium was replaced by fresh medium containing from 0.5 to 10 mM galactose before infection. Cocultures were incubated at 37°C in 5% CO2, and at selected time points non-macrophage-associated yeasts were removed by washing. Cocultures were stained with 100 μg/ml of calcofluor white (CW; Sigma) for 10 min and mounted onto microscopy slides for epifluorescence microscopy as described above.
For human acute monocytic leukemia cell line (THP-1; ATCC TIB-202)-derived macrophages, log-phase THP-1 cell cultures in suspension in high-glucose RPMI 1640 medium with GlutaMAX (Life Technologies) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies), and 10% FBS (Life Technologies) were seeded into 24-well plates on top of round cover slides at a density of 5.0 × 105 cells/well in 1 ml of the same medium. Differentiation and attachment were induced by adding 20 nM phorbol 12-myristate 13-acetate (PMA; Sigma). After 48 h of incubation at 37°C in 5% CO2, preconfluent THP-1-derived macrophage-like cell monolayers were washed, and fresh culture medium was added. Cultures were infected with 1:1 mixed yeast suspensions containing 6.0 × 105 yeast cells, prepared as described above, and the plates were centrifuged at 200 × g for 1 min. For adherence experiments, THP-1-derived macrophage-like cells were treated with cytochalasin D as described above for BMDMs. Cocultures were stained with CW and observed under epifluorescence microscopy as described.
Cytokine production.
BMDM cultures established as described above were seeded in 96-well plates at a density of 1.0 × 105 cells/well in 0.2 ml of IMDM with 10% FBS. Cultures were infected with either single-strain or 1:1 mixed yeast strain suspensions, prepared as described above, at an MOI of 3, 1, or 0.3. The plates were centrifuged at 200 × g for 1 min and incubated at 37°C in 5% CO2. After 1.5 h of incubation, 2.5 μg/ml of amphotericin B was added to all wells (including the uninfected controls) to prevent yeast overgrowth. At 24 h the plates were centrifuged, and supernatants were transferred to new plates. Cytokines were quantified by standard enzyme-linked immunosorbent assay (ELISA) techniques using purified anti-interleukin 12p40 (anti-IL-12p40) (clones 15.6) or anti-tumor necrosis factor (anti-TNF) (clone 6B8) for coating and biotinylated anti-IL-12p40 (clone C17.8) or anti-TNF (clone MP6-XT22) combined with Extravidin-alkaline phosphatase (Sigma) and p-nitrophenyl phosphate substrate (Sigma) for detection. IL-1β production was quantified by BD cytometric bead array according to the manufacturer's instructions (BD Biosciences).
Yeast adherence to epithelial cells.
Epithelial cell lines Chinese hamster ovary modified cell line Lec2 (CHO-Lec2; ATCC CRL1736), human cervix adenocarcinoma cells (CHO-HeLa; ATCC CCL-2), and human colorectal adenocarcinoma cells (Caco-2; ATCC HTB-37) were employed. CHO-Lec2 cells were cultured in high-glucose minimum essential medium α (MEM-α; Life Technologies), while HeLa and Caco-2 cells were cultured in Dulbecco's modified Eagle medium (DMEM; Life Technologies). Both media included l-glutamine and were supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin (Life Technologies), and 10% FBS (Life Technologies). Log-phase epithelial cells were seeded in 24-well plates at a density of 1.0 × 105 cells/well in 1 ml of culture medium and allowed to grow to full confluence at 37°C in 5% CO2, typically for 48 to 72 h. Epithelial cell monolayers were infected with 1:1 mixed yeast suspensions containing 3.0 × 105 yeast cells, prepared as described earlier, and the plates were centrifuged at 200 × g for 1 min. Cocultures were incubated at 37°C in 5% CO2 for 30 min, and nonadherent yeasts were removed by washing. Adherent yeasts were recovered by lysis of the epithelial cells in 0.1% Triton X-100 and plated onto YPD agar plates for quantification of CFU. YPD agar alone and YPD agar plates containing 30 μg/ml of fluconazole were used to distinguish between azole-susceptible and azole-resistant yeast strains.
RESULTS
CgPDR1 hyperactivity does not affect survival and replication inside BMDMs.
In a previous study, we found that azole resistance mediated by CgPDR1 hyperactivity in C. glabrata led to increased fitness in vivo and increased virulence in murine models of disseminated infection (13). A follow-up report revealed that the ABC transporter CgCdr1 and the mitochondrial protein Pup1 partially contributed to this phenotype (14). To investigate the mechanism behind the gain of virulence mediated by CgPDR1, we set up ex vivo coculture experiments with primary BMDMs as a model of the interaction of C. glabrata with the innate cellular immune response. We used fluorescent protein-labeled yeasts with GFP or RFP expressed from episomal plasmids with selection based on URA3 complementation of uracil auxotrophy. As shown in Fig. 1, in strains constructed on the DSY562 strain background, fluorescent protein expression did not affect growth in comparison to empty vector control strains. The same was true for all other strains (data not shown). Additionally, we stained test strains with the fluorescent dye FITC or TRITC prior to inoculation, to be able to track the division of the yeasts within BMDMs after phagocytosis. Since the fluorescent dyes do not propagate to daughter cells during replication, this allows the identification of newly divided cells. BMDM cultures were inoculated with C. glabrata strains DSY562 and DSY565, an azole-susceptible clinical isolate and its matched isolate with a GOF mutation on CgPdr1 (L280F) leading to azole resistance and increased virulence (13). Nonstained yeast cell buds resulting from intracellular growth inside BMDMs first became visible after 5 h of coincubation (Fig. 2A). At 24 h of coincubation many nonstained yeasts could be observed, and in some cases BMDMs were completely filled with C. glabrata (Fig. 2A). This result was indistinguishably observed for both strains DSY562 and DSY565. Continuing the coincubation for periods longer than 24 h allowed the continuous multiplication of the yeasts. This eventually resulted in the disruption of the phagocytic cell, which left behind a yeast microcolony on the cell culture plates. Again, there was no apparent difference between the two strains. To quantify the replication of the yeasts, we performed similar experiments in which the readout was changed to lysing the BMDMs and plating the resulting yeast suspensions onto solid media to count CFU. Ratios between counts obtained 2 h after inoculation and the inocula revealed that most yeasts (around 70% of the inocula) remained associated to BMDMs after washing the coculture. The ratios were somewhat lower for strain DSY565 than for DSY562 (Fig. 2B, 2 h ratios), although the differences did not reach statistical significance. Ratios between CFU counts at 24 h and 2 h were similar in all cases, thus illustrating a similar ability to replicate inside BMDMs for the two strains (Fig. 2C).
