

Abstract
Alkyl glycosides are attractive surfactants because of their high surface activity and good biodegradability and can be produced from renewable resources. Through enzymatic catalysis, one can obtain well-defined alkyl glycosides, something that is very difficult to do using conventional chemistry. However, there is a need for better enzymes to get a commercially feasible process. A thermostable β-glucosidase from the well-studied glycoside hydrolase family 1 from Thermotoga neapolitana, TnBgl1A, was mutated in an attempt to improve its value for synthesis of alkyl glycosides. This was done by rational design using prior knowledge from structural homologues together with a recently generated model of the enzyme in question. Three out of four studied mutations increased the hydrolytic reaction rate in an aqueous environment, while none displayed this property in the presence of an alcohol acceptor. This shows that even if the enzyme resides in a separate aqueous phase, the presence of an organic solvent has a great influence. We could also show that a single amino acid replacement in a less studied part of the aglycone subsite, N220F, improves the specificity for transglycosylation 7-fold and thereby increases the potential yield of alkyl glycoside from 17% to 58%.
INTRODUCTION
Surfactants are among the most commonly produced chemicals today, with a global market estimated to reach 30 million metric tons in 2030 (1). A group of surfactants with attractive properties are alkyl glycosides, exhibiting antimicrobial activity, biodegradability, and low toxicity (2). These nonionic surfactants find their use mainly in cosmetics, pharmaceuticals, and the food industry. Currently, industrially produced alkyl glycosides are of technical grade and have various alcohol groups, numbers of monosaccharide units, and linkages (3). For the above-mentioned areas of use, well-defined alkyl glycosides would be preferable and can be prepared by the use of protection-deprotection chemistry or, alternatively, through enzymatic synthesis using glycoside hydrolases (glycosidases).
The enzymatic approach can be followed through either the thermodynamically controlled reverse hydrolysis reaction or the kinetically controlled transglycosylation reaction (4). The latter has the possibility to overshoot the thermodynamic equilibrium and is more influenced by the properties of the enzyme (5); therefore, it is the focus of this study. However, this attractive biocatalysis requires engineering to function as an industrially feasible option. Obtaining a high synthesis yield is dependent on a high ratio of transferase to hydrolase activity of the enzyme and has been the topic of several previous studies (6–8).
For rational design of improved biocatalysts, it is advantageous to work with a well-characterized enzyme or at least an enzyme from a well-studied family, such as glycoside hydrolase family 1 (GH1). GH1 is composed of retaining glycosidases with broad substrate specificity and contains several thousand enzymes, of which 255 have been characterized, and 37 structures are available in the Carbohydrate-Active EnZymes database (9). Moreover, for industrial purposes, an enzyme of thermophilic origin can be considered favorable, since elevated temperatures can yield higher substrate solubility, lower viscosity, and thereby lower pumping costs, and limited risks of bacterial contamination (10). Consequently, a β-glucosidase from Thermotoga neapolitana, TnBgl1A, is an ideal starting point for protein engineering studies.
In this study, we have generated mutants of the enzyme with the aim of increasing the transferase/hydrolase ratio (rs/rh), using results from mutational studies of other β-glycosidases along with structural information from a recently generated model (11).
MATERIALS AND METHODS
Chemicals.
Hexyl-β-d-glucoside (HG), p-nitrophenol (pNP), and p-nitrophenol-β-d-glucoside (pNPG) were obtained from Sigma-Aldrich (St. Louis, MO), and all other chemicals were from VWR International (Stockholm, Sweden).
Mutagenesis.
The genes encoding wild-type (wt) TnBgl1A (GenBank accession number AF039487) and the TnBgl1A codon optimized for expression in Escherichia coli (GenBank accession number KC776911) were previously cloned into PET22b(+) (Novagen, Madison, WI) (12). Mutagenesis was performed using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the sequences with GenBank accession numbers KC776911 (for V168S and W322F) and AF039487 (for N220F and G222F) as the templates and the primers presented in Table 1. The resulting plasmids were transformed into E. coli Nova Blue cells for storage and into E. coli BL21 (Novagen) for expression. The complete gene was sequenced by GATC Biotech AG (Konstanz, Germany) to confirm the mutations.
Table 1.
