

Abstract
Cellular processes are tightly controlled through well-coordinated signaling networks that respond to conflicting cues, such as reactive oxygen species (ROS), endoplasmic reticulum (ER) stress signals, and survival factors to ensure proper cell function. We report here a direct interaction between inhibitor of κB kinase (IKK) and apoptosis signal-regulating kinase 1 (ASK1), unveiling a critical node at the junction of survival, inflammation, and stress signaling networks. IKK can be activated by growth factor stimulation or tumor necrosis factor alpha engagement. IKK forms a complex with and phosphorylates ASK1 at a sensor site, Ser967, leading to the recruitment of 14-3-3, counteracts stress signal-triggered ASK1 activation, and suppresses ASK1-mediated functions. An inhibitory role of IKK in JNK signaling has been previously reported to depend on NF-κB-mediated gene expression. Our data suggest that IKK has a dual role: a transcription-dependent and a transcription-independent action in controlling the ASK1-JNK axis, coupling IKK to ROS and ER stress response. Direct phosphorylation of ASK1 by IKK also defines a novel IKK phosphorylation motif. Because of the intimate involvement of ASK1 in diverse diseases, the IKK/ASK1 interface offers a promising target for therapeutic development.
INTRODUCTION
Within the intracellular networks that control stress response, cell differentiation, and apoptosis, apoptosis signal regulating kinase 1 (ASK1) plays a pivotal role as a signaling hub (1). ASK1 senses, processes, and transmits various environmental cues to intracellular signaling machinery, impacting both physiological and pathophysiological processes. In response to stress signals, such as reactive oxygen species (ROS) or infectious agents, ASK1 initiates a mitogen-activated protein kinase (MAPK) signaling cascade that ultimately results in activation of MAPKs, jun N-terminal kinase (JNK) and p38, and their corresponding biological outputs. Importantly, pathological signals, including expanded poly-Q-induced endoplasmic reticulum (ER) stress in Huntington's disease, as well as stress signals in other neurodegenerative diseases, engage ASK1 in the propagation of damage signals. Similarly, a number of other pathological signals, such as ROS, evoke sustained ASK1 activation, which triggers cellular damage in diseases such as cardiac hypertrophy and diabetes. However, how ASK1 activity is neutralized in cells under survival conditions remains to be fully elucidated. ASK1 appears to be regulated by two mechanisms: protein-protein interactions and posttranslational modifications. For example, stress signals, such as ROS, impact ASK1 by triggering reversible binding of thioredoxin and phosphorylation-induced association with 14-3-3 proteins. Thioredoxin, in its reduced form, can bind ASK1, keeping it in an inactive conformation. However, elevated ROS levels lead to oxidized cysteines in thioredoxin, inducing the release of ASK1, recruitment of TRAF2/6 to the kinase, and facilitating ASK1 activation (2). Increased ROS also triggers dissociation of 14-3-3 proteins from ASK1, relieving ASK1 inhibition (3). ASK1 binding to 14-3-3 is mediated by phosphorylated Ser967, which serves as a molecular sensor for signal integration (4). When bound to 14-3-3, ASK1 activity is inhibited, suppressing ASK1-mediated apoptosis. Stress signals reduce this phosphorylation and, subsequently, 14-3-3 binding (3, 4). Similarly, the protein phosphatase calcineurin activates ASK1 through the dephosphorylation of Ser967 (5). Conversely, increased ASK1/14-3-3 binding is correlated with decreasing ASK1 activity and increased cell survival (6, 7). By controlling the phosphorylation status of Ser967, an upstream protein kinase cascade(s) may be able to integrate diverse signaling pathways with ASK1-mediated stress responses.
Here, we report a central node at the junction of survival, inflammation, and stress signaling networks through a direct interaction between ASK1 and the inhibitor of κB kinase (IKK), which reveals a critical mechanism by which IKK neutralizes stress and apoptotic signaling by a transcription-independent mechanism. An inhibitory role of IKK in JNK signaling was preciously attributed to the NF-κB induced XIAP and GADD45 in a transcription-dependent manner (8). Discovery of the IKK/ASK1 complex as a novel signaling integration machinery may offer unique opportunities to precisely manipulate disease-evoked stress response through this newly uncovered molecular interaction interface for future therapeutic interventions.
MATERIALS AND METHODS
Reagents.
H2O2, epidermal growth factor (EGF), insulin-like growth factor 1 (IGF-1), wortmannin, and PS1145 (all from Sigma), Akt inhibitor, phosphatidylinositol ether analog, and recombinant Akt1 (all from Calbiochem), LY294002 (Alomone Labs), tumor necrosis factor alpha (TNF-α; BD Pharmingen), recombinant MEK (Cell Signaling), recombinant IKKα (Upstate Cell Signaling Solutions), IKKβ (Invitrogen), Akt (Invitrogen), histone 2B (Sigma), IκB (Abcam), and [γ-32P]ATP (Perkin-Elmer) were used in supplied solution or reconstituted according to the manufacturer's instructions.
Kinase assays.