Fig 1.

Growth curves of C. glabrata strains labeled with fluorescent proteins. The test strains were grown overnight and diluted to a density of 5.0 × 104 cells/ml in selective medium (YNB minimal medium with appropriate amino acids and bases and without uracil). Cultures were grown under constant agitation at 30°C, and growth was determined at 2-h intervals from 14 h to 24 h by measuring the absorbance at 540 nm. The test strains were isogenic strains on the DSY562 strain background harboring episomal plasmids expressing ScPGK1p-yEGFP (VSY55 △), ScPGK1p-yEmRFP (VSY57 ▽), or the empty vector (VSY59 ○).
Fig 2.

Replication of C. glabrata inside BMDMs following phagocytosis. (A) Microscopy images of three representative examples acquired following 5 h and 24 h of coincubation of C. glabrata DSY565 with BMDMs. Log-phase cultures of RFP-labeled yeasts (VSY58) were stained with FITC, as described in Materials and Methods, and added to preconfluent BMDM monolayers (established on top of round cover slides in 24-well plates) at an MOI of 1. Cocultures were incubated in IMDM at 37°C in 5% CO2 and mounted onto microscopy slides at selected time points for visualization. Since the FITC stain does not propagate to daughter cells during budding, the absence of FITC staining denotes yeast cells resulting from replication inside BMDMs. Bar, 10 μm. DIC, differential interference contrast. (B) Two-hour and 24-h inoculum CFU count ratios. Log-phase C. glabrata (VSY59 and VSY60) cultures containing either a single strain (single inoculum) or a 1:1 mix of two strains (mixed inoculum) were added to preconfluent BMDM monolayers at an MOI of 1. After 2 h of coincubation, the cultures were washed to remove noninternalized yeast cells. Immediately following the washes, as well as after 24 h of coincubation, BMDMs were lysed and dilutions of the resulting yeast suspensions were plated onto solid medium. (C) Count ratios (24-h CFU/2-h CFU) of the data displayed in panel B. Results on panels B and C are means + standard deviations (SD) of a minimum of three independent experiments. P values of <0.05 were considered significant, and differences between strains DSY562 and DSY565 are not statistically significant (P > 0.05, unpaired Student's t test).
CgPDR1 hyperactivity does not affect cytokine production by BMDMs.
The production of proinflammatory cytokines by phagocytes is an important response of the host to infection. To investigate the impact of CgPDR1 hyperactivity on cytokine production, we measured concentrations of TNF-α, IL-12, and IL-1β in the culture medium following 24 h of coincubation of C. glabrata with BMDMs. We could not detect any differences between strains DSY562 and DSY565 or the isogenic strains obtained by deleting CgPDR1 on the DSY562 strain background and reintroducing either the wild-type (DSY562 pdr1Δ::PDR1) or the hyperactive CgPDR1 allele (DSY562 pdr1Δ::PDR1L280F) (13) (Fig. 3 and data not shown). Using mixed inocula of 1:1 suspensions of strains DSY562 and DSY565 or the isogenic pair of strains at the same MOIs resulted in similar outcomes (data not shown). Given the similar host response upon stimulation with the two C. glabrata cell types, our results rule out an influence of this step of the yeast-host interaction on the differential virulence attributed to CgPDR1 hyperactivity.
Fig 3.

Cytokine release by BMDMs in response to C. glabrata strains. Preconfluent BMDM monolayers were infected with the matched clinical isolates DSY562 and DSY565 and with isogenic strains on the DSY562 strain background harboring either its original CgPDR1 wild-type allele (PDR1; SFY114) or the hyperactive CgPDR1L280F allele from DSY565 (L280F; SFY115). At 24 h, TNF-α (A) and IL-12 (B) levels in the supernatants were determined by ELISA. Results are means + SD of triplicate infections and are representative of three independent experiments. Differences between strains are not statistically significant (P > 0.05, one-way analysis of variance [ANOVA]). uninf, uninfected.
CgPDR1 hyperactivity leads to a decrease in competitive uptake of C. glabrata by BMDMs.