Primers used in this work
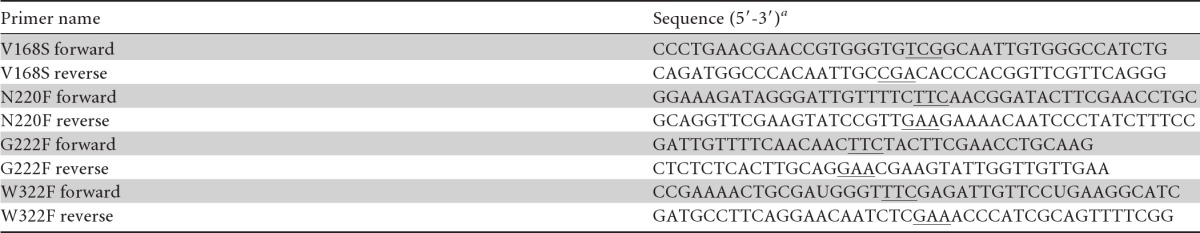
a Codons containing introduced changes are underlined.
Expression and purification.
The mutant and wild-type enzymes were produced in 0.5-liter cultivations in Erlenmeyer flasks at 37°C and pH 7 with Luria-Bertani (LB) medium containing 100 μg/ml ampicillin and were inoculated with 1% overnight precultures. After reaching an optical density at 620 nm of 0.6, expression was induced by addition of 0.5 ml 100 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG), and production was continued for 20 h. Cells were harvested by centrifugation at 5,500 × g for 10 min at 4°C, resuspended in binding buffer (20 mM imidazole, 20 mM Tris-HCl, 0.75 M NaCl, pH 7.5), and lysed by sonication 6 times for 3 min each time at 60% amplitude and a cycle of 0.5 using a 14-mm titanium probe (UP400 S; Hielscher Ultrasonic GmbH, Teltow, Germany). Heat treatment (70°C, 30 min) and centrifugation (30 min, 4°C, 15,000 × g) were used to remove most of the E. coli proteins before purification by immobilized metal affinity chromatography using an ÄKTA prime system (Amersham Biosciences, Uppsala, Sweden). The protein slurry was applied to an Histrap FF crude column (GE Healthcare) pretreated with 0.1 M copper(II) sulfate. Bound protein was eluted using elution buffer (250 mM imidazole, 20 mM Tris-HCl, 0.75 M NaCl, pH 7.5), and fractions containing protein were pooled, dialyzed against 50 mM citrate phosphate buffer, pH 5.6, overnight using a 3,500-Da-molecular-mass-cutoff dialysis membrane (Spectrum Laboratories, Rancho Dominguez, CA), and stored at −20°C until use.
Hydrolysis activity assay.
Kinetic parameters for hydrolysis (kcat and Km [Michaelis constant]) were determined at 70°C using a 96-well setup by following the hydrolysis of pNPG into pNP and glucose. In a PCR plate on ice, 190 μl 0.5 to 5 mM pNPG in 0.1 M citrate phosphate buffer, pH 5.6, was added to 10 μl enzyme solution (≈2 ng/ml). To ensure that initial reaction rates were measured, 3 plates were incubated for 5, 10, and 20 min in a ThermoMixer apparatus (HLC Biotech, Bovenden, Germany) set to 70°C at 300 rpm using a PCR plate insert before returning them to ice. One hundred twenty microliters was transferred to an enzyme-linked immunosorbent assay plate, and the plate was read at 405 nm after reaching 22°C using a Multiscan GO microplate spectrophotometer (Thermo Scientific, Hudson, NH). The pNP concentration was determined by using an 8-point external calibration curve (0.05 to 2.5 mM). Fourteen replicates per mutant, time point, and pNPG concentration yielded 252 data points for each parameter fit. The kinetic parameters were determined via nonlinear regression by the least-squares method using the Matlab (version 7.12) program (MathWorks, Natick, MA).
Transferase reaction.
The transglycosylation rates were determined by following the formation of HG from pNPG and hexanol. pNPG (2,550 μl, 34 mM) in dry hexanol was preheated in a ThermoMixer apparatus (HLC Biotech, Bovenden, Germany) set to 70°C at 800 rpm. Reactions were started by addition of 450 μl enzyme (≈10 ng/ml) in 0.1 M citrate phosphate buffer, pH 5.6. One hundred microliters of hexanol-phase samples was withdrawn at different time points and diluted with 100 μl Milli-Q H2O and 400 μl acetonitrile before analysis by high-pressure liquid chromatography (HPLC).
Reverse hydrolysis.
Kinetic parameters for reverse hydrolysis (kcat and Km) were determined at 70°C by following the formation of HG from d-glucose and hexanol. d-Glucose equivalent to 67 to 1,000 mM in the aqueous phase was preheated together with 2,550 μl dry hexanol in a ThermoMixer apparatus (HLC Biotech, Bovenden, Germany) set to 70°C at 800 rpm. Reactions were started by addition of 450 μl enzyme (≈50 ng/ml) in 0.1 M citrate phosphate buffer, pH 5.6. One hundred microliters of hexanol-phase samples was withdrawn at different time points and diluted with 100 μl Milli-Q H2O and 400 μl acetonitrile before analysis by HPLC.