For Akt kinase assays, recombinant Akt1 (20 ng), immunoprecipitated was added to purified ASK1 C-terminal fragment (0.5 μg) or recombinant histone 2B (0.5 μg) in a kinase buffer (20 mM HEPES, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM dithiothreitol [DTT], 25 mM MgCl). Reactions were started with an ATP mixture consisting of 20 mM cold ATP and 3 μCi of [γ-32P]ATP and carried out for 2 h at 30°C, after which reactions were terminated by boiling in 6× sodium dodecyl sulfate (SDS) sample buffer. Two kinase assay formats were used for IKK: an antibody-based assay to specifically monitor phosphorylated S967 and a radioisotope incorporation assay. (i) For antibody-based IKK kinase assays, active recombinant IKKβ (0.5 μg) was added to immunoprecipitated ASK1 in a kinase buffer (8 mM morpholinepropanesulfonic acid, 200 mM EDTA, 15 mM MgCl2) with 100 mM ATP. Kinase reactions were performed at 37°C for 10 to 30 min. All kinase reactions were terminated by boiling in SDS sample buffer. Ser967 phosphorylation was determined by immunoblotting. (ii) For the direct radiolabeling in vitro IKK kinase assay, a purified ASK1 C-terminal fragment (0.5 μg) or recombinant IκB (0.5 μg) was incubated with recombinant IKKβ (5 to 40 ng) in a kinase buffer. Reactions were started with an ATP mixture consisting of 20 mM cold ATP and 3 μCi of [γ-32P]ATP and carried out for 10 min to 2 h, after which reactions were terminated by boiling them in 6× SDS sample buffer. For in vitro kinase assays, samples were run on an SDS-PAGE gel, and [γ-32P]ATP incorporation into substrates was measured by exposure on film and scintillation counting. The total protein levels were assessed by Coomassie blue staining.
Endogenous ASK1 immunoprecipitation.
COS7 or murine embryonic fibroblast (MEF) cells were grown on 10-cm plates to confluence and harvested with 0.2% NP-40 lysis buffer. Lysates were cleared with 30 μl of 50% slurry of protein G-Sepharose. ASK1 was immunoprecipitated with 5 μl of an ASK1-specific antibody (F-9; Santa Cruz) and 25 μl of a 50% slurry of protein G-Sepharose at 4°C for 4 to 6 h, rotating slowly. Samples were washed 500 μl of lysis buffer, and beads were isolated by quick centrifugation. The supernatant was discarded, and the process was repeated three times. The protein levels were assessed by immunoblotting.
Purification of ASK1 C terminus from Escherichia coli.
One colony of bacteria harboring an expression plasmid for glutathione S-transferase (GST)–ASK1 C-terminal fragment (ASK1K939-T1374) was grown in 500 ml of ZYP-5052 autoinduction medium [1% N-Z-amine AS, 0.5% yeast extract, 50 mM Na2HPO4, 50 mM KH2PO4, 25 mM (NH4)2SO4, 2 mM MgSO4, 0.2× metals, 0.5% glycerol, 0.05% glucose, 0.2% α-lactose] overnight at 37°C with shaking. The sample was spun down at 14,000 rpm for 15 min, and the pellet was resuspended in 10 ml of 1× phosphate-buffered saline (PBS; 1 M DTT, 100 mM phenylmethylsulfonyl fluoride [PMSF], 5 M NaCl) and sonicated six times for 30 min each time at 4°C on ice. The sample was centrifuged for 10 min at 14,000 rpm at 6°C. The supernatant was mixed with 2 ml of a 50% slurry of glutathione-Sepharose (GE Healthcare) in 1× PBS and rotated slowly at 4C for 2 h. Samples were eluted with elution buffer (20 mM reduced glutathione, 0.25 mM PMSF, 5 mM DTT, 0.1% Triton X-100). Samples were concentrated by centrifugation and dialyzed into storage buffer (10 mM HEPES, 150 mM NaCl, 10% glycerol).
Apoptosis assays.
Caspase activity was evaluated using a CaspACE assay (Promega) according to the manufacturer's protocol. In brief, DEVD-pNA substrate was used to test the effect of cell lysates on the liberation of p-nitroaniline from the substrate in a colorimetric assay. The caspase-3 specific activity was determined by normalizing each sample to the protein concentration. An annexin V assay was evaluated according to the manufacturer's protocol (BD Pharmingen). The percentages of annexin V- and 7-aminoactinomycin D (7-AAD)-positive cells were determined by using a Guava flow cytometer using Guava Nexin software.
Time-resolved FRET.
Cell lysates containing overexpressed proteins were mixed on 384-well black plates. Lysates were serially diluted in a reaction buffer (20 mM Tris-HCl [pH 7.0], 50 mM NaCl, 0.01% NP-40). GST-Terbium (HTRF)-conjugated antibody was used to couple GST-IKKβ as a FRET donor. Venus-ASK1 served as a fluorescence resonance energy transfer (FRET) acceptor. Samples were incubated at room temperature for 1 h, and the FRET signal was measured on the Envision spectrophotometer (excitation wavelength, 337 nm; emission wavelength, 520 nm).
PC12 neurite outgrowth assay.
To induce ASK1ΔN expression, PC12 cells were cultured in Dulbecco modified Eagle medium (DMEM) with 1% horse serum. All cell treatments were carried out for 3 days, after which cells were fixed with 2% paraformaldehyde for 30 min and solubilized with 0.1% Triton X-100 for 20 min. Fixed and solubilized cells were incubated with Alexa Fluor 488-phalloidin (Invitrogen) in 5% bovine serum albumin (BSA) at a final concentration of 1:1,000 overnight. Nuclei were counterstained with 5 μg of Hoechst dye (Invitrogen)/ml. Cells were washed three times with 1× PBS between each step. Fluorescence was measured with an ImageExpress spectrophotometer (Molecular Devices), and neurite outgrowth was quantified by using MetaExpress software (Molecular Devices).
Statistical analysis.
The data in bar graphs in the figures are presented as means with the standard deviations represented by error bars. Additional information is given in the figure legends.
RESULTS
Diverse growth factor-initiated pathways impinge on ASK1 at Ser967.
Reversible phosphorylation of ASK1 at Ser967 serves as a sensing mechanism, integrating diverse environmental cues to illicit critical biological responses through ASK1 (3, 6). Stress signals often induce dephosphorylation of Ser967 and promote cell death and, indeed, Ser967 was dephosphorylated upon serum withdrawal (Fig. 1a) and by exposure to ROS, stimuli known to increase ASK1 kinase activity (3, 9, 10). Consistent with this notion, serum withdrawal enhanced ASK1-mediated apoptosis (Fig. 1b).
