

Abstract
Functional prediction of carbohydrate-active enzymes is difficult due to low sequence identity. However, similar enzymes often share a few short motifs, e.g., around the active site, even when the overall sequences are very different. To exploit this notion for functional prediction of carbohydrate-active enzymes, we developed a simple algorithm, peptide pattern recognition (PPR), that can divide proteins into groups of sequences that share a set of short conserved sequences. When this method was used on 118 glycoside hydrolase 5 proteins with 9% average pairwise identity and representing four characterized enzymatic functions, 97% of the proteins were sorted into groups correlating with their enzymatic activity. Furthermore, we analyzed 8,138 glycoside hydrolase 13 proteins including 204 experimentally characterized enzymes with 28 different functions. There was a 91% correlation between group and enzyme activity. These results indicate that the function of carbohydrate-active enzymes can be predicted with high precision by finding short, conserved motifs in their sequences. The glycoside hydrolase 61 family is important for fungal biomass conversion, but only a few proteins of this family have been functionally characterized. Interestingly, PPR divided 743 glycoside hydrolase 61 proteins into 16 subfamilies useful for targeted investigation of the function of these proteins and pinpointed three conserved motifs with putative importance for enzyme activity. Furthermore, the conserved sequences were useful for cloning of new, subfamily-specific glycoside hydrolase 61 proteins from 14 fungi. In conclusion, identification of conserved sequence motifs is a new approach to sequence analysis that can predict carbohydrate-active enzyme functions with high precision.
INTRODUCTION
Transforming gene sequence data into valid information, based on which new biological understanding can be built, is becoming increasingly important, as the amounts of gene sequences available are increasing dramatically, and it has been revealed and documented that even very distantly related gene sequences may code for proteins with similar function. It is most essential for such progress to improve the ability to predict function from sequence. This is most often attempted by sequence alignment, based on finding regions with similar sequences in two or more biological polymers. Typically, protein alignment is used for identification of conserved regions that can have functional importance to find proteins with similar characteristics. This approach is very successful in comparing closely related sequences. However, the outcome of alignment of very distantly related sequences can be notoriously difficult to interpret and may be unreliable. Advanced methods for sequence alignment misalign 11 to 19% of the sequences analyzed (1). This problem is even more pronounced when many divergent sequences are compared (2).
The Carbohydrate-Active Enzyme database (CAZy) (http://www.cazy.org/) is a great resource for understanding glycoside hydrolase (GH) evolution and biology (3). As pointed out by the team at CAZy, it is not straightforward to predict the enzymatic activity of glycoside hydrolases based on their sequence (4). Therefore, CAZy divides the glycoside hydrolases into 131 protein families, GH1 to GH131, based on sequence and structural information (5–7). Due to convergent evolution of glycoside hydrolase function (8), most of the GH families comprise enzymes with different functions, and up to 28 different enzyme activities have been described for proteins belonging to a single GH family. It is therefore not possible to predict the activity of a glycoside hydrolase simply by assigning it to a GH family.
Likewise, prediction of function is complicated by the fact that proteins with the same enzymatic function belong to different GH families. Many of the glycoside hydrolase families include enzymes that are important for lignocellulose turnover in nature and are potentially interesting enzymes for industrial conversion of biomass. One example is the endo-1,4-β-d-glucanase (EC 3.2.1.4), which can be found in 17 different GH families.
An obvious approach to functional prediction of glycoside hydrolases is to analyze a single GH family by alignment and elucidation of phylogenetic relationships. However, the low sequence identity between proteins even in a single GH family makes alignment difficult to perform and unreliable. Furthermore, enzymes with the same function often develop from different ancestors by convergent evolution, obscuring the correlation between phylogeny and enzyme activity (8). In a recent report, Aspeborg and coworkers analyzed the proteins in GH5 family and delineated 51 functionally relevant subfamilies (9). This work is a tremendous effort and implies the use of several algorithms for alignment, phylogenetic tree building, and considerable manual curation by experts in the fields of sequence analysis and glycoside hydrolase biology.
The notion that similar enzymes often share a few short motifs, e.g., around the active site, even when the overall sequences are very different (10), suggests an alternative approach to functional prediction. The hypothesis for the present work was that the function of glycoside hydrolases could be predicted by recognizing short, conserved sequence motifs in functionally characterized glycoside hydrolases and that the presence of such motifs could be used to predict the function of other glycoside hydrolases.
Although the relevant conserved sequence motifs are important, they will normally occur only once within each protein, e.g., a conserved motif forming an active site. Therefore, we developed a method designated peptide pattern recognition (PPR), which consists of two steps: (i) finding a limited number of n-mer short sequences that are highly conserved in a collection of glycoside hydrolases and (ii) selecting glycoside hydrolases that contain more than a threshold number of the n-mer short sequences. This approach implies that any input glycoside hydrolase that is unrelated to the other input glycoside hydrolases will be discarded. The level of relatedness of the sequences in a group depends on the length of the n-mer short sequences and the number of n-mers used to define the group. In the present report, settings that included sequences with as low as 20% identity in the same group resulted in a functionally meaningful subdivision of the glycoside hydrolases. The sequences that are not included in the group can be used to perform another round of analysis to find a new group, and so on. The output of PPR is a group of related glycoside hydrolases selected from the input and a list of the n-mer short sequences that are most conserved in this group.
This method can be used to predict the enzyme activity of glycoside hydrolases if the identified short, conserved sequence motifs are functionally important: uncharacterized glycoside hydrolases can be expected to have the same activity as functionally described glycoside hydrolases that share the same short, conserved sequence motifs.
To test the hypothesis, we applied PPR to glycoside hydrolase families 5 and 13. The complete sequences of the proteins were analyzed without removing signal peptides, carbohydrate-binding modules, or other sequences not directly related to catalytic activity. Despite this lack of curation, the PPR analysis provided subfamilies that could predict the function of the GH5 proteins with 97% accuracy and that of the GH13 proteins with 82% accuracy. This enables a shortcut for targeted discovery, where a specific function (e.g., endo-1,4-β-d-glucanase [EC 3.2.1.4]) is aimed for. Furthermore, the analysis of 8,138 GH13 proteins holding 28 described functions demonstrated the ability of the presently described method to handle large data sets.
Despite their importance for natural and industrial degradation of lignocellulosic material, only a few GH61 proteins have been characterized enzymatically (11–16). A search for short, conserved sequence motifs in 743 GH61 proteins divided the proteins into 16 subfamilies. Assuming that these subfamilies are functionally relevant, as was found for the subfamilies generated for the GH5 and GH13 proteins, this subdivision provides a guide for characterization of the enzyme activity of the GH61 family toward lignocellulose as well as other substrates. Furthermore, the lists of conserved peptides pinpoint putative functionally important amino acids in the GH61 proteins and were used to identify new GH61 proteins from 14 different fungi.