HPLC analysis.
Transferase and reverse hydrolysis reactions were monitored using reverse-phase HPLC (L-7100 pump, L-7000 interface, L-7250 autosampler with a 20-ml injection loop, and L-7400 UV detector; LaChrom; Hitachi Ltd., Tokyo, Japan) with an HPLC equipped with an evaporative light-scattering detector (500 ELSD; Alltech Associates Inc., Deerfield, IL) with an evaporator temperature of 94°C and a nebulizer gas flow of 2.5 standard liters per minute and a Kromasil 100-5C18 column (4.6 μm by 250 mm; Kromasil; EkaChemicals AB, Separation Products, Bohus, Sweden). A gradient was applied from 50% to 70% methanol in Milli-Q H2O over 5 min and kept at 70% methanol for 1 min before returning to initial conditions for reequilibration. A constant flow rate of 1.0 ml/min was used. pNPG elutes after 3.5 min and was followed at 405 nm as well as with the ELSD detector. HG and pNP both have a retention time of 7.5 min, but HG does not absorb at 405 nm and pNP is too volatile to be detected by the ELSD detector. Concentrations were determined by use of 8-point external standard curves.
Structural analysis.
Discussion of homologous positions is based on sequence alignments obtained using the ClustalW program (13) and superimposed structures obtained using PyMOL software (Delano Scientific, Palo Alto, CA).
RESULTS AND DISCUSSION
Active-site mutants.
The glycone (−1) subsite of GH1 glycosidases has been extensively investigated, while less focus has been directed toward the aglycone (+1) subsite, arguably due to the few structures with ligands in the aglycone binding region determined (14). Among the few GH1 structures solved with ligands occupying the aglycone subsites are p-nitrophenyl-β-thioglucoside in maize Glu1 (Zea mays Glu1 [ZmGlu1]) (15) and cellotetraose in rice β-glucosidase (16). In rice, the glycone subsite is formed by direct interactions, while the aglycone subsites mainly consist of water-mediated hydrogen bonds. The binding site (Fig. 1) is often described as a slot with two walls, where one (here called platform) is highly conserved and the other (here called roof) is more variable (16–18). Apart from structural data, several mutational studies have been aimed at understanding the substrate specificity of β-glycosidases, with a majority focusing on hydrolysis reactions.
Fig 1.

p-Nitrophenyl-β-d-glucopyranoside docked into the crystal structure of TnBgl1A (atom coordinates are from T. S. Kulkarni, S. Khan, T. Mahmood, A. Sundin, S. Lindahl, C. Turner, D. T. Logan, and E. Nordberg Karlsson, unpublished data). In the left zoomed image, the catalytic residues E164 and E349 are red, the slot platform and roof are blue, and the selected mutation sites are green (except for W322, which is blue). In the right zoomed image, overlaid aglycone platform tryptophan orientations for other family 1 glycoside hydrolases from eukaryotes (Protein Data Bank accession numbers 3AI0, 2E9L, 1E1F, 1V03, and 2RGM) are green and those from prokaryotes (Protein Data Bank accession numbers 1E4I, 1OIN, 1NP2, 3AHX, 1GON, and 1VFF) are blue.
The importance of aromatic residues in determining the transferase/hydrolase ratio has been observed in several glycoside hydrolases (19–22). Furthermore, a comparison of natural glycosyltransferases and glycoside hydrolases has shown that transferases tend to have tyrosine and phenylalanine and hydrolases have alanine and valine at a position corresponding to the +1 subsite of TnBgl1A (23). Therefore, in this study, we have focused on introducing the aromatic amino acid phenylalanine into the +1 subsite with the aim to increase the hydrophobicity of the aglycone subsite, favoring hexanol over water and thereby increasing the transferase/hydrolase ratio of the thermostable β-glucosidase TnBgl1A. The first position chosen for insertion of a phenylalanine was N220, a less conserved position of the +1 subsite. The homologous position in rice β-glucosidase, D243, has been shown to participate in water-mediated interactions with short polyglucosides (16). Another interesting position chosen was G222, where mutation to glutamine has resulted in increased transglycosylation (M. Megyeri et al., unpublished data). The last position chosen was W322, to which the main aglycone interactions in the structurally homologous Zea mays and Sorghum bicolor β-glucosidases through aromatic stacking interactions have been attributed (24). The exception to this strategy was V168S, which instead was chosen on the basis of a multiple-sequence alignment identifying position 168 to be involved in a second-layer interaction with subsite +1. To investigate the significance of this position, the hydrophobic valine was changed to an amino acid of opposite polarity occurring in structural homologues from GH1, serine (14).