Fig 1.

Phosphorylation of ASK1 at Ser967 is dynamically regulated. (a to f) COS7 cells were transfected with HA-ASK1 or HA-ASK1 S967A. (a) Ser967 phosphorylation was monitored after serum starvation for the indicated times. (b) Serum starvation-induced apoptosis was measured by staining with annexin V and 7-AAD after 24 h. The data represent means plus the standard deviations (n = 5). (c) COS7 cells were serum starved for 24 h and treated with the indicated doses of IGF-1, EGF, PDGF, TGF-β, or vehicle (PBS plus 0.1% BSA) for 20 min. (d and e) After 24 h of serum starvation, the cells were treated with IGF-1 or vehicle as indicated. (f and g) COS7 cells were treated with H2O2 (1 mM) in the presence of IGF-1 (100 ng/ml) or vehicle (PBS plus 0.1% BSA). The level of pSer967 of overexpressed (f) or immunoprecipitated endogenous ASK1 (g) was measured. pS967 and total ASK1 protein were detected by Western blotting (a, c to g) and quantified with ImageJ (g).
To reveal pathways that control this molecular sensing system, we examined factors that might enhance phosphorylation of ASK1 Ser967. Reintroduction of serum after serum starvation restored ASK1 Ser967 phosphorylation, leading to the hypothesis that there may be a prosurvival kinase-activating component(s) within serum, resulting in the phosphorylation of Ser967 and, subsequently, 14-3-3 binding and ASK1 inhibition. Correspondingly, serum blocked apoptosis induced by wild-type ASK1 but not an unphosphorylatable ASK1 mutant (S967A) (Fig. 1b), indicating that serum's prosurvival function occurs at least in part by controlling the phosphorylation status of ASK1 Ser967. In order to identify this kinase component, well-characterized growth factors were tested: EGF, IGF-1, platelet-derived growth factor (PDGF), and transforming growth factor β (TGF-β). Treatment of cells with EGF, IGF-1, and PDGF, but not TGF-β, resulted in marked phosphorylation of Ser967 (Fig. 1c), suggesting that these growth factors play a key role in regulating this event. Further kinetics studies revealed that IGF-1 promoted Ser967 phosphorylation in both a time and dose dependent manner (Fig. 1d and e). Notably, IGF-1 was also able to block ROS-induced dephosphorylation of ASK1 (Fig. 1f). Furthermore, IGF-1 could indeed impair stress induced dephosphorylation of endogenous ASK1 (Fig. 1g), confirming that our findings were relevant under physiological conditions.
Akt/PKB is an upstream kinase for Ser967 of ASK1.
IGF-1 is known to induce the activation of Akt/protein kinase B (PKB). Akt has a major prosurvival role in many settings and downregulates ASK1 activity through phosphorylation of Ser83 (9). Similar to Ser83, the region surrounding Ser967 of ASK1 fits nicely within the defined phosphorylation motif of Akt (RXRXXSXX), suggesting the site could potentially be modified by Akt. In order to investigate whether Akt mediates IGF-1 induced Ser967 phosphorylation, constitutively active Akt (Akt ΔPH) was utilized. Expression of wild-type or Akt ΔPH resulted in dramatic increase in Ser967 phosphorylation, which is correlated with decreased ASK1 activity as shown with reduced phosphorylation at Thr838 (Fig. 2a).
Fig 2.

Akt/PKB promotes phosphorylation of ASK1 at Ser967. (a to c) COS7 cells were transfected with HA-ASK1, wild-type Akt (Akt WT), constitutively active Akt (Akt ΔPH), and/or catalytically inactive Akt (Akt KM). (a) At 48 h after transfection, the cells were serum starved for 24 h. (b) COS7 cells transfected with HA-ASK1 (WT) or HA-ASK1 S967A (SA) grown in the presence of serum were treated with wortmannin (200 nm), LY294002 (30 μM), Akt inhibitor 124005 (40 μM), or dimethyl sulfoxide (DMSO; vehicle) for 30 min. (c) At 48 h after transfection, the cells were serum starved for 24 h and treated with IGF-1 (100 ng/ml) for 20 min. (d) An in vitro radiolabeling kinase assay for ASK1 phosphorylation was performed with recombinant Akt. The total protein levels were analyzed by Coomassie blue staining. The pS967 ASK1 and total protein levels in panels a to c were analyzed by Western blotting.
To complement the results obtained by Akt overexpression, we used a pharmacological approach to examine the role of Akt in Ser967 phosphorylation. The PI3K inhibitors, wortmannin and LY294002, or the specific Akt inhibitor 124005 were used to manipulate Akt activity. Both inhibition of PI3K signaling and direct inhibition of Akt reduced phosphorylation levels of Ser967 (Fig. 2b), confirming a role for Akt in the phosphorylation of this site.
To further examine whether Akt mediates growth factor-evoked Ser967 phosphorylation, cells overexpressing various Akt proteins (wild-type Akt, Akt ΔPH, or Akt KM) were treated with IGF-1. Akt KM is a catalytically inactive Akt mutant (K179M). Although active Akt enhanced phosphorylation of ASK1 Ser967, overexpression of the dominant-negative Akt blunted IGF-1's ability to increase ASK1 phosphorylation (Fig. 2c). These results suggest a critical role of Akt in bridging the growth factor effects with suppression of ASK1 activity.