Identification of short, conserved sequence motifs is a new method implemented as PPR for analysis of glycoside hydrolases that can predict function from sequence with a high level of accuracy and delineate function-related subfamilies. This method moves the prediction of function from sequence one step closer by finding the functionally and structurally most essential peptides of a protein.
MATERIALS AND METHODS
PPR algorithm and implementation.
A list of protein sequences was used as the input for the algorithm, with each protein on the list used as a seed protein (see Fig. S1 in the supplemental material for the flow diagram):
Make all the n-mer peptides that occur in the sequence of the seed protein.
Select all proteins that contain more than a cutoff value of the peptides.
Make all the n-mer peptides that occur in these proteins, and select the N most frequently occurring peptides. N is a predefined number of n-mer peptides. The selected peptides should occur in at least two of the proteins (score, ≥2). Peptide cutoff is a specific frequency (e.g., if more than 20 out of 100 proteins contain a peptide, the frequency is 0.2). Each peptide should have a frequency higher than the peptide cutoff to be included on the list. A peptide cutoff of >0.2 was used in all studies unless otherwise indicated.
Go back to step 2 until no new peptides are made in the following round.
All peptides that occurred in only one protein were excluded from the analysis to reduce the number of calculations.
When groups had been made from each of the seed proteins, the group including the highest number of proteins was selected.
The output was a peptide pattern defined as a list of the N most frequently occurring n-mer peptides in the largest group of proteins and a group of proteins defined as all the proteins that include more than the cutoff value of the n-mer peptides.
The score of a protein was defined as the number of peptides (from the list of n-mers) included in the protein sequence.
The frequency of a peptide was defined as the number of proteins that contain a peptide divided by the total number of proteins in the group.
The algorithm was executed more than once on the same input list of proteins by removing the largest group of proteins from the input after each run. In this way, several peptide patterns and protein groups were extracted from the input sequences.
The possibility of a specific number of peptides occurring at random and the possibility of a protein containing a high number of the peptides can be calculated as follows, where comb(a, b) means a!/[b! × (a − b)!]: there are 206 = 6.4 × 107 different hexamers. Choosing 100 of these to create a list of hexapeptides can be done in comb(6.4 × 107, 100) = 4.4 × 10622 different ways. A 305-amino-acid-long protein consists of up to 300 different hexamers. This gives comb(6.4 × 107, 300) = 2.3 × 101,727 different combinations of 300 hexamers. The probability that a 305-amino-acid-long protein contains at least 10 of 100 hexamers by chance is comb(100, 10) × comb(206, 300 − 10)/comb(206, 300) = 7.6 × 10−41. The parameters number of peptides, peptide length, and number of peptides that a protein should contain to be part of a group (cutoff) can be adjusted to increase or decrease this probability. Increasing N will increase the probability, whereas increasing peptide length or cutoff will decrease the probability. Higher probability will lead to larger groups including more proteins, and lower probability will have the opposite effect.
Short, conserved sequence motifs and assignment of subfamilies for GH5 proteins.
Characterized eukaryotic GH5 proteins from CAZy (http://www.cazy.org/) (3) were used as the input for PPR with different parameters. GenBank and Joint Genome Institute accession numbers for the proteins can be found in the supplemental material.
For each round of PPR, the largest group of proteins was removed as a subfamily, and the analysis was repeated with the rest of the proteins.
Each protein subfamily was assigned a function corresponding to the function of the most abundant enzyme type in the subfamily.
Next, all 118 GH5 proteins were given a score for each subfamily-specific peptide lists by
Finding all the peptides from the list that were present in the sequence of the protein.
Summing the frequency of these peptides. This gave the subfamily-specific frequency score.
The proteins were assigned to the subfamily with the highest subfamily-specific frequency score.
Finally, the function of the proteins, as reported in CAZy, was compared to the function assigned to the subfamily.
Generation of function-specific peptide lists from GH5 proteins.
The eukaryotic GH5 proteins were divided into four lists of proteins with the same function: endo-1,4-β-d-glucanase (EC 3.2.1.4), glucan 1,3-β-glucosidase (EC 3.2.1.58), glucan endo-1,6-β-glucosidase (EC 3.2.1.75), and mannan endo-β-1,4-mannosidase (EC 3.2.1.78). Half of the protein sequences chosen at random from each list were used for step 3 of the PPR algorithm: make all the n-mer peptides that occur in these proteins, and find the N most frequently occurring peptides, where N is a predefined number of n-mer peptides. In this way, peptide patterns were created for the four enzyme classes. No peptide cutoff was used for this analysis.
The other half of the GH5 proteins were assigned a function by calculating the frequency score for each function-specific peptide list and assigning the protein to the function with the highest function-specific frequency score.
Short, conserved sequence motifs and assignment of subfamilies for GH13 and GH61.
The implementation of PPR where each protein was used to generate a group of proteins in each repetition of the algorithm is computationally intensive. To reduce the number of computations and reach similar results, we developed the following procedure: for each protein used as the seed protein,
Make all the n-mer peptides that occur in the sequence of the seed protein.
Count the number of proteins that contain more than a cutoff value of the peptides.
Assign this number to the seed protein.
Rank all the seed proteins according to this number, with the seed protein with the highest number first.
Use the 100 highest-ranked proteins on this list as seed proteins for PPR analysis.
Select the largest group of proteins as a subfamily, and remove the proteins from the list of proteins and from the list of seed proteins.
Repeat the PPR analysis.
This procedure significantly reduced the number of calculations when many proteins were used as the input, as only 100 seed proteins were used in each round of PPR instead of all the proteins and were used for PPR analysis of 8,138 GH13 proteins downloaded from CAZy and for 743 GH61 proteins (accession numbers in the supplemental material) that were downloaded from CAZy or found by CDD-search (10) in the protein database at the NCBI (http://www.ncbi.nlm.nih.gov/protein/).
This version of PPR is available for download [http://vbn.aau.dk/en/publications/peptide-pattern-recognition(1400c5df-fa69-4701-8d67-ec5c38cc963b).html].
Distribution of hexapeptides in GH61 proteins.
The position of a conserved hexapeptide was defined as the median of the position in all the proteins that contained the hexapeptide sequence.
Design of primers.
Conserved hexapeptides were reverse translated according to the genetic code. Positions containing any nucleotide (A, C, G, or T) were replaced with inosine (see Table S1 in the supplemental material). Degenerate nucleotides at the 3′ end of the primers were removed from the sequence of the primers. The degeneracy of the primer that results from reverse translation of each hexapeptide was calculated based on the genetic code and replacing positions containing any nucleotide (A, C, G, or T) with inosine (see Table S1 in the supplemental material). In addition, the relative position of the hexapeptides in the proteins was estimated as the median of the distance of the peptide to the N terminus of each protein in the group that contained the peptide.
Sequences for primers were selected based on three criteria: (i) they should have high frequency in the group of GH61 proteins; (ii) they should give an amplicon of at least 40 bp, excluding primer sequences, in order to be able to obtain sufficient sequence information to identify the PCR product; and (iii) the primers should have the smallest possible redundancy, and redundant bases at the 3′ end were not allowed.