Papps.
A water content of 15% was chosen on the basis of the optimal total reaction rate from previous studies of β-glycosidase-catalyzed synthesis of alkyl glucosides (25–27). Very large reaction volumes would be needed for sampling from the aqueous phase without affecting the reaction. To circumvent this problem and calculate total amounts from samples taken only from the hexanol phase, apparent partitioning coefficients (Papps) were determined for pNPG, pNP, and HG over the studied concentration range (Fig. 2). Papp, defined as the concentration in the hexanol phase divided by the concentration in the aqueous phase at equilibrium, was determined to be 2.6 ± 0.3 for HG, 85 ± 5 for pNP, and 0.60 ± 0.02 for pNPG (data not shown).
Fig 2.
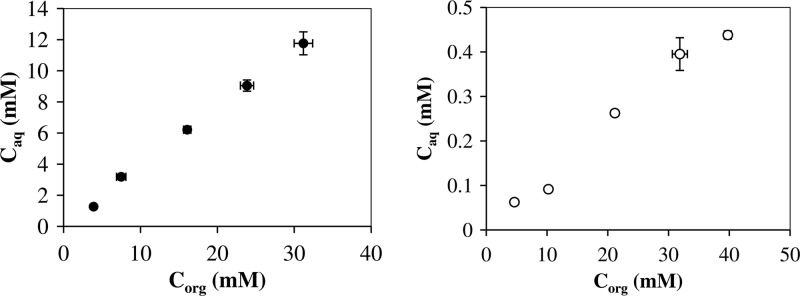
Apparent partitioning of hexyl-β-glucoside (•) and p-nitrophenyl-β-d-glucoside (○) plotted as concentration in the aqueous phase (Caq) versus concentration in the hexanol phase (Corg). Error bars are 1σ.
Hydrolysis.
Kinetic parameters for hydrolysis were determined with the model substrate pNPG at 70°C using a 96-well setup and are presented in Table 2. The N220F, V168S, and W322F mutations increased catalytic efficiency (kcat/Km), and the V168S and W322F mutations did so through decreased Km values, suggesting an effect on pNPG binding. However, since excess substrate is commonly used in practical applications, kcat is judged to be the most important parameter. Therefore, the effect of N220F, which showed a large increase in kcat, was more significant.
Table 2.
Kinetic constants for hydrolysis of p-nitrophenol-β-d-glucopyranoside
| TnBgl1A variant | Vmaxa (μmol min−1 mg−1) | kcata (s−1) | Kma (mM) | kcat/Kma (s−1 mM−1) |
|---|---|---|---|---|
| wt | 179 ± 4 | 154 ± 3 | 0.45 ± 0.05 | 342 ± 39 |
| V168S | 204 ± 7 | 175 ± 6 | 0.20 ± 0.02 | 875 ± 80 |
| N220F | 732 ± 30 | 628 ± 25 | 1.28 ± 0.16 | 491 ± 64 |
| G222F | 191 ± 6 | 164 ± 5 | 0.77 ± 0.09 | 213 ± 26 |
| W322F | 87 ± 4 | 74 ± 3 | 0.17 ± 0.02 | 452 ± 58 |
Data are presented as averages with ranges representing 95% confidence intervals.
Transglycosylation.
The reaction system chosen for transglycosylation in this study uses pNPG as the glycosyl donor and hexanol as the acceptor and is depicted in Fig. 3. In a first step, the glycosyl enzyme is formed and pNP is released. The free enzyme is regenerated by transferring the glucose unit onto a glycosyl acceptor, in this case, either water or hexanol. The produced HG can also be accepted as a glycosyl donor, opening the possibility for secondary hydrolysis. Furthermore, pNPG could theoretically function as a glycosyl acceptor, yielding p-nitrophenyl-β-glucobiose, an unwanted but common side reaction product during transglycosylation (28, 29). However, in this study no such side reaction product was detected using HPLC or thin-layer chromatography (limit of detection for pNPG, <0.2 mM; data not shown). A typical time course for wild-type TnBgl1A-catalyzed transglycosylation between pNPG and hexanol is presented in Fig. 4. For each mutant, initial reaction rates for hydrolysis and synthesis were calculated on the basis of the first 30 min of the reactions. All samples were taken from the hexanol phase, and the apparent partitioning has been accounted for in the calculations of initial reaction rates.
Fig 3.