The ability to evade phagocytosis is a strategy employed by different pathogenic microorganisms to escape the host's immunity. To specifically investigate this step of the interaction between yeasts and phagocytes, we set up competition experiments using fluorescent protein-labeled yeasts in coculture with BMDMs. The competition design was chosen because of its ability to uncover subtle phenotypic differences that might otherwise be missed in single-strain experiments. Mixed suspensions including GFP-expressing DSY562 and RFP-expressing DSY565 strains and vice versa were used to inoculate preconfluent BMDM monolayers. Cocultures were stained at different time points with the chitin-binding dye calcofluor white (CW), which does not penetrate viable phagocytes and thus stains noninternalized yeasts only. We next observed yeast cells under epifluorescence microscopy (Fig. 4A). Counting internalized GFP- and RFP-labeled yeast cells revealed lower numbers of phagocytosed DSY565 cells, expressed as percentages of each strain in the total number of yeasts, or percent internalization ratios (PIR; Fig. 4B). This difference was statistically significant for all time points, ranging from 30 min up to 4 h of coincubation. PIR values were around 60% and 40% for DSY562 and DSY565, respectively. No significant influence of the incubation time was found. Similar experiments performed with a DSY562-derived isogenic strain pair resulted in similar PIR differences after 2 h of incubation (Fig. 4C), thus confirming that the L280F GOF mutation on CgPdr1 was responsible for the difference. A difference was also observed when using 3-fold lower or higher MOIs (Fig. 4C). Even at an MOI of 6, the difference between the two strains was still observed. Control experiments testing the same strain labeled with GFP or RFP resulted in PIRs of around 50% (Fig. 4D) and thus ruled out an influence of the fluorescent protein. To investigate the role of the CgPdr1 targets CgCdr1 and Pup1, which were previously found to contribute to CgPdr1-mediated gain of virulence (14), we performed additional experiments comparing strain DSY565 with the corresponding mutants on the same strain background. Significant differences in PIR were found between DSY565 and the cdr1Δ pup1Δ double mutant, but no difference was detected for either of the two single mutants (Fig. 4E). Taken together, our results revealed a bias toward decreased phagocytosis of strains containing the hyperactive CgPDR1L280F allele in competition with strains with the wild-type CgPDR1. The CgPdr1 targets CgCDR1 and PUP1 may both contribute to this difference.
Fig 4.

Phagocytosis of C. glabrata by BMDMs. (A) Representative microscopy images acquired following 2 h of coincubation of C. glabrata strains with BMDMs. Log-phase cultures of yeast strains DSY562 and DSY565 labeled with GFP and RFP (VSY55 to VSY58) were mixed and added to preconfluent BMDM monolayers (established on top of round cover slides) at an MOI of 1 for each of the two yeast strains (total MOI, 2). At selected time points, cultures were stained with calcofluor white (CW) and the round cover slides were mounted onto microscopy slides for visualization. The chitin-binding stain CW does not penetrate BMDMs and thus stains noninternalized yeasts only. Bar, 20 μm. DIC, differential interference contrast. (B) Quantification of competitive uptake of matched clinical isolates DSY562 and DSY565 by BMDMs. Cultures were prepared and visualized as described for panel A. A minimum of 100 BMDMs and internalized green and red yeast cells were counted and expressed as percent internalization ratios (PIRs; percentage of yeasts of each strain in the total number of yeasts from both competitor strains). (C) PIRs determined as described for panel B after 2 h of coincubation. Yeasts are isogenic strains on the DSY562 strain background harboring either its original CgPDR1 wild-type allele (PDR1) or the hyperactive CgPDR1L280F allele from DSY565 (L280F; VSY103 to VSY106). Strains were inoculated at an MOI of 0.3, 1, or 3 for each of the two yeast strains (total MOI, 0.6, 2, or 6). (D) Control PIRs determined as described for competitions between the same clinical isolate labeled with the two different fluorescent proteins. (E) PIRs determined as described for competitions between clinical isolate DSY565 (VSY56 and VSY58) and either cdr1Δ, pup1Δ, or cdr1Δ pup1Δ double mutant strains constructed on the same strain background (VSY68 to VSY73). Results on panels B through E are means + SD of a minimum of three independent experiments performed in duplicate (with fluorescent protein labeling swapping between strains). Statistical analysis of the differences between PIRs on panel B was performed using repeated-measures 2-way ANOVA on data transformed to global ranks, followed by Bonferroni's posttest for selected pairwise comparisons (no significant influence of the incubation time was found; for strain, P < 0.0001; for time, P = 1.000; for the time-strain interaction, P = 0.1230). For panels C, D, and E, differences between PIRs in strain pairs were analyzed using the Mann-Whitney U test. P values of <0.05 were considered significant, and asterisks represent P value ranges (ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001).
CgPDR1 hyperactivity leads to decreased adherence to BMDMs.
Given the described differences in phagocytosis between distinct C. glabrata isolates, we next addressed whether or not the CgPDR1 allele was able to influence the adherence step preceding phagocytosis. We performed similar coculture experiments in a medium containing the actin polymerization inhibitor cytochalasin D to inhibit phagocytosis. Preconfluent BMDM cultures were incubated for 30 min in cytochalasin D and then inoculated with mixed suspensions of the yeast strains as for the above-described phagocytosis experiments. Cytochalasin D has been used before without causing any general adverse effects to mammalian cells or Candida species yeasts (30). After 1 h of coincubation, nonadherent yeasts were washed, and cultures were observed under epifluorescence microscopy. CW staining was used to confirm that yeasts adhering to BMDMs were not internalized. Visual counts of GFP- and RFP-labeled yeast cells were lower for strains exhibiting the hyperactive allele CgPDR1L280F. The determined percent adherence ratios (PAR; Fig. 5A) closely resembled corresponding PIRs. Again, experiments with mixed inocula of the same strain separately labeled with GFP and RFP revealed no difference and thus no influence of the fluorescent protein labeling on adherence (PARs of around 50%; Fig. 5B). Unlike what was observed for phagocytosis, testing cdr1Δ and pup1Δ mutants on the DSY565 strain background did not reveal significant differences of adherence (Fig. 5C). This suggests that CgCdr1 and Pup1 may interfere with the phagocytosis step only.
Fig 5.