A tail of six bases (CTGGAC) was added to the 5′ end of all primer sequences, as this improves the performance of short primers (17–19).
Reverse primers were designed to be complementary to the DNA sequence encoding the hexapeptide and according to the same rules.
The primers were synthesized and purified by high-performance liquid chromatography (HPLC) by Sigma-Aldrich (United Kingdom).
Fungi.
Fungi (Table 1) were purchased from the Centraalbureau voor Schimmelcultures, The Netherlands, and grown on 6% wheat bran (Finax, Denmark)–1.5% agar (Sigma-Aldrich, United Kingdom) plates at the recommended temperature.
Table 1.
List of fungi used for PCR
| Order | Fungus | CBS no.a |
|---|---|---|
| Sordariales | Chaetomium senegalense | 728.84 |
| Sordariales | Chaetomium thermophilum | 180.67 |
| Sordariales | Corynascus thermophilus | 406.69 |
| Sordariales | Melanocarpus albomyces | 638.94 |
| Sordariales | Remersonia thermophila | 540.69 |
| Sordariales | Scytalidium indonesiacum | 259.81 |
| Sordariales | Scytalidium thermophilum | 620.91 |
| Onygenales | Malbranchea cinnamomea | 115.68 |
| Eurotiales | Talaromyces byssochlamydoides | 151.75 |
| Eurotiales | Talaromyces emersonii | 393.64 |
| Eurotiales | Talaromyces leycettanus | 398.68 |
| Eurotiales | Talaromyces thermophilus | 236.58 |
| Eurotiales | Thermoascus aurantiacus | 891.70 |
| Eurotiales | Thermomyces lanuginosus | 632.91 |
Strain registration number at the Centraalbureau voor Schimmelcultures.
DNA purification.
Fungal mycelium was scraped off the top of a wheat bran agar plate, frozen in liquid nitrogen, and ground with a mortar and pestle. DNA was extracted from the homogenized mycelium with the Fungal DNA minikit (Omega Bio-Tek, USA) according to the manufacturer's instructions.
PCR.
A mix of 100 ng total fungal DNA in 1× Run PCR buffer; 2 mM each dATP, dCTP, dGTP, and dTTP; 400 nM forward primer; 400 nM reverse primer; and 1 U Run DNA polymerase (A&A Biotechnology, Poland) in a total volume of 20 μl was used for PCR on a MyCycler instrument (Bio-Rad, USA) with the following thermal profile: an initial denaturation step at 95°C for 5 min; 40 cycles of 95°C for 20 s, 54°C for 30 s, and 72°C for 60 s; and a final extension step at 72°C for 5 min.
PCR products were analyzed by agarose gel electrophoresis, and selected DNAs were cut out and purified with the Qiaquick kit (Qiagen, Germany).
One microliter of the purified PCR product was reamplified in a 50-μl reaction mixture under the same conditions as the original PCR except that only 15 to 20 cycles of PCR were performed.
Sequencing and analysis.
PCR products were cycle sequenced by Eurofins-MWG (Germany) or StarSEQ (Germany) with one of the degenerate primers used for PCR.
The resulting sequences were translated into amino acid sequences and used for a BLAST search (20) against the nonredundant protein sequence database at the NCBI and inspected for conserved domains (10) in the CDD database at the NCBI to identify sequences encoding GH61-like proteins.
A sequence alignment was made with ClustalW (21) and adjusted manually. Phylogenetic trees were made with MUSCLE, PhyML, and TreeDyn at Phylogeny.fr (22).
Statistical analysis.
P values were calculated by one of three different methods, as indicated. Combinatorics means that the P value was calculated as the number of positive outcomes divided by the total number of outcomes. Simulation means that the P value was estimated by running a computer simulation of the distribution 108 or 109 times. The P values estimated by simulations are given with 99% confidence. χ2 test means that the observed result was compared to the expected distribution, and the P value was calculated by Pearson's χ2 test.
Nucleotide sequence accession numbers.
The DNA sequences of the products of PCR amplification of GH61 from 14 fungi were deposited in the GenBank/EMBL/DDBJ database under accession numbers HF565034 to HF565047. The DNA and translated amino acid sequences can be found in the supplemental data.
RESULTS
Rationale and theory of PPR.
The members of a group of proteins with similar structure and function are characterized by having a number of identical or closely related short sequence motifs (10). These sequence motifs may be conserved for a number of reasons, for example, if they are located in the active site of an enzyme, in structurally important regions, or in regions involved in binding to other proteins. Pinpointing the conserved peptides will yield a new conceptual understanding of biological functions.
PPR was designed to find such conserved, short sequences by counting the number of proteins that contain each peptide of length n. The output of PPR consists of a list of peptides and a group of proteins that are mutually dependent: a list of peptides consists of the peptides most frequently occurring in the group of proteins, and a group of proteins is defined as the proteins containing a high number of these peptides. Thus, to identify protein groups and peptides, PPR is executed as a cyclical algorithm until no new proteins and peptides are found.
Division of eukaryotic GH5 proteins into function-related subfamilies by recognition of short, conserved sequence motifs.
The amino acid sequences of 118 functionally characterized eukaryotic GH5 proteins were downloaded from CAZy (see accession numbers in the supplemental material). This collection included proteins with four enzymatic activities and with very divergent sequences. The average pairwise identity was only 9%, and only 23% of the pairwise sequence comparisons produced any significant alignment. Phylogenetic tree analysis separated the GH5 proteins into five phylogenetic clusters: two clusters of EC 3.2.1.4 enzymes and one cluster for each of the other three enzyme classes (see Fig. S2 in the supplemental material). However, 13 of the 118 proteins (11%) were placed outside the clusters.
To investigate if short, conserved sequence motifs could be found in this sequence collection and if the motifs could be used for separation of the proteins into different enzyme classes, the proteins were analyzed by PPR, the tool for finding short, conserved sequence motifs. Three critical parameters for the analysis were (i) the length of the sequence motifs, (ii) the number of motifs used to define a group, and (iii) the number of motifs that should be found in a protein in order to include the protein in the group (cutoff value).
To establish a set of useful conditions, we performed PPR analysis of the GH5 proteins with different values for these three parameters. Each analysis resulted in a number of protein subfamilies that were assigned a function corresponding to the function of the most abundant enzyme type in the subfamily. All 118 GH5 proteins were scored against the subfamily-specific peptide lists and placed into the subfamily with the highest score. The experimentally reported function of the proteins was compared to the function predicted by PPR. The parameters tested were a peptide length of 3 to 10 amino acids, peptide lists with 30 to 200 conserved peptides, and a cutoff from 5 to 40 peptides. The cutoff and the number of conserved peptides are important for group size, as a higher cutoff for the same peptide list leads to the inclusion of fewer proteins, whereas a longer peptide list for the same cutoff includes more proteins. It is convenient to describe this relationship as stringency and define stringency as the cutoff divided by the number of conserved peptides per group.