Reaction scheme for the conversion of p-nitrophenyl-β-d-glucoside to hexyl-β-glucoside (transglycosylation), with conversion to glucose (hydrolysis) being the side reaction. When d-glucose is used as the glycosyl donor, formation of hexyl-β-glucoside is possible through a condensation reaction (reverse hydrolysis). E-COOH, enzyme.
Fig 4.
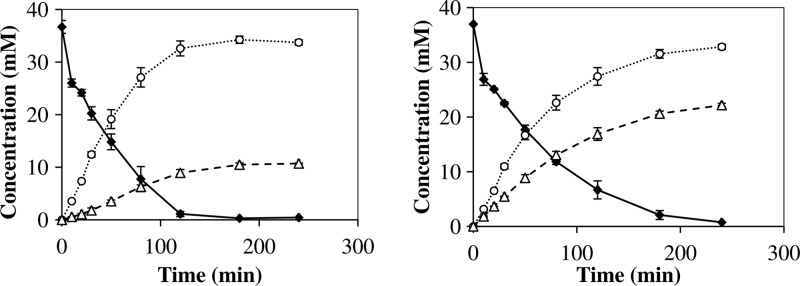
Time course for synthesis of hexyl-β-glucoside (Δ) and p-nitrophenol (○) from p-nitrophenyl-β-d-glucoside (•) catalyzed by TnBgl1A wt (left) and N220F (right). All concentrations refer to the hexanol phase, and error bars are 1σ.
In Table 3, the total initial reaction rate is presented along with the ratio of transferase over hydrolase activity (rs/rh) and the calculated theoretical yield in the absence of secondary hydrolysis, as was done by van Rantwijk et al. (4). All mutations led to reduced total reaction rates, in contrast to those for the hydrolytic reactions studied in an aqueous environment. In addition, the N220F mutation resulted in a major increase in selectivity (7-fold increase of rs/rh), increasing the potential yield of alkyl glycoside from 17% to 58%. This suggests that the mutation strategy was successful in creating an environment more suited for hexanol in the active site, with hexanol thereby outcompeting water as a glycosyl acceptor.
Table 3.
Total activity, rs/rh, and expected yield for the selected mutants
| TnBgl1A variant | Total initial reaction ratea (μmol min−1 mg−1) | rs/rha | Expected yield (%) |
|---|---|---|---|
| wt | 285 ± 21 | 0.20 ± 0.03 | 17 |
| V168S | 262 ± 18 | 0.20 ± 0.02 | 17 |
| N220F | 163 ± 11 | 1.38 ± 0.20 | 58 |
| G222F | 199 ± 16 | 0.22 ± 0.03 | 18 |
| W322F | 141 ± 11 | 0.12 ± 0.08 | 11 |
Data are presented as averages with ranges representing 95% confidence intervals.
The N220/G222 position.
The N220 position has also shown significance in structural homologues of TnBgl1A. D261N in ZmGlu1, corresponding to position N220, reduced hydrolytic activity toward pNPG (30). Furthermore, in Sorghum bicolor the homologous position, N259, is crucial for aglycone recognition (24), and a mutation in the analogous position in human cytosolic β-glucosidase, F225S, removed all hydrolytic activity (31). Moreover, in the closely related organism Thermus thermophilus, the N282T mutation increased transglycosylation, and it was proposed that it did so by affecting N219 (position N220 in TnBgl1A) to increase the interaction with the glycosyl donor. However, this mutation also increased self-condensation. This was countered by introducing a second mutation, A221W, corresponding to amino acid G222 in TnBgl1A, to form π stacking interactions with the pNP-containing acceptor (29).
Additionally, the G222 position has shown significance in ZmGlu1, where the M263F mutation reduced the kcat for pNPG but increased it for o-nitrophenol-β-d-glucoside (30). G222 is located near the narrowest part of the active-site cleft, and previous findings suggest steric hindrance is a mechanism for the reduced catalytic activity that occurs after insertion of a bulkier amino acid in this position (32). In the present study, G222F increased the Km for d-glucose, but no significant increase of specificity for transglycosylation in the presence of hexanol was shown with G222F, likely because it is too far away from the catalytic residues to influence acceptor preference.
In short, our study further supports the influence of N220 and G222 on aglycone specificity and additionally shows that N220 influences not only glycosyl donor binding but also glycosyl acceptor specificity.
Reverse hydrolysis.