Adherence of C. glabrata to BMDMs. The test strains were matched clinical isolates DSY562 and DSY565 and isogenic strains on the DSY562 strain background harboring either its original CgPDR1 wild-type allele (PDR1) or the hyperactive CgPDR1L280F allele from DSY565 (L280F). (A) Quantification of competitive adherence to BMDMs. Experimental conditions were similar to those described for phagocytosis experiments in legend to Fig. 4C, with the exception that they were performed with 1 h of coincubation in medium containing 1.0 μM cytochalasin D to inhibit phagocytosis. Percent adherence ratios (PARs) represent percentages of yeasts of each strain in the total number of yeasts from both competitor strains. (B) Control PARs determined as described for competitions between the same clinical isolate labeled with the two different fluorescent proteins. (C) PARs determined as described for competitions between clinical isolate DSY565 and either cdr1Δ, pup1Δ, or cdr1Δ pup1Δ double mutant strains constructed on the same strain background. Results are means + SD of a minimum of three independent experiments performed in duplicate (with fluorescent protein labeling swapping between strains). Statistical analyses of the differences between PARs for each pair of competitor strains were performed using the Mann-Whitney U test for panel A and the unpaired Student t test for panels B and C. P values of <0.05 were considered significant, and asterisks represent P value ranges (ns, P > 0.05; *, P < 0.05; **, P < 0.01).
Additional hyperactive CgPDR1 alleles reproduce interaction phenotypes with BMDMs.
To determine if the discovered adherence and phagocytosis phenotype was dependent on CgPDR1L280F or rather related to CgPDR1 hyperactivity per se, we tested additional hyperactive CgPDR1 alleles in the same phagocytosis/adherence assay. A group of isogenic strains was used, constructed by reintroducing in a DSY562 pdr1Δ background either the wild-type (DSY562 pdr1Δ::CgPDR1) or the hyperactive CgPDR1 alleles with GOF mutations L280F from DSY565 (DSY562 pdr1Δ::CgPDR1L280F), R376W from DSY739 (DSY562 pdr1Δ::CgPDR1R376W), and T588A from DSY2234 (DSY562 pdr1Δ::CgPDR1T588A) (13). These two additional hyperactive alleles tested in competition with the wild type were found to cause a decrease similar to that caused by PDR1L280F in uptake (Fig. 6A) and adherence to BMDMs (Fig. 6B). Allele PDR1R376W has been previously shown to cause a similar pattern of target gene overexpression to PDR1L280F, based mainly on CgCDR1, while allele PDR1T588A displays a different pattern, rather based on CgCDR2 overexpression (13). Nevertheless, and just as previously observed for gain of virulence in mouse models of systemic infection, these two additional alleles both recapitulated the CgPDR1L280F-mediated phenotypes in what regards their interaction with BMDMs.
Fig 6.

Phagocytosis and adherence of C. glabrata to BMDMs. The test strains were isogenic strains constructed on the DSY562 strain background harboring either its original CgPDR1 wild-type allele (Cgpdr1Δ::CgPDR1; PDR1, VSY103 and VSY105) or hyperactive alleles CgPDR1L280F from DSY565 (L280F, VSY104 and VSY106), CgPDR1R376W from DSY739 (R376W, VSY136 and VSY138) and CgPDR1T588A from DSY2234 (T588A, VSY137 and VSY139). (A) Quantification of competitive uptake by BMDMs expressed as percent internalization ratios (PIRs; percentages of yeasts of each strain in the total number of yeasts from both competitor strains). Cultures were prepared and visualized as described above for Fig. 4C. (B) Quantification of competitive adherence to BMDMs expressed as percent adherence ratios (PARs). Experimental conditions were similar to those described for Fig. 5A. Results are means + SD of a minimum of three independent experiments performed in duplicate (with fluorescent protein labeling swapping between strains). P values of <0.05 were considered significant, and asterisks represent P value ranges (*, P < 0.05, Mann-Whitney U test).
CgPDR1-mediated differences in the interaction with BMDMs are reproduced with THP-1-derived macrophage-like cells.
To investigate whether the uncovered phenotype could be reproduced using different phagocytic cells, we repeated the coculture experiments with human macrophages. We used the acute monocytic leukemia cell line THP-1 differentiated into adherent macrophage-like cells. Phagocytosis experiments yielded results similar to those obtained with BMDMs. In fact, resulting PIRs were indistinguishable from those obtained following coincubation with BMDMs (Fig. 7A). Adherence experiments to THP-1-derived phagocytes treated with cytochalasin D resulted in considerably lower numbers of adherent yeasts than those seen with BMDMs. Under our experimental conditions, adhesion indexes (AI; representing the number of adherent yeasts per 100 phagocytic cells) to BMDMs approached 300 and 200 for the wild type and the hyperactive CgPDR1L280F allele, respectively. These indexes were about 8-fold lower with THP1-derived cells. However, a decreased adherence of strains containing the hyperactive allele was still observed, with PAR values similar to PIRs (Fig. 7B). Repeating the experiments with THP-1-derived phagocytic cells fixed in formaldehyde instead of using cytochalasin D-treated cultures produced similar results (data not shown).
Fig 7.

Phagocytosis and adherence of C. glabrata to THP-1-derived macrophage-like cells. The test strains were matched clinical isolates DSY562 and DSY565 and isogenic strains on the DSY562 strain background harboring either its original CgPDR1 wild-type allele (PDR1) or the hyperactive CgPDR1L280F allele from DSY565 (L280F). (A) Quantification of competitive uptake by THP-1-derived phagocytes as percent internalization ratios (PIRs; percentage of yeasts of each strain in the total number of yeasts from both strains). Experimental conditions were those described for the phagocytosis experiments in the legend to Fig. 4. (B) Quantification of competitive adherence to THP-1-derived phagocytes as percent adherence ratios (PARs; percentage of yeasts of each strain in the total number of yeasts from both strains). Experimental conditions were similar to those described for the adherence experiments shown in Fig. 5. Results are means + SD of a minimum of three independent experiments performed in duplicate (with fluorescent protein labeling swapping between strains). P values of <0.05 were considered significant, and asterisks represent P value ranges (*, P < 0.05, Mann-Whitney U test).