The number of GH5 proteins that were assigned to subfamilies with the same function depended on the stringency of the PPR analysis with a broad maximum: stringencies of between 0.10 and 0.33 correctly classified 78 to 86% of the GH5 proteins when averaging the results for peptide lengths from 3 to 10 amino acids (Fig. 1). However, the best correlation between classification and enzymatic activity was obtained with peptides with lengths of 4 to 6 amino acids. Hexamer peptides at a stringency of 0.14 (cutoff = 10; number of peptides = 70) gave the highest level of correct classification of all the conditions tested. These parameters generated nine subfamilies containing 97 of the GH5 proteins (Fig. 2). Both endo-1,4-β-d-glucanase (EC 3.2.1.4) and mannan endo-β-1,4-mannosidase (EC 3.2.1.78) were divided into several subfamilies, whereas glucan 1,3-β-glucosidase (EC 3.2.1.58) and glucan endo-1,6-β-glucosidase (EC 3.2.1.75) were placed into a single subfamily for each enzyme type. Except for one glucan endo-1,6-β-glucosidase from Schizosaccharomyces pombe, the subfamilies correlated with the function of the proteins (P < 2 × 10−8, determined by simulation). Cross comparison of the hexapeptide lists for the subfamilies showed that only 4 of the 477 conserved peptides were shared between two subfamilies (Fig. 2).
Fig 1.

Correlation of PPR stringency and peptide length to correct prediction of the function of GH5 proteins. The correct prediction of the function of 118 GH5 proteins as a function of the stringency (cutoff/number of peptides) was calculated as an average of the prediction rates obtained by performing PPR analysis with peptide lengths of 3, 4, 5, 6, 8, and 10 amino acids. Likewise, correct prediction rates for all stringencies were calculated for each peptide length, as indicated.
Fig 2.
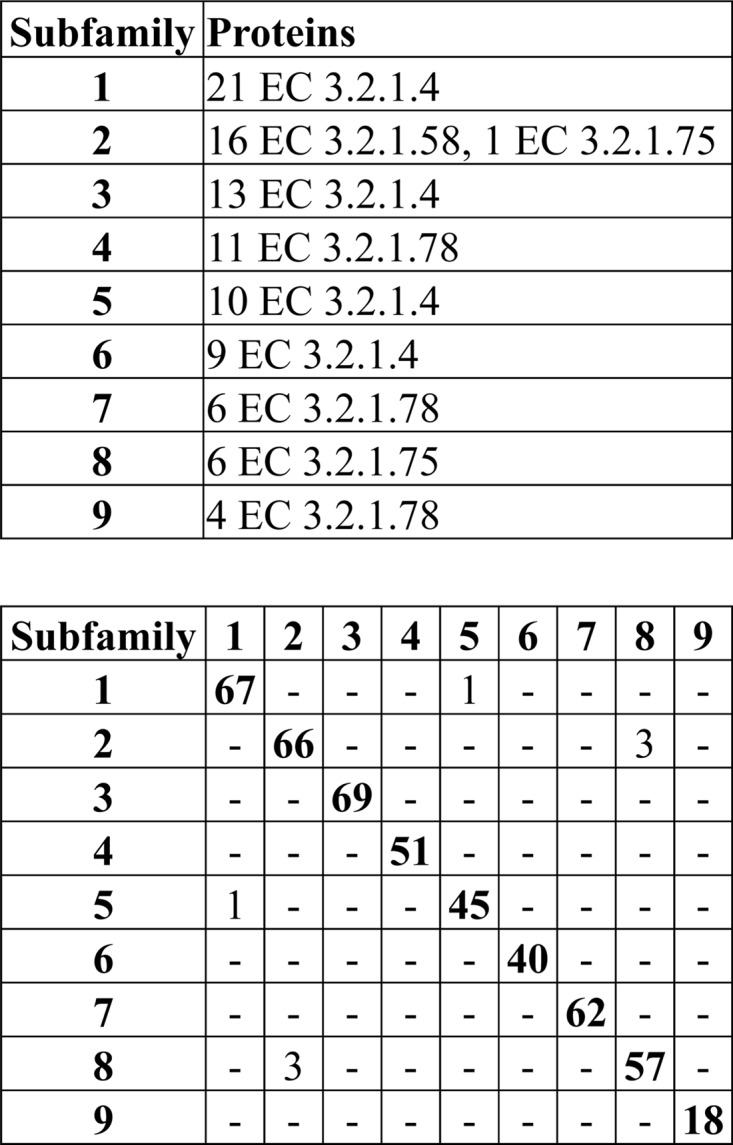
Analysis of the proteins by EC number and cross comparison of the peptides in each GH5 subfamily.
Scoring of the 118 GH5 proteins with the subfamily-specific peptide lists assigned the correct function to 115 of the proteins (Table 2), corresponding to successful prediction of the function of 97% of the proteins (P = 9 × 10−48 by χ2 test). This is comparable to the precision of functional separation of the GH5 enzymes by phylogenetic analysis.
Table 2.
Prediction of the function of the 118 GH5 proteins by the peptides lists for each protein subfamily
| Classification | No. of predictions |
P valuea | |
|---|---|---|---|
| Correct | Wrong | ||
| EC 3.2.1.4 | 59 | 1 | 5 × 10−20 |
| EC 3.2.1.58 | 18 | 1 | 1 × 10−17 |
| EC 3.2.1.75 | 7 | 0 | 2 × 10−7 |
| EC 3.2.1.78 | 31 | 1 | 2 × 10−14 |
| Total | 115 | 3 | 2 × 10−56 |
Calculated by combinatorics.
One of the proteins classified into subfamily 1 (P = 6 × 10−263 by combinatorics) was the GH5 endo-1,4-β-d-glucanase (EC 3.2.1.4) from Thermoascus aurantiacus (GenBank accession number AAL1642.1). Eleven functionally important amino acid residues can be inferred from the crystal structure of this enzyme (23, 24). Except for the disulfide bridge, all of these functionally important amino acids, including the two catalytic glutamates, were found in the 70 most frequent hexapeptides for subfamily 1 (P = 4 × 10−8 by combinatorics), indicating that the short, conserved motifs identified for the subfamily are related to endo-1,4-β-d-glucanase function, as predicted (Table 3).
Table 3.
Conserved amino acids in the structure of Thermoascus aurantiacus (Eurotiales) GH5 endo-1,4-β-d-glucanase with indication of the number of conserved peptides from subfamily 1 that contain these residues
| Residue | No. of peptides | Highest frequency | Sequencea |
|---|---|---|---|
| G44 | 1 | 0.38 | GMNIFR |
| E133 | 5 | 0.71 | FDTNNE |
| W170 | 6 | 0.52 | WTGAWT |
| W174 | 4 | 0.67 | TGAWTW |
| Y200 | 6 | 0.95 | MHQYLD |
| E240 | 3 | 0.57 | GEFAGG |
| W273 | 2 | 0.76 | WAAGPW |
| W278 | 5 | 0.76 | WAAGPW |
| W279 | 4 | 0.76 | AAGPWW |
The conserved amino acid residue is underlined.