Given the evidence that Akt directly phosphorylates BAD and Forkhead transcription factors within 14-3-3 consensus-binding motifs (11–13), it seemed likely that Akt also phosphorylates ASK1 directly. We tested this notion with an in vitro kinase assay. Active, recombinant Akt was utilized to phosphorylate ASK1 as a substrate. Surprisingly, no transfer of phosphate from ATP to Ser967 ASK1 was detected (Fig. 2d). After confirming the activity of recombinant Akt by its ability to phosphorylate the known substrate histone 2B, we came to the conclusion that Akt does not likely phosphorylate ASK1 directly at Ser967.
IKK mediates growth factor and Akt-induced phosphorylation of ASK1.
After discovering that Akt is likely to be upstream of the kinase responsible for phosphorylation of ASK1 at Ser967, we investigated kinase substrates of Akt. Notably, Akt activates the prominent prosurvival kinase, IKK (14–16). Although IKK plays a vital role in NF-κB activation, the importance of IKK in the regulation of other signaling events is becoming increasingly evident (17). Thus, we tested the premise that IKK phosphorylates ASK1. Cotransfection of COS7 cells with wild-type IKK (IKK WT) prevented serum starvation-induced dephosphorylation of ASK1 (Fig. 3a). Similarly, when cells were cotransfected with a catalytically inactive form of IKK (IKK K44M), a decrease of phosphorylated Ser967 was observed even in the presence of serum (Fig. 3b), suggesting that the kinase activity of IKK is critical in maintaining ASK1 Ser967 phosphorylation.
Fig 3.

IKK phosphorylates ASK1 at Ser967. (a and b) COS7 cells were transfected with HA-ASK1 and GST-IKKβ (IKK WT) or catalytically inactive GST-IKKβ (IKK KM). At 24 h after transfection, the medium was replaced with DMEM (a) or DMEM containing 10% fetal bovine serum (b). (c and d) An in vitro antibody-based (c) or radiolabeling (d) kinase assay was performed by adding increasing doses of active recombinant IKKβ to immunoprecipitated ASK1 (c) or purified recombinant ASK1 C-terminal fragment (K939-T1374) (d) as monitored by both 32P incorporation into substrate (exposure on film [upper]) and scintillation counting (lower). CBS, Coomassie blue staining; CPM, counts per minute. (E) MEF cells (WT, IKKα−/−, and IKKβ−/−) were serum starved (24 h) and treated with IGF-1 (100 ng/ml) for 20 min. Endogenous ASK1 was immunoprecipitated to monitor pS967 status and 14-3-3 association. (f) Immunoprecipitation of endogenous ASK1 from COS7 cells serum starved and treated with PS1145 for 24 h prior to treatment with IGF-1 (100 ng/ml) for 20 min. (g and h) pS967 ASK1 was monitored in COS7 cells transfected with HA-ASK1, GST-IKKβ (IKK WT), catalytically inactive GST-IKKβ (IKK KM), constitutively active HA-Akt (Akt ΔPH), and/or catalytically inactive HA-Akt (Akt KM) and serum starved for 24 h (g) or treated with IGF-1 (75 ng/ml) or vehicle (PBS plus 0.1% BSA) for 20 min after pretreatment with LY294002 (30 μM) or vehicle (DMSO) for 2 h (h). (i) The presence of ASK1 was assessed in GST-IKKβ complexes isolated from COS7 cells transfected with HA-ASK1 and GST-IKKβ 48 h after transfection (left). Interaction was also assessed by time-resolved fluorescence resonance energy transfer (TR-FRET). Lysates of COS7 cells transfected with either Venus-ASK1 or GST-IKKβ were mixed as indicated, and fluorescence was measured with the Envision spectrophotometer (right). For panels a to i, pS967ASK1 and total protein levels were assessed by Western blotting.
To determine whether IKK can directly catalyze the phosphorylation of ASK1 at Ser967, an in vitro kinase assay was performed. Interestingly, IKKβ, as well as IKKα, phosphorylated immunoprecipitated ASK1 in vitro (Fig. 3c), a phenomenon not observed with purified Akt (Fig. 2d). Furthermore, recombinant IKKβ phosphorylated purified recombinant C-terminal fragment of ASK1 in a dose-dependent manner in a reconstituted biochemical system (Fig. 3d), confirming ASK1 as a direct substrate of IKKβ, along with IκB.
To validate a role of IKK as a Ser967 kinase, a genetic and a pharmacological approach were used. Knockout of IKKα or IKKβ decreased IGF-1-induced ASK1 Ser967 phosphorylation and reduced 14-3-3 interaction (Fig. 3e). In addition, incubation of cells with a small molecule IKK inhibitor, PS1145, abolished IGF-1-induced Ser967 phosphorylation (Fig. 3f). Thus, it is likely that IKK acts downstream of IGF-1 and Akt to phosphorylate ASK1 at Ser967.
To confirm the order of IKK with respect to Akt within the IGF-1 to Ser967 signaling axis, we carried out a series of combination experiments with various constitutively active and dominant-negative mutants. Although dominant-negative IKK KM inhibited Ser967 phosphorylation induced by the constitutively active Akt ΔPH, dominant-negative Akt KM failed to block active IKK-catalyzed Ser967 phosphorylation (Fig. 3g and h), placing Akt upstream of IKK. As shown in Fig. 1, IGF-1 treatment restored Ser967 phosphorylation after serum starvation through activation of a Ser967 kinase. Importantly, inhibition of Akt or IKK reduced the effect of IGF-1 on Ser967 phosphorylation (Fig. 3h). However, active IKK overcame the effect of phosphatidylinositol 3-kinase and Akt inhibitors, restoring phosphorylation of ASK1 at Ser967 (Fig. 3h). Further, IGF-1 treatment showed increased levels of p-IKK, a marker of IKK activity, indicating that IKK is indeed activated in response to growth factors (data not shown). Together, these data establish a novel signaling axis by which IKK acts downstream of Akt to phosphorylate ASK1 at Ser967.