CgPDR1 hyperactivity favors adherence to epithelial cells.
Given the decreased uptake by and adherence to phagocytes in C. glabrata strains with hyperactive CgPDR1 alleles, we wondered whether similar differences might be found in the interaction with other host cell types. Adherence to host epithelial surfaces is a key step in the host-pathogen interaction and is required for the successful establishment of colonization and infection by yeasts. This way, we tested the role of CgPDR1 on the adherence to epithelial cells, using cell lines CHO-Lec2, HeLa, and Caco-2. Lec2 cells present N-acetyl lactosamine (Galβ1-4 GlcNAc) as the terminal glycosylation of complex N-linked carbohydrates, which increases binding of C. glabrata. This type of cells was used initially to test C. glabrata adherence and the role of specific adhesins in this process (31). We found that most yeasts remained adherent to Lec2 cell monolayers (up to around 60% of the inocula), as previously reported (31), while adherence to human HeLa or Caco-2 cells was lower (20 to 30% of the inocula). The CgPDR1 allele containing the GOF caused an increased adherence to all tested epithelial cell lines in comparison to the wild-type allele in an isogenic strain background (Fig. 8). A similar tendency was found when testing the clinical isolates DSY562 and DSY565 (data not shown).
Fig 8.

Adherence of C. glabrata to epithelial cells. Confluent cell monolayers in 24-well plates of epithelial cell lines CHO-Lec2, CaCo-2, and HeLa were inoculated with 3.0 × 105 log-phase yeast cells in a 1:1 mix of two competitor strains. The test strains were isogenic strains on the DSY562 strain background harboring the DSY562 CgPDR1 wild-type allele (PDR1, VSY101) or the hyperactive CgPDR1L280F allele from DSY565 (L280F, VSY102). Yeasts were allowed to adhere for 30 min, and the cultures were washed to remove nonadherent yeast cells. Epithelial cell monolayers were disrupted, and the resulting yeast suspensions were plated onto YPDA medium to count CFU. Quantification of adherent yeasts is expressed as percent adherence ratios (PARs; percentage of yeasts of each strain in the total number of yeasts from both strains). Results are means + SD of a minimum of three independent experiments. P values of <0.05 were considered significant, and asterisks represent P value ranges (*, P < 0.05, unpaired Student's t test).
Galactose reverses differences on competitive uptake and adherence to BMDMs.
The fact that adherence of C. glabrata to CHO-Lec2 epithelial cells is increased in comparison to other epithelial cells, including parent CHO cells, suggests a central role for yeast cell wall lectins binding host cell N-acetyl lactosamine. Following the observation that our strains did indeed adhere strongly to Lec2 cells, we wondered whether a similar process might mediate the interaction with phagocytes. We thus tested the effect of supplementing the culture medium with galactose, serving here as a surrogate for the ligand N-acetyl lactosamine (a disaccharide of β1-4-bound N-acetylglucosamine and galactose). Repeating the competitive coincubation of C. glabrata strains bearing different CgPDR1 alleles with BMDMs in the presence of galactose revealed an influence of this monosaccharide on adherence and uptake by BMDMs. Galactose completely eliminated differences in PIR and PAR at concentrations above 1.0 mM and 0.5 mM, respectively (Fig. 9A and B). This resulted from the fact that phagocytic and adhesion indexes (PI and AI, respectively, representing the number of yeasts associated to 100 BMDMs) were lowered for strain DSY562 to match those measured for DSY565, which were not affected by galactose (data not shown). Thus, galactose seems to competitively inhibit adherence of the strain bearing the wild-type CgPDR1 allele to macrophages, while not affecting adherence of the strain bearing the hyperactive CgPDR1 allele.
Fig 9.
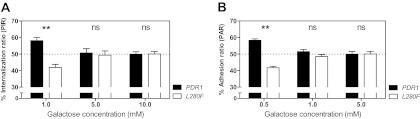
Phagocytosis and adherence of C. glabrata to BMDMs in medium supplemented with galactose. The test strains were the isogenic strain pair on the DSY562 strain background harboring the DSY562 CgPDR1 wild-type allele (PDR1) or the hyperactive CgPDR1L280F allele from DSY565 (L280F). (A) Quantification of competitive uptake by BMDMs expressed as percent internalization ratios (PIRs; percentages of yeasts of each strain in the total number of yeasts from both competitor strains). Experimental conditions were similar to those described for Fig. 4C, with the exception that they were performed in medium supplemented with different concentrations of galactose. (B) Quantification of competitive adherence to BMDMs expressed as percent adherence ratios (PARs). Experimental conditions were similar to those described for Fig. 5A, with the exception that they were performed in medium supplemented with different concentrations of galactose. Results are means + SD of a minimum of three independent experiments performed in duplicate (with fluorescent protein labeling swapping between strains). P values of <0.05 were considered significant, and asterisks represent P value ranges (ns, P > 0.05; **, P < 0.01, Kruskal-Wallis nonparametric test, followed by Dunn's post hoc test for multiple comparisons between pairs of competitor strains).
DISCUSSION
The influence of the development of drug resistance in yeast pathogens on virulence or even the interaction with cells of the innate immune system specifically is an emerging subject (32). As a follow-up to the elucidation by our group that GOF mutations in CgPDR1 mediate not only azole resistance but also gain of virulence in C. glabrata (13), we investigated in this work the impact of such mutations on the interaction with mammalian host cells. We employed a model based on competitive coincubation of mammalian cell cultures with mixed inocula of two C. glabrata strains harboring different CgPDR1 alleles. We used yeasts expressing the fluorescent protein GFP or RFP to allow strain identification. A similar system has successfully been employed by Keppler-Ross et al. to study the recognition of yeasts by macrophages (33). The competitive design was chosen because of its potential to uncover slight phenotypic differences that might otherwise go undetected.