Function-specific conserved sequence motifs in GH5 proteins with the same activity.
Another way to find short, conserved sequences is to take proteins with a known function and make the best possible peptide pattern for these proteins by finding the N most frequently occurring n-mers in the protein sequences. The list of n-mers will be useful for characterization of the proteins with known function and for finding other proteins with the same function whenever the conserved sequences are related to protein function.
To test this hypothesis, we randomly chose half of the 118 GH5 proteins for each of the four enzyme classes. Next, we made list of conserved n-mers with different lengths and with different numbers of conserved n-mers on the list. The parameters were n-mer length from 2 to 10 amino acids and number of n-mers from the 10 to the 3,200 most conserved n-mers. Finally, the four generated peptide lists were used to predict the function of the other half of the GH5 proteins. Each analysis was repeated 10 times.
The best result was prediction of the function of 98% of the proteins with 92% accuracy, giving a correct prediction rate (predicted proteins × correct predictions) of 90% (see Fig. S3 in the supplemental material). Interestingly, the highest prediction rates were achieved with a large number (400 to 3,200) of 3- to 5-mer peptides. When the analysis was performed with all possible peptides of a given length, it was possible to achieve a correct prediction rate of 92% with 4-mer peptides (Table 4). However, even a limited number of conserved sequences correctly predicted the function of a large number of the GH5 proteins; e.g., lists of 20 conserved 4-mers predicted the function of 69% of the proteins with 96% accuracy (see Fig. S3 in the supplemental material), indicating that the short, conserved sequence motifs found in the GH5 proteins with identical enzymatic activity are indeed functionally relevant.
Table 4.
Prediction of the function of GH5 proteins by function-specific peptide lists
| Peptide length (amino acids) | No. of peptides | % correct predictionsa | % proteins predictedb | % proteins correctly predictedb | P valuec |
|---|---|---|---|---|---|
| 2 | 400 | 39 | 98 | 38 | 10−35 |
| 3 | 8 × 103 | 93 | 98 | 91 | 10−57 |
| 4 | 1.6 × 105 | 94 | 98 | 92 | 7 × 10−60 |
| 5 | 3.2 × 106 | 96 | 93 | 89 | 8 × 10−59 |
| 6 | 6.4 × 107 | 99 | 89 | 88 | 6 × 10−62 |
| 8 | 2.6 × 1010 | 98 | 81 | 79 | 2 × 10−53 |
| 10 | 5.1 × 1011 | 100 | 75 | 75 | 3 × 10−54 |
Percentage of all predictions.
Percentage of all proteins.
P values for correctly predicted proteins were calculated by combinatorics.
Comparison of PPR with benchmark method for analysis of the GH13 family.
The GH13 family, which is the GH family with the largest number of described enzymatic functions, has been classified into 35 subfamilies (25) that can be found in CAZy. The generation of the subfamilies implied the sequential use of several algorithms for sequence alignment, clustering, and removal of sequences with insufficient similarity. The analysis included 1,691 GH13 sequences, was very time-consuming, and would have been difficult to perform on the 2,456 GH13 sequences in CAZy at the time of publication (25). In comparison to this highly accurate but time-consuming analysis, automated PPR analysis based on short, conserved motifs of 8,138 GH13 proteins yielded 50 subfamilies. The PPR analysis of 8,138 GH13 proteins took 7 h with a script written in Ruby, which is a relatively slow programming language, and was executed on a powerful desktop computer (IntelR Core i7-2600 CPU at 3.40 GHz, with 16 GB RAM). It took less than 25 min to perform a PPR analysis of 1,691 sequences chosen at random from the 8,138 GH13 proteins. Cross comparison of 5,442 proteins that were assigned to both a CAZy subfamily (25) (http://www.cazy.org/) and a PPR subfamily showed a high correlation between CAZy and PPR subfamilies (R2 = 0.871; P < 2 × 10−7, determined by simulation). On average, 90% of the proteins in each CAZy subfamily were assigned to one PPR subfamily (Fig. 3).
Fig 3.

Correlation between PPR subfamilies and CAZy subfamilies of 5,457 GH13 proteins. Shown is a cross comparison of the proteins in the PPR subfamilies with the same proteins in CAZy subfamilies. The PPR subfamilies were arranged to give the highest number of shared proteins between subfamilies along the diagonal of the diagram.
Several GH13 proteins contain multiple domains and more than one catalytic domain that can confuse analysis. The CAZy subfamilies of GH13 accounted for this protein structure, whereas the classification based on short, conserved motifs did not. Nevertheless, short, conserved motifs found with PPR classified 442 of 540 experimentally characterized GH13 proteins (82%) into subfamilies that were assigned the same function as the protein (P < 2 × 10−7, determined by simulation) (see Table S2 in the supplemental material).
PPR peptide lists for pinpointing interesting amino acids in the GH61 protein family.
The GH61 proteins are important for fungal biomass degradation. However, the highly heterogeneous sequences of the GH61 proteins make them difficult to compare and analyze in a comprehensive way. A CDD-search of the protein databases available at the NCBI combined with all the GH61 proteins in CAZy identified 763 proteins with a GH61 domain. We used PPR to find short, conserved sequence motifs in these GH61 proteins.
PPR made 16 subfamilies containing 493 of the 763 GH61 proteins (see Table S3 in the supplemental material). After 16 rounds, the subfamilies became too small (fewer than 10 proteins) to define any common peptide pattern for the remaining proteins. Most of the conserved hexapeptides for the 16 subfamilies were specific for the subfamily, but almost all the conserved peptides were found in the N-terminal half of the GH61 proteins (P = 5 × 10−190 by χ2 test) (Fig. 4), with two peaks at residues 100 to 120 (P = 8 × 10−42 by χ2 test) and 160 to 200 (P = 2 × 10−158 by χ2 test). The entire lists of conserved peptides can be found in Table S4 in the supplemental material.
Fig 4.

Cross comparison and distribution of the conserved hexapeptides in the GH61 sequences. The distribution of hexapeptides for each subfamily was calculated as the number of hexapeptides mapping to each 20-amino-acid interval, as described in Materials and Methods. The accumulated hexapeptide frequency (vertical axis) was calculated as the sum of the distribution of all the subfamilies in each 20-amino-acid interval. The horizontal axis designates the amino acid intervals.
Within subfamilies, there were large sequence differences between the proteins. In the largest group, the average sequence identity between pairs of proteins was 48% and varied from 27 to 99.6%. However, 10% of the pairwise sequence comparisons did not yield any significant alignment. Sequence differences between subfamilies were even larger, and the average sequence identity between the 16 proteins with the highest score in each subfamily was 29% and varied from 22 to 45%, with 10% of the comparisons not yielding any significant alignment.