IKK directly interacts with ASK1.
To provide further evidence of cross talk between IKK and ASK1, we examined the possibility of a direct interaction between these two kinases. With a GST-affinity pulldown assay, ASK1 is found in the IKK protein complex (Fig. 3i). Then, we utilized time-resolved fluorescence resonance energy transfer (TR-FRET) technology, which can detect protein interactions within the distance of 100Å. Indeed, incubation of IKK with ASK1 led to an increase in FRET signal (Fig. 3i). These results indicate that ASK1 and IKK likely interact directly.
The IGF-1/Akt/IKK signaling antagonizes H2O2-induced ASK1 activation.
If IKK is directly coupled to ASK1-mediated signaling, stimulation of IKK by survival signals and upstream regulators is expected to counteract ASK1 activation induced by stress signals. To test this model, we examined the role of IKK in the cross talk between IGF-1 and ROS induced-ASK1 pathways. As shown in Fig. 1, H2O2 triggered dephosphorylation of ASK1 at Ser967, which can be reversed by IGF-1. Like IGF-1, the expression of Akt ΔPH, as well as IKK, effectively blocked dephosphorylation of Ser967 by H2O2, supporting a role for Akt and IKK in mediating the IGF-1 effect (Fig. 4a and see also Fig. 3h). Consistent with our model, IGF-1 addition is associated with IKK activation (p-IKK) and inhibition of ASK1 as shown with reduced pThr838 and the downstream pJNK. Similarly, serum starvation reduced pSer967 of ASK1 (Fig. 4b). Because pSer967 mediates 14-3-3 binding, reduced pSer967 should impact 14-3-3 recruitment. Indeed, serum starvation of cells was correlated with the decreased 14-3-3 association with ASK1 (Fig. 4b). Significantly, the overexpression of IKK reversed Ser967 phosphorylation as well as 14-3-3 association with ASK1 (Fig. 4b), keeping ASK1 in an inhibited state as seen with reduced pJNK. Thus, the IGF-1/Akt/IKK kinase cascade supports cell survival in part by suppressing ASK1 activity through enhanced phosphorylation of Ser967, counteracting the action of prodeath and stress signals such as ROS and serum starvation.
Fig 4.
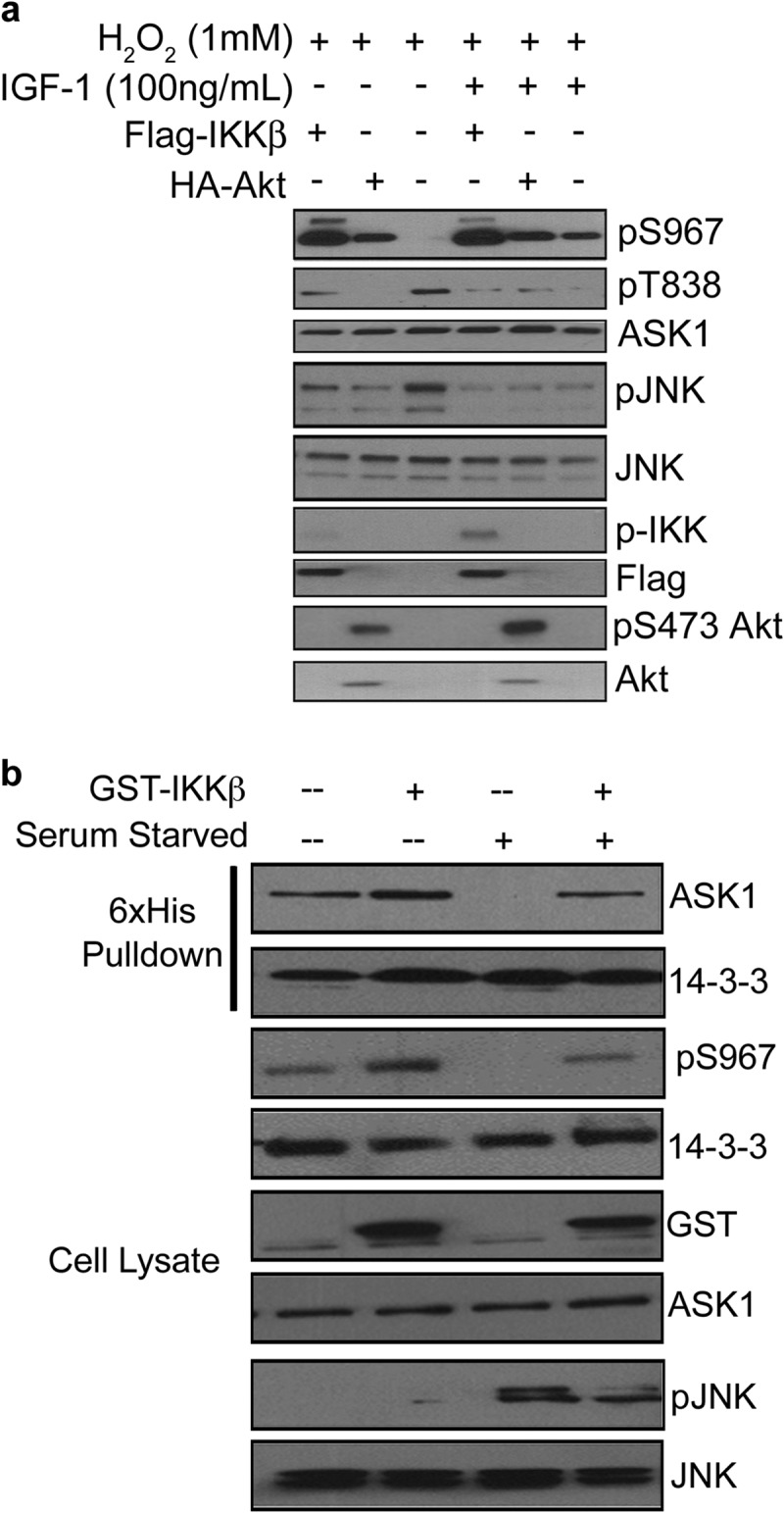
The IGF-1/Akt/IKK pathway counteracts stress-induced ASK1 activity. (a) COS7 cells transfected with GST-IKKβ or HA-Akt and treated with H2O2 (1 mM) as indicated and pSer967 ASK1 was monitored by Western blotting. (b) COS7 cells transfected with HA-ASK1, 6×His-14-3-3γ, and/or GST-IKKβ. Cells were serum starved (16 h) and 6×His-14-3-3 complexes were isolated by nickel-chelating resin. Dissociation of HA-ASK1 and 6×His-14-3-3 was monitored by Western blotting.