To get further data on what effects can be attributed to the presence of hexanol, kinetic parameters for reverse hydrolysis were determined under conditions that were the same as those used in the transglycosylation study, apart from using d-glucose as a glycosyl donor. In a first step, the glycosyl enzyme is formed and water is released. The free enzyme is regenerated by transferring the glucose unit onto a hexanol molecule, forming HG (Fig. 3). The apparent kinetic parameters with regard to glucose are presented in Table 4. All samples were taken from the hexanol phase, and the apparent partitioning has been accounted for in the calculations of the initial reaction rates. In agreement with hydrolysis and transglycosylation data, W322F lowered the catalytic efficiency substantially. Another significant effect was the increase in the Michaelis constant for N220F, which suggests an unfavorable change in the active-site environment for the hydrophilic d-glucose molecule and could explain why favoring hexanol over water at the active site did not show the same beneficial effect on the reaction seen for transglycosylation (Table 3).
Table 4.
Apparent kinetic constants for reverse hydrolysis between d-glucose and hexanol when the concentration of hexanol is kept constant
| TnBgl1A variant | Vmaxa (μmol min−1 mg−1) | kcata (s−1) | Kma (mM) | kcat/Kma (s−1 μM−1) |
|---|---|---|---|---|
| wt | 1.67 ± 0.15 | 1.43 ± 0.13 | 219 ± 61 | 6.5 ± 1.9 |
| V168S | 1.17 ± 0.12 | 1.00 ± 0.10 | 199 ± 73 | 5.0 ± 1.9 |
| N220F | 1.55 ± 0.18 | 1.33 ± 0.15 | 357 ± 104 | 3.7 ± 1.2 |
| G222F | 1.73 ± 0.15 | 1.48 ± 0.13 | 245 ± 66 | 6.1 ± 1.7 |
| W322F | 0.30 ± 0.05 | 0.26 ± 0.04 | 219 ± 122 | 1.2 ± 0.7 |
Data are presented as averages with ranges representing 95% confidence intervals.
Hexanol influence.
In a pure aqueous environment, three of the mutants displayed increased catalytic activity, while in the presence of an alcohol, no such effects in either reverse hydrolysis, transglycosylation, or the undesired hydrolytic side reactions were seen. This shows that even if the enzyme is in a separate aqueous phase, hexanol has a major influence. Most striking was the case of N220F, for which the hydrolytic activity was four times that of the wild type in aqueous solution (Table 2), while in the presence of hexanol, the N220 enzyme had only half the hydrolysis rate of the wild type (Table 3).
Importance of the platform.
As expected from structural homologues (24), W322F significantly reduced hydrolysis in TnBgl1A. Moreover, the equal reduction of transglycosylation suggests that it is the formation of the glycosyl enzyme that is affected by reducing interactions with pNPG. In enzymes from eukaryotes, this interaction is suggested to be a π stacking interaction (15, 24, 33), but due to the orientation of the tryptophan (Fig. 1), it has to be another interaction in prokaryotic enzymes (34–36). Regardless of the type of interaction, our data affirm that the functional significance of the amino acid is conserved.
Conclusions.
This work shows that in addition to the well-documented platform and roof of the aglycone binding site, N220 and G222 form a third site with a major influence on substrate binding. By introducing a single mutation at this site, we were able to increase the specificity toward transglycosylation 7-fold and increase the potential yield of alkyl glycoside from 17% to 58%, thereby taking another step toward industrially useful β-glycosidases for formation of alkyl glycosides. However, a further improvement of rs/rh is needed before sufficient process economics for producing alkyl glycosides through enzymatic transglycosylation can be reached.
ACKNOWLEDGMENTS
We thank Tania Pozzo for allowing us to use the G222F construct that she prepared for a previous study.
This work was supported by the Swedish Research Council (VR) and the EU FP7 program AMYLOMICS.