An example of classification of a protein sequence is GH61E from the ascomycete Thielavia terrestris of the order Sordariales. This protein included 26 of the conserved peptides from subfamily 3 with an accumulated frequency of 9.12 (P = 5 × 10−126 by combinatorics). Only 0 to 4 hexapeptides from other subfamilies were found in the sequence of GH61E (see Table S5 in the supplemental material). The spatial position of the most conserved hexapeptides for GH61 subfamily 3 could be depicted on the tertiary structure of GH61E (26). Many of the residues are hidden within the structure, but three sequence stretches consisting of amino acids from the conserved hexapeptides were found on the surface of the crystal structure (Fig. 5). One is located on the metal ion-binding surface of GH61 and includes Gln-151 and Tyr-153, which are involved in binding to the divalent cation (Fig. 5). The other two sequences are located on other surfaces of the protein and are conserved within subfamilies but differ considerably between subfamilies (Fig. 5). To our knowledge, these conserved protein surfaces have not been described in connection with GH61 function.
Fig 5.

Mapping of conserved amino acid residues on the surface of GH61E (Protein Data Bank [PDB] accession number 3EII). Conserved amino acids in GH61 subfamily 3 mapping to the surface of the GH61E structure (12) are indicated in yellow. Shown is an alignment of GH61E and the conserved amino acid residues of the GH61 subfamilies in the regions depicted on the surface of the GH61E structure. Residues that are highly conserved between subfamilies are indicated in red. Numbering above the alignment indicates amino acid positions relative to the start residue in GH61E.
The few GH61 proteins that have been enzymatically characterized possess cellulose oxidase activity rather than glycoside hydrolase activity as such (14, 16, 26–28). These enzymes are further classified as type 1 or type 2 depending on whether the oxidation products are modified at the reducing or the nonreducing end (14). These proteins are classified into 6 of the 16 GH61 subfamilies (see Table S6 in the supplemental material). The two type 1 proteins (subfamilies 1 and 5) and the two type 2 proteins (subfamilies 2 and 4) are in different subfamilies, but this result is not statistically significant (P = 0.77 by combinatorics) for only four proteins distributed into subfamily 16.
To test whether the conserved sequences could be used to design efficient degenerate primers, we designed degenerate primers for subfamily 1 (see Table S1 in the supplemental material). As the two most conserved hexapeptides (occurring in 80 and 78% of the proteins) could be used for the design of reverse primers, we did not find it necessary to design a third reverse primer. Three forward primers were designed. One of the hexapeptides used for forward primer design (SHHGPV) contained one serine residue that is encoded by 6 different codons at the N terminus and does not contribute significantly to specificity. Therefore, this primer was made by reverse translation of the peptide HHGPV. In in silico PCR, the three forward and two reverse primers were able to amplify 85 of the 130 proteins classified as belonging to subfamily 1, 1 protein classified as belonging to subfamily 4, and none of the other 657 GH61 proteins. This suggested that the peptide patterns were suitable for designing subfamily-specific primers.
The primers were used in all six possible combinations for PCR of DNA from 14 fungi from three different orders of ascomycota; at least one of the primer sets gave an amplification product with the expected size, and for some fungi, all the primer sets gave a positive product (Fig. 6). For each fungus, the longest amplicon that had the expected size was sequenced and analyzed for open reading frames. All the amplicons yielded a sequence that encoded a novel, putative GH61 family protein. Although the sequences are only partial, it was possible to classify all except one as belonging to subfamily 1 (Fig. 7). The unassigned sequence (from Chaetomium senegalense) was the shortest of the sequences (37 amino acids) but was 78% identical to the new sequence from Remersonia thermophila, indicating that this GH61 protein from C. senegalense also belongs to subfamily 1 (Fig. 7).
Fig 6.

Amplification of a GH61 protein with subfamily 1-specific primers. PCR was performed with the 6 possible combinations of the 5 primers constructed for GH61 subfamily 1 on Chaetomium thermophilum DNA, and the product was analyzed on a 2% agarose gel. Numbers and bars to the right of the gel indicate the migration of the bands in the DNA size marker, and numbers above the lanes indicate the combination of primers.
Fig 7.
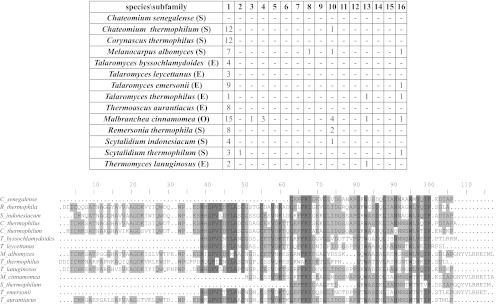
Characterization of the PCR products from 14 fungi. The number of conserved peptides from the GH61 subfamily was counted in each PCR product from the 14 fungi from the orders Sordariales (S), Helotiales (H), and Eurotiales (E). Furthermore, all the sequences were aligned. Sequences originating from the primers were discarded before analysis.
DISCUSSION
Sequence analysis by identification of short, conserved sequence motifs is efficient for predicting protein function, structure, and distant relationships. The simplicity allows for making subgroupings of GH families much faster than was previously possible, as shown for the GH13 family in the present study.
We investigated two different approaches to use short, conserved sequence motifs to recognize functionally identical proteins and compared them by predicting the function of GH5 proteins. The highest level of accuracy in prediction was obtained by using the subfamilies obtained by a PPR analysis to assign functions to the proteins. This finding is in agreement with the notion that it is difficult to find a common pattern of peptides for all GH5 proteins with the same function, and it therefore makes sense to divide each enzymatic function into subgroups. Nevertheless, the simple approach of making lists of all the peptides that were found in enzymes of a given type and using these lists to predict function could correctly predict the function of over 90% of the enzymes. Therefore, this method may be useful for some applications, although it does not provide subfamilies of proteins with related sequences.
Functional prediction of the eukaryotic GH5 proteins can also be inferred by building a phylogenetic tree with ClustalW. Therefore, identification of short, conserved peptides does not perform better than standard methods for elucidating the function of this small number of proteins. However, alignment-based analysis becomes more difficult when many highly divergent sequences are used as the input and requires many hours of manual curation by experts in the field to yield good results (9, 25, 29). Although the search for short, conserved sequences was performed by the computer algorithm PPR, our results show that the subfamilies generated by this approach correlated to a large extent with the subfamilies generated by alignment-based methods by the team at CAZy (25). Glycoside hydrolases can be classified with the PFAM system, but CAZy is generally used as a reference because it is considered to be the most precise classification system (3). Improvements of glycoside hydrolase classification have focused on exploiting PFAM for automatic annotation of proteins into CAZy families, thereby pinpointing the importance of CAZy as the state of the art (30, 31) and as the classification system that PPR should be compared to.
The advantage of using a one-step approach such as PPR compared to a multistep procedure that needs much hands-on time and the use of techniques to guide the result in the right direction is obvious. Moreover, PPR did not have any problems in handling the more than 8,000 GH13 proteins in the CAZy database today, where the limit for the alignment-based method was judged to be around 2,000 protein sequences (25). Due to its computational simplicity, PPR is well suited for analysis of large numbers of sequences. This is a major strength of this approach compared to sequence alignment and makes it especially suitable for handling the GH families that often comprise large numbers of divergent sequences.