Our first approach regarding labeling of the yeasts with fluorescent proteins relied on the use of URA3 CgCEN-ARS plasmid pCgACU-5 (28). Three tandemly fused GFP sequence copies from plasmid pBS-3xGFP-TRP1 (34) and yeast-enhanced monomeric red fluorescent protein (yEmRFP) from plasmid yEpGAP-Cherry (29) were cloned in the vector under the control of the S. cerevisiae glyceraldehyde-3-phosphate dehydrogenase isozyme 3 gene (TDH3) promoter (pCgACU-TDH3p-3xGFP). However, expressing 3 copies of GFP from this plasmid led to a significant growth defect in our C. glabrata strains (data not shown). We tried a number of modifications, including expressing one copy of GFP only, reducing the TDH3 promoter size, using the alternative GFP gene from pYESmtGFP (35), linearizing the plasmid for integration at the CgCEN8 locus, or replacing the TDH3 promoter by the TEF3 or the ACT1 promoters. None of these approaches yielded satisfactory results, since constructed strains all showed either a similar growth defect or a lack of optimal GFP expression (data not shown). The problem was finally solved by using plasmid pGRB2.3 (see Materials and Methods), a similar centromere sequence and autonomously replicating sequence (CEN-ARS) plasmid containing GFP expressed under the control of the S. cerevisiae 3-phosphoglycerate kinase gene (PGK1) promoter in plasmid pGRB2.2 (27). Our results highlight the need for carefully engineering strains to avoid in vitro fitness defects that would otherwise bias competition assays with mammalian cells.
C. glabrata is known to be able to persist in mice following intravenous (i.v.) infection for long periods of time without leading to a fatal infection. This is the case even in immunocompetent animals (16, 19). In fact, C. glabrata is able to survive and replicate inside macrophages following phagocytosis (22, 23). Recently, Seider et al. referred to C. glabrata as a facultative intracellular pathogen, based on their finding that it actively modifies the phagolysosome environment and ultimately replicates and leads to macrophage lysis (23). We observed a similar replication behavior in our experimental setting, with the first yeast sister buds becoming visible after 5 h of coincubation and a 6-fold increase in intracellular dividing yeasts after 24 h. Just as reported by Seider et al., incubating the cocultures for longer periods eventually resulted in the lysis of macrophages. It has previously been suggested that overexpression of efflux pumps may allow C. neoformans to counteract phagolysosome acidification and thus increase fungal intracellular survival in macrophages (36). Importantly, this does not seem to be the case in the context of our C. glabrata strains. Even though the L280F GOF mutation leads to a 50-fold overexpression of CgCDR1 and a 4-fold overexpression of CgCDR2 (13), we did not observe any influence on survival and replication within macrophages. CgPDR1 did not affect the macrophage response in terms of cytokine production either. CgPDR1 did, however, modulate adherence to and phagocytosis by primary murine macrophages. GOF mutations on this gene led to decreased adherence and phagocytosis. This observation was not restricted to murine macrophages and was also observed in human THP1-derived macrophage-like cells. To rule out an influence of the growth conditions in selective minimal growth medium, we repeated competitive phagocytosis tests with log-phase yeasts grown for two generations in rich medium (YPD). There was no detectable loss of the episomal plasmids bearing the fluorescent proteins, and the results closely reproduced those found using the selective minimal medium (data not shown).
Since CgPdr1 target genes CgCDR1 and PUP1 are both partially implicated in the CgPDR1-mediated gain of virulence in C. glabrata (14), we also tested the influence of these genes on phagocytosis by and adherence to BMDMs. Interestingly, the double knockout of CgCDR1 and PUP1 led to an increase in phagocytosis in competition with the parent strain but did not influence binding to macrophages. These results recall a previously suggested distinction between adherence to macrophages and the subsequent actin-mediated phagocytosis (37) and ultimately suggest that CgCDR1 and PUP1 participate in the actin-mediated phagocytosis step only.
Unlike what we observed in the interaction with phagocytes, CgPDR1 hyperactivity does not seem to decrease adherence to mammalian epithelial cells but rather seems to increase it. The phenotype was slightly more evident when testing CHO-Lec2 cells than HeLa or Caco-2 cells, possibly because of the increased adherence of C. glabrata to the former cell line. However, the difference in adherence was observed in all cases. This result is a priori consistent with the previously described gain of virulence in our strains, since adherence to host epithelial surfaces is thought to be a virulence mechanism in C. glabrata (38). This way, azole-resistant C. glabrata cells harboring CgPDR1 GOF mutations employ a complementary strategy to achieve enhanced virulence. The decreased ability of innate immune cells (such as macrophages) to capture C. glabrata, representing an immune evasion mechanism, together with the increased adherence to epithelial cells would allow C. glabrata to both persist in and cause damage to the host. The combination of these opposite adherence profiles contributes to the resulting enhanced virulence caused by CgPDR1 GOF mutations. According to previously published data, C. glabrata pathogenesis appears to rely on immune evasion strategies to persist within mammalian hosts (16). Prevention of phagocytosis may be the most basic mechanism involved in the process (20). Yeasts prevent phagocytosis by masking immunostimulatory PAMPs from host recognition by PRRs (20). Prominent examples include β-glucan shielding with a mannoprotein coat in C. albicans, production of an α-(1,3)-glucan layer that overlays the immunostimulatory β-glucan in Histoplasma capsulatum, modification of cell wall glucan polymer linkage from a β-(1,3)-glucan to an α-(1,3)-glucan in Paracoccidioides brasiliensis, or increase in the size of the Cryptococcus neoformans antiphagocytic polysaccharide capsule (20). These mechanisms all have in common that they rely on cell wall carbohydrate modifications on the fungus side to mask it from recognition by the host's C-type lectin receptor Dectin-1. Recently, Kuhn and Vyas have presented evidence of the reverse paradigm, whereby a fungal lectin mediates adherence to a mammalian cell carbohydrate ligand (37). The lectin is the glycosylphosphatidylinositol (GPI)-anchored cell wall adhesin EPA1, which is a member of an extensive family of related EPA genes previously established as a major mediator of adhesion to epithelial cells in vitro (25, 31) and a probable contributor to colonization in vivo and virulence (22, 25). The regulation of EPA1 is complex (25, 39–41), and its expression is known to be highly heterogeneous between different C. glabrata strains (apparently due to differences in subtelomeric transcriptional silencing) (42).