Footnotes
Published ahead of print 22 March 2013
REFERENCES
- 1. Anonymous. 2012. Asia to boost global surfactants demand. Focus Surfactants 2012: 3 doi:10.1016/S1351-4210(12)70034-3 [Google Scholar]
- 2. Matsumura S, Imai K, Yoshikawa S, Kawada K, Uchibor T. 1990. Surface activities, biodegradability and antimicrobial properties of n-alkyl glucosides, mannosides and galactosides. J. Am. Oil Chem. Soc. 67: 996–1001 [Google Scholar]
- 3. von Rybinski W, Hill K. 1998. Alkyl polyglycosides—properties and applications of a new class of surfactants. Angew. Chem. Int. Ed. Engl. 37: 1328–1345 [DOI] [PubMed] [Google Scholar]
- 4. van Rantwijk F, Woudenberg-van Oosterom M, Sheldon RA. 1999. Glycosidase-catalysed synthesis of alkyl glycosides. J. Mol. Catal. B Enzym. 6: 511–532 [Google Scholar]
- 5. Kasche V. 1986. Mechanism and yields in enzyme catalysed equilibrium and kinetically controlled synthesis of β-lactam antibiotics, peptides and other condensation products. Enzyme Microb. Technol. 8: 4–16 [Google Scholar]
- 6. Hansson T, Adlercreutz P. 2001. Enhanced transglucosylation/hydrolysis ratio of mutants of Pyrococcus furiosus beta-glucosidase: effects of donor concentration, water content, and temperature on activity and selectivity in hexanol. Biotechnol. Bioeng. 75: 656–665 [DOI] [PubMed] [Google Scholar]
- 7. Kone FM, Le Bechec M, Sine JP, Dion M, Tellier C. 2009. Digital screening methodology for the directed evolution of transglycosidases. Protein Eng. Des. Sel. 22: 37–44 [DOI] [PubMed] [Google Scholar]
- 8. Mladenoska I, Grey CE, Winkelhausen E, Kuzmanova S, Adlercreutz P. 2007. Competition between transglycosylation and hydrolysis in almond β-glucosidase-catalyzed conversion of p-nitrophenyl-β-d-glucoside in monophasic water/alcohol mixtures. Biocatal. Biotransform. 25: 382–385 [Google Scholar]
- 9. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37: D233–D238 doi:10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vieille C, Zeikus GJ. 2001. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 65: 1–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khan S, Pozzo T, Megyeri M, Lindahl S, Sundin A, Turner C, Karlsson EN. 2011. Aglycone specificity of Thermotoga neapolitana beta-glucosidase 1A modified by mutagenesis, leading to increased catalytic efficiency in quercetin-3-glucoside hydrolysis. BMC Biochem. 12: 11 doi:10.1186/1471-2091-12-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turner C, Turner P, Jacobson G, Almgren K, Waldeback M, Sjoberg P, Karlsson EN, Markides KE. 2006. Subcritical water extraction and β-glucosidase-catalyzed hydrolysis of quercetin glycosides in onion waste. Green Chem. 8: 949–959 [Google Scholar]
- 13. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 14. Marana SR. 2006. Molecular basis of substrate specificity in family 1 glycoside hydrolases. IUBMB Life. 58: 63–73 [DOI] [PubMed] [Google Scholar]
- 15. Czjzek M, Cicek M, Zamboni V, Burmeister WP, Bevan DR, Henrissat B, Esen A. 2001. Crystal structure of a monocotyledon (maize ZMGlu1) beta-glucosidase and a model of its complex with p-nitrophenyl beta-d-thioglucoside. Biochem. J. 354: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chuenchor W, Pengthaisong S, Robinson RC, Yuvaniyama J, Svasti J, Cairns JR. 2011. The structural basis of oligosaccharide binding by rice BGlu1 beta-glucosidase. J. Struct. Biol. 173: 169–179 [DOI] [PubMed] [Google Scholar]
- 17. Czjzek M, Cicek M, Zamboni V, Bevan DR, Henrissat B, Esen A. 2000. The mechanism of substrate (aglycone) specificity in beta-glucosidases is revealed by crystal structures of mutant maize beta-glucosidase-DIMBOA, -DIMBOAGlc, and -dhurrin complexes. Proc. Natl. Acad. Sci. U. S. A. 97: 13555–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mendonca LM, Marana SR. 2008. The role in the substrate specificity and catalysis of residues forming the substrate aglycone-binding site of a beta-glycosidase. FEBS J. 275: 2536–2547 [DOI] [PubMed] [Google Scholar]
- 19. Hansson T, Kaper T, van Der Oost J, de Vos WM, Adlercreutz P. 2001. Improved oligosaccharide synthesis by protein engineering of beta-glucosidase CelB from hyperthermophilic Pyrococcus furiosus. Biotechnol. Bioeng. 73: 203–210 [DOI] [PubMed] [Google Scholar]