The ability of PPR to accept unrelated protein sequences as an input contributes significantly to the ease of analysis. Unrelated proteins are separated into groups of similar proteins that can be described by short, conserved peptides. Not only different proteins but also different parts of the input will be separated, as demonstrated for the GH61 proteins, where only a few conserved peptides mapped to the variable C-terminal half (26, 32). In practical protein analyses, the exclusion of irrelevant proteins and sequence trimming often present a significant preanalytical effort and are subject to errors.
The GH61 family is a large gene family with low sequence similarity, but PPR divided the family into manageable subfamilies with conserved peptides useful for analysis of the subfamilies. Despite the interest in GH61 proteins, only a few have been characterized enzymatically. In analogy to the GH5 analysis, the 16 subfamilies of GH61 proteins probably reflect significantly fewer different substrate affinities and enzymatic activities. However, the division of GH61 proteins into subfamilies provides a platform for functional and structural analysis of this interesting family of copper monooxygenases.
Although sequence similarity is low, the homology between known GH61 structures and the structure of the bacterial homologue CBM33 is rather high (15, 26, 32, 33). In this context, it is interesting that the conserved peptides for the GH61 subfamilies included a number of conserved amino acids in the GH61 proteins on the copper-binding surface and on two other surface areas of the GH61 proteins. All 16 GH61 subfamilies had conserved amino acids within each subfamily in these regions, indicating that they may be important for function. Interestingly, some of the residues were not conserved between subfamilies and may indicate different functions or substrate preferences for some of the subfamilies. However, the functionally characterized GH61 proteins belong to only 6 of the 16 subfamilies, and much additional work is necessary to investigate the enzymatic activity of the GH61 proteins from all 16 subfamilies.
Interestingly, PPR classified GH5 and GH13 proteins into groups of proteins with the same enzymatic activity when using the same parameters as those used for constructing the GH61 subfamilies. For both the GH5 and the GH13 subfamilies, proteins with the same function were found in several subfamilies. This can be explained by the hypothesis that the same function can evolve in phylogenetically distant sequences (8). In analogy to the other two GH families, the 16 groups of GH61 proteins probably reflect significantly fewer different functions.
Expression of the 10 GH61 genes from the white-rot fungus Heterobasidion irregulare on lignocellulose showed that expression of some of these proteins is induced by growth on a lignocellulosic substrate compared to a substrate with malt extract (34). The most induced of the GH61 proteins (GH61H) belongs to subfamily 5, as does one of the characterized GH61 proteins with polysaccharide monooxygenase activity (14), indicating that subfamily 5 is induced during lignocellulose degradation. However, only the relative expression levels between growth on lignocellulose and growth on malt substrate have been reported for the 10 GH61 proteins from H. irregulare (34). This makes it difficult to draw any conclusions about the absolute level of expression of the genes and, thus, about the importance of the individual GH61 proteins for lignocellulose degradation. For example, a highly and constitutively expressed GH61 protein can contribute more to substrate degradation than a protein that is highly induced from a low level of basal expression. Nevertheless, the expression and upregulation of several GH61 proteins from H. irregulare on a lignocellulosic substrate point out that several GH61 subfamilies take part in lignocellulose degradation (34). The H. irregulare GH61 proteins can be classified as subfamilies 2, 4, 5, 13, and 15.
New GH61 genes, assigned to specific protein subfamilies, can be identified by performing an in silico search for genes encoding proteins that contain the conserved peptides from one or all of the GH61 subfamily-specific peptide lists, or the peptides can be used to design probes or primers for finding new GH61 proteins, as demonstrated in the present study. In addition to providing a means to find new GH61 proteins, it is also possible to use the peptide lists to focus exclusively on new GH61 proteins belonging to a specific subfamily. Such a search can be done on assembled genomes, transcriptomes, or unassembled sequencing data, e.g., from metatranscriptomic analyses.
Another way to use the list of conserved peptides is to pinpoint amino acids that are important for function. The nickel-binding surface of the GH61 proteins is important for substrate interactions, and differences in the amino acid residues mapping to this surface may reflect interactions with different substrates (11, 15, 16, 32, 35). The PPR analysis pinpointed a number of conserved amino acids in the GH61 proteins on the nickel-binding surface and on two other surface areas of the GH61 proteins. All 16 GH61 subfamilies had conserved amino acids in these regions, indicating that they may be important for function. However, some of the residues were not conserved between subfamilies and may indicate different functions or substrate preferences for some of the subfamilies.
Identification of short, conserved sequence motifs that occur once within each sequence (e.g., by PPR) is a new approach to sequence analysis. It should not be confounded with alignment-independent approaches based on word frequency methods, as used in text analyses (36). These methods look for short sequences (words) within a protein sequence and count the number of times each word is repeated in the sequence. The similarity between two or more sequences is calculated by comparing the frequency of each word within each sequence (37, 38).
Variations include dividing the sequences into subsequences with different chemical properties (for example, hydrophilic and hydrophobic) (39). These methods require less computation than alignment and can be used for comparison of distantly related sequences (36) but will often overlook short amino acid motifs conserved in proteins with the same function, because such motifs normally occur only once within each protein, e.g., a conserved motif forming an active site, and do not have a high weight in word frequency methods (10). In contrast, the present method and the PPR algorithm were developed exactly to find such motifs by giving a high level of importance to sequences found in many proteins but ignoring the number of times each motif occurs within a single sequence. This makes PPR fundamentally different from traditional word frequency methods.
Identification of short, conserved sequence motifs is not the only alignment-independent method for sequence analysis. Chaos game representation is a method that creates a picture for each biological sequence and compares the resulting pictures (40). However, this method can accommodate only four different words and is therefore suitable for nucleotide sequence comparison but difficult to adapt to protein sequences made up of 20 different words/amino acids (38, 41).
In conclusion, the present report presents a new, alignment-independent method for comparison and analysis of glycoside hydrolases. The potency of the method was demonstrated by the ability to predict glycoside hydrolase function, to identify functionally relevant subfamilies, and to pinpoint conserved sequences in large numbers of highly divergent proteins. Although the present study is focused on glycoside hydrolases, identification of short, conserved motifs may also be a useful approach for elucidation of structure-function relationships in other protein families with the same characteristics and may be applicable at the DNA and RNA levels.
Supplementary Material
ACKNOWLEDGMENTS
We thank Bernard Henrissat and Mette Lange for constructive and insightful suggestions and Helle Taiger Oskarsson for help with preparation of the manuscript.
This work was supported by Danish Strategic Research Council project 2101-08-0041 (BioRef) and by Novozymes A/S. Both authors are designated as inventors on a patent application on PPR filed by Aalborg University. Part of the authors' research is financed through a transfer of the commercial rights of the PPR patent application to Novozymes A/S.
Footnotes
Published ahead of print 22 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03803-12.