Preliminary data collected in our lab support the hypothesis that CgPDR1 controls the expression of cell wall adhesins in our strains. On one hand, whole-genome transcriptional analyses showed that GOF mutations in CgPDR1 regulate several GPI-anchored cell wall adhesins, including some in the EPA family (14). We observed a difference in adherence not only to phagocytes but to epithelial cells as well, even though the latter was less striking. The phenotype was also affected by galactose, which is known to be the terminal monosaccharide residue of Epa ligands (43). We found that galactose could revert CgPDR1-mediated differences in phagocytosis when added to the medium in concentrations in the order of 5.0 mM; Cormack et al. describe 10 mM as the concentration required to inhibit 50% of the adherence to HEp-2 epithelial cells (31). In our experiments, galactose eliminated differences between the two strains by partially inhibiting adherence of the C. glabrata strain bearing the wild-type CgPDR1 allele while not affecting the competitor drug-resistant strain. This suggests that decreased adherence and phagocytosis by macrophages in strains carrying hyperactive CgPDR1 alleles may be caused by the decreased expression of a galactose-binding lectin on the yeast cell wall. Furthermore, preliminary data in our strains suggest that there are no qualitative or quantitative differences in carbohydrate composition in the cell wall (data not shown).
Cell wall adhesins mediate increases in adherence to mammalian cells, as described for epithelial cells (22, 25, 31) and, more recently, to phagocytes (37). In the case of our work, GOF mutations in CgPDR1 led to decreased adherence and phagocytosis by macrophages but in contrast seemed to favor increased adherence to epithelial cells, thus suggesting a more complex situation requiring future investigations. We plan a multisystem approach based upon genomic, transcriptomic, and proteomic analyses of cell wall components in our strains, as well as in other model C. glabrata strains, in the search for candidate CgPDR1-regulated genes mediating interaction with mammalian cells.
In conclusion, starting from the known drug resistance and gain of virulence mediated by GOF mutations in the zinc cluster transcription factor CgPDR1 (13), we showed in this work that C. glabrata may employ a strategy based on CgPDR1-mediated evasion from adherence and phagocytosis by murine and human phagocytes. Furthermore, the same alterations favor adherence to epithelial cells. This shows that CgPDR1 modulates the interaction with mammalian cells and may contribute to the previously described gain of virulence in different ways. The zinc cluster transcription factor CgPDR1 thus stands out as another regulator of host-pathogen interactions in C. glabrata.
ACKNOWLEDGMENTS
We are grateful to N. Dean (Stony Brook University, USA), K. Kuchler (Medical University of Vienna, Austria), B. Cormack (Johns Hopkins University, USA), and B. Westermann (Universität Bayreuth, Germany) for the kind gifts of plasmids used in this study. We thank members of the Sanglard Lab for helpful discussions.
This work was supported by a grant to D.S. from the Swiss National Research Foundation (31003A-127378).
Footnotes
Published ahead of print 4 March 2013
REFERENCES
- 1. Pfaller MA, Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol. 36:1–53 [DOI] [PubMed] [Google Scholar]
- 2. Lockhart SR, Iqbal N, Ahlquist AM, Farley MM, Harrison LH, Bolden CB, Baughman W, Stein B, Hollick R, Park BJ, Chiller T. 2012. Species identification and antifungal susceptibility of Candida bloodstream isolates from population-based surveillance in two U.S. cities: 2008-2011. J. Clin. Microbiol. 50:3435–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moretti ML, Trabasso P, Lyra L, Fagnani R, Resende MR, de Oliveira Cardoso LG, Schreiber AZ. 2012. Is the incidence of candidemia caused by Candida glabrata increasing in Brazil? Five-year surveillance of Candida bloodstream infection in a university reference hospital in southeast Brazil. Med. Mycol. [Epub ahead of print.] doi:10.3109/13693786.2012.708107 [DOI] [PubMed] [Google Scholar]
- 4. Pfaller MA. 2012. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 125:S3–S13 [DOI] [PubMed] [Google Scholar]
- 5. Sanglard D, Ischer F, Bille J. 2001. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45:1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Izumikawa K, Kakeya H, Tsai H, Grimberg B, Bennett J. 2003. Function of Candida glabrata ABC transporter gene, PDH1. Yeast 20:249–261 [DOI] [PubMed] [Google Scholar]
- 8. Bennett JE, Izumikawa K, Marr KA. 2004. Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrob. Agents Chemother. 48:1773–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Torelli R, Posteraro B, Ferrari S, La Sorda M, Fadda G, Sanglard D, Sanguinetti M. 2008. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol. Microbiol. 68:186–201 [DOI] [PubMed] [Google Scholar]
- 10. Vermitsky J-P, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61:704–722 [DOI] [PubMed] [Google Scholar]
- 11. Tsai H, Krol A, Sarti K, Bennett J. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paul S, Schmidt JA, Moye-Rowley WS. 2011. Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot. Cell 10:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268 doi:10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferrari S, Sanguinetti M, Torelli R, Posteraro B, Sanglard D. 2011. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One 6:e17589 doi:10.1371/journal.pone.0017589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roetzer A, Gabaldon T, Schuller C. 2011. From Saccharomyces cerevisiae to Candida glabrata in a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol. Lett. 314:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jacobsen ID, Brunke S, Seider K, Schwarzmüller T, Firon A, d'Enfert C, Kuchler K, Hube B. 2010. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect. Immun. 78:1066–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arendrup M, Horn T, Frimodt-Moller N. 2002. In vivo pathogenicity of eight medically relevant Candida species in an animal model. Infection 30:286–291 [DOI] [PubMed] [Google Scholar]