IKK suppresses ASK1-mediated neurite outgrowth.
To probe the effect of IKK activation on the biological function of ASK1, we utilized an established ASK1 functional assay in PC12 cells. In this PC12 cell system, ASK1ΔN, a constitutively active ASK1 mutant, is induced through the removal of tetracycline to promote neurite outgrowth (18) (Fig. 5a). ASK1-induced neurite outgrowth serves as a biological readout for ASK1 function (Fig. 5a). For our purpose, PC12 cells were treated with TNF-α to activate IKK upon ASK1 induction, and the IKK activity was monitored by NF-κB translocation. IKK activation significantly decreased ASK1-induced neurite outgrowth (Fig. 5a and b). Furthermore, treatment of cells with the IKK inhibitor PS1145 restored ASK1-induced neurite outgrowth to control levels. These data together suggest that activation of IKK can suppress ASK1-mediated function.
Fig 5.

IKK suppresses ASK1-mediated functions. (a) IKK activation inhibits ASK1ΔN-mediated neurite outgrowth. PC12 cells, cultured in the presence or absence (induction of ASK1ΔN) of tetracycline, were treated with TNF-α (10 ng/ml) and/or PS1145 (10 μM) as indicated. After treatment, cells were fixed and stained with phalloidin. (b) Quantification of neurite outgrowth in panel a. (c) COS7 cells were transfected with HA-ASK1 WT, HA-ASK1 S967A, or the catalytically inactive HA-ASK1 K709R and serum starved (24 h). Caspase-3 activity was measured as pmol of p-nitroaniline released/h/μg of protein. The first bar indicates activity of the blank. The data represent means plus the the standard deviations (n = 3). pS967 ASK1 and total protein levels were detected by Western blotting. (d) Model of ASK1 regulation by IKK.
IKK inhibits ASK1-mediated apoptosis in a Ser967-dependent manner.
ASK1 is a critical mediator of apoptosis signaling and, by inducing phosphorylation at Ser967, IKK would inhibit ASK1-mediated apoptosis. To test this hypothesis, COS7 cells were transfected with ASK1 to induce apoptosis with or without IKK. Caspase-3 protease activity was monitored as the readout for apoptotic cell death, and phospho-Ser967 levels in cell lysates were monitored by Western blotting in order to reveal the level of Ser967 phosphorylation during the treatment. Consistent with a prosurvival role of IKK, expression of IKK inhibited ASK1-induced apoptosis (Fig. 5c). However, it is possible that this inhibition is due to known prosurvival effects of IKK independent of ASK1. In order to address this possibility, we expressed the ASK1 mutant S967A to induce apoptosis. IKK had minimal effects on the apoptotic activity of this unphosphorylatable mutant (Fig. 5c), suggesting that IKK inhibits ASK1 apoptotic activity primarily through inducing phosphorylation at Ser967. Thus, phosphorylation of ASK1 at Ser967 may be a major mechanism through which IKK exerts its prosurvival and antistress regulatory role in the cell.
DISCUSSION
The Ser/Thr protein kinases, IKKα and IKKβ, were discovered as critical mediators of the NF-κB signaling pathway, which regulate immune responses, inflammation, and programmed cell death (17, 23, 24). Diverse environmental signals can trigger the activation of IKK through various transmembrane receptors such as TNF-α receptors, Toll-like receptors, and the IGF-1 receptor (17). Although the paradoxical activation of both prosurvival and prodeath pathways by TNF-α was puzzling at the beginning, the intricate cross talk between the NF-κB and JNK pathways illustrates the demand for a well-orchestrated signaling network for meaningful biological readouts. It has been established that IKK activation inhibits JNK signaling through NF-κB induced XIAP and GADD45 in a transcription-dependent manner (8). Our report unravels a direct action of activated IKK on ASK1, an upstream regulator of JNK, which establishes a crucial point of cross talk between IKK-mediated immune response, inflammatory, and prosurvival pathways and ASK1-mediated stress responses. Our data support a dual regulatory mechanism of IKK on the ASK1/JNK stress signaling axis (Fig. 5d). Activation of IKK leads to (i) phosphorylated IκB to induce a transcription-dependent inhibition of JNK through NF-κB and to (ii) phosphorylation and direct inhibition of ASK1, an activator of JNK, ensuring a precisely regulated signaling output.
ASK1 serves as a central signaling hub in mediating stress response and apoptosis induced by a number of stimuli, including cytokines, ROS, ER stressors, infectious agents, and cancer chemotherapeutics (1). Due to its physiological importance, ASK1's activity is tightly controlled. One critical control mechanism is through the reversible phosphorylation of Ser967, which senses the activation state of upstream protein kinases and dictates the docking of the 14-3-3 effector protein for ASK1 inhibition. Our results revealed IKK as an upstream kinase that controls phosphorylation of Ser967, constituting a new node in this complex of signaling networks. Even though we only presented the evidence for the importance of the IGF-1/Akt/IKK cascade in the regulation of ASK1, it is envisioned that other signals that can lead to IKK activation may be able to impact the ASK1-mediated JNK and p38 signaling in a wide range of biological cellular context. Thus, the IKK/ASK1 association may serve as a central integration mechanism for diverse, opposing signaling pathways, such as functional interactions between IKK-transmitted growth factor, cytokine, and pathogenic signals and ASK1-transmitted stress signals, including ROS and ER stress.