- 20. Kuriki T, Kaneko H, Yanase M, Takata H, Shimada J, Handa S, Takada T, Umeyama H, Okada S. 1996. Controlling substrate preference and transglycosylation activity of neopullulanase by manipulating steric constraint and hydrophobicity in active center. J. Biol. Chem. 271: 17321–17329 [DOI] [PubMed] [Google Scholar]
- 21. Matsui I, Yoneda S, Ishikawa K, Miyairi S, Fukui S, Umeyama H, Honda K. 1994. Roles of the aromatic residues conserved in the active center of Saccharomycopsis alpha-amylase for transglycosylation and hydrolysis activity. Biochemistry 33: 451–458 [DOI] [PubMed] [Google Scholar]
- 22. Kelly RM, Leemhuis H, Rozeboom HJ, van Oosterwijk N, Dijkstra BW, Dijkhuizen L. 2008. Elimination of competing hydrolysis and coupling side reactions of a cyclodextrin glucanotransferase by directed evolution. Biochem. J. 413: 517–525 [DOI] [PubMed] [Google Scholar]
- 23. Saab-Rincón G, del-Río G, Santamaría RI, López-Munguía A, Soberón X. 1999. Introducing transglycosylation activity in a liquefying α-amylase. FEBS Lett. 453: 100–106 [DOI] [PubMed] [Google Scholar]
- 24. Verdoucq L, Moriniere J, Bevan DR, Esen A, Vasella A, Henrissat B, Czjzek M. 2004. Structural determinants of substrate specificity in family 1 beta-glucosidases: novel insights from the crystal structure of sorghum dhurrinase-1, a plant beta-glucosidase with strict specificity, in complex with its natural substrate. J. Biol. Chem. 279: 31796–31803 [DOI] [PubMed] [Google Scholar]
- 25. Hansson T, Andersson M, Wehtje E, Adlercreutz P. 2001. Influence of water activity on the competition between β-glycosidase-catalysed transglycosylation and hydrolysis in aqueous hexanol. Enzyme Microb. Technol. 29: 527–534 [Google Scholar]
- 26. Panintrarux C, Adachi S, Araki Y, Kimura Y, Matsuno R. 1995. Equilibrium yield of n-alkyl-β-d-glucoside through condensation of glucose and n-alcohol by β-glucosidase in a biphasic system. Enzyme Microb. Technol. 17: 32–40 [Google Scholar]
- 27. Turner P, Svensson D, Adlercreutz P, Karlsson EN. 2007. A novel variant of Thermotoga neapolitana beta-glucosidase B is an efficient catalyst for the synthesis of alkyl glucosides by transglycosylation. J. Biotechnol. 130: 67–74 [DOI] [PubMed] [Google Scholar]
- 28. Feng HY, Drone J, Hoffmann L, Tran V, Tellier C, Rabiller C, Dion M. 2005. Converting a {beta}-glycosidase into a {beta}-transglycosidase by directed evolution. J. Biol. Chem. 280: 37088–37097 [DOI] [PubMed] [Google Scholar]
- 29. Tran V, Hoffmann L, Rabiller C, Tellier C, Dion M. 2010. Rational design of a GH1 beta-glycosidase to prevent self-condensation during the transglycosylation reaction. Protein Eng. Des. Sel. 23: 43–49 [DOI] [PubMed] [Google Scholar]
- 30. Verdoucq L, Czjzek M, Moriniere J, Bevan DR, Esen A. 2003. Mutational and structural analysis of aglycone specificity in maize and sorghum beta-glucosidases. J. Biol. Chem. 278: 25055–25062 [DOI] [PubMed] [Google Scholar]
- 31. Berrin JG, Czjzek M, Kroon PA, McLauchlan WR, Puigserver A, Williamson G, Juge N. 2003. Substrate (aglycone) specificity of human cytosolic beta-glucosidase. Biochem. J. 373: 41–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zouhar J, Vevodova J, Marek J, Damborsky J, Su XD, Brzobohaty B. 2001. Insights into the functional architecture of the catalytic center of a maize beta-glucosidase Zm-p60.1. Plant Physiol. 127: 973–985 [PMC free article] [PubMed] [Google Scholar]
- 33. Chuenchor W, Pengthaisong S, Robinson RC, Yuvaniyama J, Oonanant W, Bevan DR, Esen A, Chen CJ, Opassiri R, Svasti J, Cairns JR. 2008. Structural insights into rice BGlu1 beta-glucosidase oligosaccharide hydrolysis and transglycosylation. J. Mol. Biol. 377: 1200–1215 [DOI] [PubMed] [Google Scholar]
- 34. Guasch A, Vallmitjana M, Pérez R, Querol E, Pérez-Pons JA, Coll M. 1999. Cloning, overexpression, crystallization and preliminary X-ray analysis of a family 1 β-glucosidase from Streptomyces. Acta Crystallogr. D Biol. Crystallogr. 55: 679–682 [DOI] [PubMed] [Google Scholar]
- 35. Jeng WY, Wang NC, Lin MH, Lin CT, Liaw YC, Chang WJ, Liu CI, Liang PH, Wang AH. 2011. Structural and functional analysis of three beta-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J. Struct. Biol. 173: 46–56 [DOI] [PubMed] [Google Scholar]
- 36. Sanz-Aparicio J, Hermoso JA, Martinez-Ripoll M, Lequerica JL, Polaina J. 1998. Crystal structure of beta-glucosidase A from Bacillus polymyxa: insights into the catalytic activity in family 1 glycosyl hydrolases. J. Mol. Biol. 275: 491–502 [DOI] [PubMed] [Google Scholar]