- 18. Brieland J, Essig D, Jackson C, Frank D, Loebenberg D, Menzel F, Arnold B, DiDomenico B, Hare R. 2001. Comparison of pathogenesis and host immune responses to Candida glabrata and Candida albicans in systemically infected immunocompetent mice. Infect. Immun. 69:5046–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brunke S, Hube B. 2012. Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell. Microbiol. [Epub ahead of print.] doi:10.1111/cmi.12091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seider K, Heyken A, Lüttich A, Miramón P, Hube B. 2010. Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr. Opin. Microbiol. 13:392–400 [DOI] [PubMed] [Google Scholar]
- 21. Roetzer A, Gratz N, Kovarik P, Schuller C. 2010. Autophagy supports Candida glabrata survival during phagocytosis. Cell. Microbiol. 12:199–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaur R, Ma B, Cormack BP. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 104:7628–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, Majer O, Barz D, Haas A, Kuchler K, Schaller M, Hube B. 2011. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J. Immunol. 187:3072–3086 [DOI] [PubMed] [Google Scholar]
- 24. Roetzer A, Klopf E, Gratz N, Marcet-Houben M, Hiller E, Rupp S, Gabaldón T, Kovarik P, Schüller C. 2011. Regulation of Candida glabrata oxidative stress resistance is adapted to host environment. FEBS Lett. 585:319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Domergue R, Castano I, De Las Penas A, Zupancic M, Lockatell V, Hebel JR, Johnson D, Cormack BP. 2005. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308:866–870 [DOI] [PubMed] [Google Scholar]
- 26. Sanglard D, Ischer F, Monod M, Bille J. 1996. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob. Agents Chemother. 40:2300–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frieman MB, McCaffery JM, Cormack BP. 2002. Modular domain structure in the Candida glabrata adhesin Epa1p, a beta1,6 glucan-cross-linked cell wall protein. Mol. Microbiol. 46:479–492 [DOI] [PubMed] [Google Scholar]
- 28. Kitada K, Yamaguchi E, Arisawa M. 1996. Isolation of a Candida glabrata centromere and its use in construction of plasmid vectors. Gene 175:105–108 [DOI] [PubMed] [Google Scholar]
- 29. Keppler-Ross S, Noffz C, Dean N. 2008. A new purple fluorescent color marker for genetic studies in Saccharomyces cerevisiae and Candida albicans. Genetics 179:705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dalle F, Wachtler B, L'ollivier C, Holland G, Bannert N, Wilson D, Labruere C, Bonnin A, Hube B. 2010. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell. Microbiol. 12:248–271 [DOI] [PubMed] [Google Scholar]
- 31. Cormack BP, Ghori N, Falkow S. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285:578–582 [DOI] [PubMed] [Google Scholar]
- 32. Lewis RE, Viale P, Kontoyiannis DP. 2012. The potential impact of antifungal drug resistance mechanisms on the host immune response to Candida. Virulence 3:368–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keppler-Ross S, Douglas L, Konopka JB, Dean N. 2010. Recognition of yeast by murine macrophages requires mannan but not glucan. Eukaryot. Cell 9:1776–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee W, Oberle J, Cooper J. 2003. The role of the lissencephaly protein PacI during nuclear migration in budding yeast. J. Cell Biol. 160:355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westermann B, Neupert W. 2000. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast 16:1421–1427 [DOI] [PubMed] [Google Scholar]
- 36. Sanguinetti M, Posteraro B, La Sorda M, Torelli R, Fiori B, Santangelo R, Delogu G, Fadda G. 2006. Role of AFR1, an ABC transporter-encoding gene, in the in vivo response to fluconazole and virulence of Cryptococcus neoformans. Infect. Immun. 74:1352–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhn DM, Vyas VK. 2012. The Candida glabrata adhesin Epa1p causes adhesion, phagocytosis, and cytokine secretion by innate immune cells. FEMS Yeast Res. 12:398–414 [DOI] [PubMed] [Google Scholar]
- 38. Kaur R, Domergue R, Zupancic ML, Cormack BP. 2005. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 8:378–384 [DOI] [PubMed] [Google Scholar]
- 39. Castaño I, Pan S-J, Zupancic M, Hennequin C, Dujon B, Cormack BP. 2005. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 55:1246–1258 [DOI] [PubMed] [Google Scholar]
- 40. Gallegos-Garcia V, Pan SJ, Juarez-Cepeda J, Ramirez-Zavaleta CY, Briones-Martin-Del-Campo M, Martinez-Jimenez V, Castano I, Cormack B, De Las Penas A. 2012. A novel downstream regulatory element cooperates with the silencing machinery to repress EPA1 expression in Candida glabrata. Genetics 190:1285–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Las Peñas A, Pan S-J, Castaño I, Alder J, Cregg R, Cormack BP. 2003. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 17:2245–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Halliwell SC, Smith MCA, Muston P, Holland SL, Avery SV. 2012. Heterogeneous expression of the virulence-related adhesin Epa1 between individual cells and strains of the pathogen Candida glabrata. Eukaryot. Cell 11:141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zupancic ML, Frieman M, Smith D, Alvarez RA, Cummings RD, Cormack BP. 2008. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68:547–559 [DOI] [PubMed] [Google Scholar]