Although ASK1 and IKK can be activated by TNF-α, ASK1 is primarily activated through TNFR1 signaling (19), whereas IKK can become activated in response to TNFR1 and TNFR2 signaling (20). Although TNFR1 is ubiquitously expressed, TNFR2 expression is more limited (21). Previous research has shown, however, that TNF-α can promote cell survival in PC12 cells through TNFR2 (22). In our model, TNF-α may be preferentially activating TNFR2, leading to the activation of IKK and subsequent ASK1 inhibition. Previous studies have also indicated that TNF-α signaling through TNFR2 can lead to JNK inhibition. The mechanism by which this occurs, however, is poorly understood. Our data suggest that the ASK1/IKK signaling node may be responsible for this, highlighting a novel pathway by which IKK promotes cell survival.
Considering the critical roles of both ASK1 and IKK in a range of diseases, the functional interaction between IKK and ASK1 has significant mechanistic implications and offer promising therapeutic potentials. For example, the enhanced generation of ROS that activates ASK1 has been associated with the toxic action of amyloid-β in Alzheimer's disease and angiotensin II-induced cardiac hypertrophy and injury (25, 26). Our demonstration that IKK-mediated signaling blunts ROS-induced ASK1 activation suggests a possible neuroprotective mechanism of the IKK-ASK1 interaction. In a similar vein, IKK-ASK1 association may counter the action of ER stress-induced, ASK1-mediated neuronal cell death, as in poly-Q diseases, including Huntington's disease and spinocerebellar ataxias (27). Blocking ASK1 activity by IKK activation may alleviate the effect of poly-Q-induced ER stress. On the other hand, ASK1 also mediates apoptotic cell death triggered by a number of cancer therapeutics through ER stress, elevated ROS, or other mechanisms (28–32). Therefore, pathways that control ASK1 activity, such as the Akt/IKK signaling axis, are expected to have direct impact on tumor response to therapeutic agents. Indeed, tumor resistance to a number of anticancer agents, such as cisplatin and taxol, has been associated with the upregulated Akt and IKK function, which may be in part due to the enhanced IKK-ASK1 interaction and the reduced ASK1 response. Thus, the IKK-ASK1 interaction may provide a molecular target for potential therapeutic interventions. It is envisioned that lowering the IKK inhibitory effect on ASK1 by an IKK-ASK1 protein-protein interaction inhibitor may sensitize tumor cells to therapeutic agent-induced ASK1 activation and cell death to enhance therapeutic efficacy. Such a strategy may be particularly promising in many solid and hematological malignancies where the IKK complex is dysregulated (33).
Although only a few targets of IKKα and IKKβ have been confirmed outside the NF-κB signaling pathway, such as DOK1, FOXO3a, and TSC1, these IKK substrates indeed play pivotal roles in the functional effects of this kinase family (32–36). Our discovery of ASK1 as an IKK substrate, through a novel phosphorylation motif around Ser967, immediately widens the reach of IKK to the critical regulation of the cell's response to diverse stress signals, such as ROS and ER stress. In this way, the IKK/ASK1 signaling node may serve as a critical integration point that mechanistically dictates the flow of conflicting pathways within stress response networks, allowing growth factor-activated IKK to counter ROS-triggered ASK1 activation and apoptosis.
ACKNOWLEDGMENTS
This study was supported in part by the U.S. National Institutes of Health grants P01 CA116676 (F.R.K. and H.F.), the Georgia Cancer Coalition (F.R.K. and H.F.), the Georgia Research Alliance (H.F.), and a PhRMA Foundation Predoctoral Fellowship in Pharmacology/Toxicology (M.C.P.).