REFERENCES
- 1. Kim C, Lee B. 2007. Accuracy of structure-based sequence alignment of automatic methods. BMC Bioinformatics 8: 355 doi:10.1186/1471-2105-8-355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang W, Umbach DM, Li L. 2006. Accurate anchoring alignment of divergent sequences. Bioinformatics 22: 29–34 [DOI] [PubMed] [Google Scholar]
- 3. Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37: D233–D238 doi:10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henrissat B, Davies G. 1997. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7: 637–644 [DOI] [PubMed] [Google Scholar]
- 5. Henrissat B. 1991. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280(Part 2): 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henrissat B, Bairoch A. 1993. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 293(Part 3): 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henrissat B, Bairoch A. 1996. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 316(Part 2): 695–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davies GJ, Sinnott ML. 2008. Sorting the diverse: the sequence-based classifications of carbohydrate-active enzymes. Biochem. J. doi:10.1042/BJ20080382 [Google Scholar]
- 9. Aspeborg H, Coutinho PM, Wang Y, Brumer H, III, Henrissat B. 2012. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12: 186 doi:10.1186/1471-2148-12-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. 2011. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39: D225–D229 doi:10.1093/nar/gkq1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beeson WT, Phillips CM, Cate JH, Marletta MA. 2012. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134: 890–892 [DOI] [PubMed] [Google Scholar]
- 12. Forsberg Z, Vaaje-Kolstad G, Westereng B, Bunæs AC, Stenstrøm Y, MacKenzie A, Sørlie M, Horn SJ, Eijsink VGH. 2011. Cleavage of cellulose by a CBM33 protein. Protein Sci. 20: 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langston JA, Shaghasi T, Abbate E, Xu F, Vlasenko E, Sweeney MD. 2011. Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77: 7007–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Phillips CM, Beeson WT, Cate JH, Marletta MA. 2011. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6: 1399–1406 [DOI] [PubMed] [Google Scholar]
- 15. Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen J-CN, Johansen KS, Krogh KBRM, Jørgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter CP, Dupree P, Xu F, Davies GJ, Walton PH. 2011. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U. S. A. 108: 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Westereng B, Ishida T, Vaaje-Kolstad G, Wu M, Eijsink VGH, Igarashi K, Samejima M, Ståhlberg J, Horn SJ, Sandgren M. 2011. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6: e27807 doi:10.1371/journal.pone.0027807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andersen MD, Busk PK, Svendsen I, Møller BL. 2000. Cytochromes P-450 from cassava (Manihot esculenta Crantz) catalyzing the first steps in the biosynthesis of the cyanogenic glucosides linamarin and lotaustralin. Cloning, functional expression in Pichia pastoris, and substrate specificity of the isolated recombinant enzymes. J. Biol. Chem. 275: 1966–1975 [DOI] [PubMed] [Google Scholar]
- 18. Balcells I, Cirera S, Busk PK. 2011. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 11: 70 doi:10.1186/1472-6750-11-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. 2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33: e179 doi:10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- 22. Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J- F, Guindon S, Lefort V, Lescot M, Claverie J- M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36: W465–W469 doi:10.1093/nar/gkn180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lo Leggio L, Larsen S. 2002. The 1.62 A structure of Thermoascus aurantiacus endoglucanase: completing the structural picture of subfamilies in glycoside hydrolase family 5. FEBS Lett. 523: 103–108 [DOI] [PubMed] [Google Scholar]
- 24. Van Petegem F, Vandenberghe I, Bhat MK, Van Beeumen J. 2002. Atomic resolution structure of the major endoglucanase from Thermoascus aurantiacus. Biochem. Biophys. Res. Commun. 296: 161–166 [DOI] [PubMed] [Google Scholar]
- 25. Stam MR, Danchin EGJ, Rancurel C, Coutinho PM, Henrissat B. 2006. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of alpha-amylase-related proteins. Protein Eng. Des. Sel. 19: 555–562 [DOI] [PubMed] [Google Scholar]
- 26. Harris PV, Welner D, McFarland KC, Re E, Navarro Poulsen J-C, Brown K, Salbo R, Ding H, Vlasenko E, Merino S, Xu F, Cherry J, Larsen S, Lo Leggio L. 2010. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49: 3305–3316 [DOI] [PubMed] [Google Scholar]
- 27. Bey M, Zhou S, Poidevin L, Henrissat B, Coutinho PM, Berrin J-G, Sigoillot J-C. 2013. Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl. Environ. Microbiol. 79: 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R. 2012. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5: 79 doi: 10.1186/1754-6834-5-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lombard V, Bernard T, Rancurel C, Brumer H, Coutinho PM, Henrissat B. 2010. A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem. J. 432: 437–444 [DOI] [PubMed] [Google Scholar]
- 30. Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC. 2010. CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20: 1574–1584 [DOI] [PubMed] [Google Scholar]
- 31. Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. 2012. dbCAN: a Web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40: W445–W451 doi:10.1093/nar/gks479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karkehabadi S, Hansson H, Kim S, Piens K, Mitchinson C, Sandgren M. 2008. The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 A resolution. J. Mol. Biol. 383: 144–154 [DOI] [PubMed] [Google Scholar]
- 33. Vaaje-Kolstad G, Houston DR, Riemen AHK, Eijsink VGH, Van Aalten DMF. 2005. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 280: 11313–11319 [DOI] [PubMed] [Google Scholar]
- 34. Yakovlev I, Vaaje-Kolstad G, Hietala AM, Stefańczyk E, Solheim H, Fossdal CG. 2012. Substrate-specific transcription of the enigmatic GH61 family of the pathogenic white-rot fungus Heterobasidion irregulare during growth on lignocellulose. Appl. Microbiol. Biotechnol. 95: 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vaaje-Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sørlie M, Eijsink VGH. 2010. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330: 219–222 [DOI] [PubMed] [Google Scholar]
- 36. Vinga S, Almeida J. 2003. Alignment-free sequence comparison—a review. Bioinformatics 19: 513–523 [DOI] [PubMed] [Google Scholar]
- 37. Dai Q, Liu X, Yao Y, Zhao F. 2011. Numerical characteristics of word frequencies and their application to dissimilarity measure for sequence comparison. J. Theor. Biol. 276: 174–180 [DOI] [PubMed] [Google Scholar]
- 38. Davies MN, Secker A, Freitas AA, Timmis J, Clark E, Flower DR. 2008. Alignment-independent techniques for protein classification. Curr. Proteomics 5: 217–223 [Google Scholar]
- 39. Strope PK, Moriyama EN. 2007. Simple alignment-free methods for protein classification: a case study from G-protein-coupled receptors. Genomics 89: 602–612 [DOI] [PubMed] [Google Scholar]
- 40. Jeffrey HJ. 1990. Chaos game representation of gene structure. Nucleic Acids Res. 18: 2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deschavanne P, Tufféry P. 2008. Exploring an alignment free approach for protein classification and structural class prediction. Biochimie 90: 615–625 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.