Footnotes
Published ahead of print 25 March 2013
REFERENCES
- 1. Shiizaki S, Naguro I, Ichijo H. 2012. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv. Biol. Regul. 53:135–144 [DOI] [PubMed] [Google Scholar]
- 2. Takeda K, Noguchi T, Naguro I, Ichijo H. 2008. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 48:199–225 [DOI] [PubMed] [Google Scholar]
- 3. Goldman EH, Chen L, Fu H. 2004. Activation of apoptosis signal-regulating kinase 1 by reactive oxygen species through dephosphorylation at serine 967 and 14-3-3 dissociation. J. Biol. Chem. 279:10442–10449 [DOI] [PubMed] [Google Scholar]
- 4. Zhang L, Chen J, Fu H. 1999. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. U. S. A. 96:8511–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Q, Wilkins BJ, Lee YJ, Ichijo H, Molkentin JD. 2006. Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth. Mol. Cell. Biol. 26:3785–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cockrell LM, Puckett MC, Goldman EH, Khuri FR, Fu H. 2010. Dual engagement of 14-3-3 proteins controls signal relay from ASK2 to the ASK1 signalosome. Oncogene 29:822–830 [DOI] [PubMed] [Google Scholar]
- 7. Kim I, Shu CW, Xu W, Shiau CW, Grant D, Vasile S, Cosford ND, Reed JC. 2009. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 284:1593–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. 2001. Inhibition of JNK activation through NF-κB target genes. Nature 414:313–317 [DOI] [PubMed] [Google Scholar]
- 9. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. 2001. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 21:893–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. 1998. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17:2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Masters SC, Yang H, Datta SR, Greenberg ME, Fu H. 2001. 14-3-3 inhibits Bad-induced cell death through interaction with serine-136. Mol. Pharmacol. 60:1325–1331 [DOI] [PubMed] [Google Scholar]
- 12. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241 [DOI] [PubMed] [Google Scholar]
- 13. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868 [DOI] [PubMed] [Google Scholar]
- 14. Kane LP, Shapiro VS, Stokoe D, Weiss A. 1999. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 9:601–604 [DOI] [PubMed] [Google Scholar]
- 15. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. 1999. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 401:82–85 [DOI] [PubMed] [Google Scholar]
- 16. Salminen A, Kaarniranta K. 2010. Insulin/IGF-1 paradox of aging: regulation via AKT/IKK/NF-κB signaling. Cell Signal. 22:573–577 [DOI] [PubMed] [Google Scholar]
- 17. Liu F, Xia Y, Parker AS, Verma IM. 2012. IKK biology. Immunol. Rev. 246:239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takeda K, Hatai T, Hamazaki TS, Nishitoh H, Saitoh M, Ichijo H. 2000. Apoptosis signal-regulating kinase 1 (ASK1) induces neuronal differentiation and survival of PC12 cells. J. Biol. Chem. 275:9805–9813 [DOI] [PubMed] [Google Scholar]
- 19. Shinoda S, Skradski SL, Araki T, Schindler CK, Meller R, Lan JQ, Taki W, Simon RP, Henshall DC. 2003. Formation of a tumour necrosis factor receptor 1 molecular scaffolding complex and activation of apoptosis signal-regulating kinase 1 during seizure-induced neuronal death. Eur. J. Neurosci. 17:2065–2076 [DOI] [PubMed] [Google Scholar]
- 20. McFarlane SM, Pashmi G, Connell MC, Littlejohn AF, Tucker SJ, Vandenabeele P, MacEwan DJ. 2002. Differential activation of nuclear factor-κB by tumour necrosis factor receptor subtypes. TNFR1 predominates, whereas TNFR2 activates transcription poorly. FEBS Lett. 515:119–126 [DOI] [PubMed] [Google Scholar]
- 21. Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. 1997. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J. Neuroimmunol. 75:104–112 [DOI] [PubMed] [Google Scholar]
- 22. Takei Y, Laskey R. 2008. Tumor necrosis factor alpha regulates responses to nerve growth factor, promoting neural cell survival but suppressing differentiation of neuroblastoma cells. Mol. Biol. Cell 19:855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. 1997. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science 278:860–866 [DOI] [PubMed] [Google Scholar]
- 24. DiDonato JA, Mercurio F, Karin M. 2012. NF-κB and the link between inflammation and cancer. Immunol. Rev. 246:379–400 [DOI] [PubMed] [Google Scholar]
- 25. Kadowaki H, Nishitoh H, Urano F, Sadamitsu C, Matsuzawa A, Takeda K, Masutani H, Yodoi J, Urano Y, Nagano T, Ichijo H. 2005. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 12:19–24 [DOI] [PubMed] [Google Scholar]
- 26. Nako H, Kataoka K, Koibuchi N, Dong YF, Toyama K, Yamamoto E, Yasuda O, Ichijo H, Ogawa H, Kim-Mitsuyama S. 2012. Novel mechanism of angiotensin II-induced cardiac injury in hypertensive rats: the critical role of ASK1 and VEGF. Hypertens. Res. 35:194–200 [DOI] [PubMed] [Google Scholar]
- 27. Reddy PH, Williams M, Tagle DA. 1999. Recent advances in understanding the pathogenesis of Huntington's disease. Trends Neurosci. 22:248–255 [DOI] [PubMed] [Google Scholar]
- 28. Liao PC, Tan SK, Lieu CH, Jung HK. 2008. Involvement of endoplasmic reticulum in paclitaxel-induced apoptosis. J. Cell. Biochem. 104:1509–1523 [DOI] [PubMed] [Google Scholar]
- 29. Tsai SL, Suk FM, Wang CI, Liu DZ, Hou WC, Lin PJ, Hung LF, Liang YC. 2007. Anti-tumor potential of 15,16-dihydrotanshinone I against breast adenocarcinoma through inducing G1 arrest and apoptosis. Biochem. Pharmacol. 74:1575–1586 [DOI] [PubMed] [Google Scholar]
- 30. Yang WH, Fong YC, Lee CY, Jin TR, Tzen JT, Li TM, Tang CH. 2011. Epigallocatechin-3-gallate induces cell apoptosis of human chondrosarcoma cells through apoptosis signal-regulating kinase 1 pathway. J. Cell. Biochem. 112:1601–1611 [DOI] [PubMed] [Google Scholar]
- 31. Lee YK, Hwang JT, Kwon DY, Surh YJ, Park OJ. 2010. Induction of apoptosis by quercetin is mediated through AMPKα1/ASK1/p38 pathway. Cancer Lett. 292:228–236 [DOI] [PubMed] [Google Scholar]
- 32. Chen Z, Seimiya H, Naito M, Mashima T, Kizaki A, Dan S, Imaizumi M, Ichijo H, Miyazono K, Tsuruo T. 1999. ASK1 mediates apoptotic cell death induced by genotoxic stress. Oncogene 18:173–180 [DOI] [PubMed] [Google Scholar]
- 33. Arkan MC, Greten FR. 2011. IKK- and NF-κB-mediated functions in carcinogenesis. Curr. Top. Microbiol. Immunol. 349:159–169 [DOI] [PubMed] [Google Scholar]
- 34. Lee S, Andrieu C, Saltel F, Destaing O, Auclair J, Pouchkine V, Michelon J, Salaun B, Kobayashi R, Jurdic P, Kieff ED, Sylla BS. 2004. IκB kinase beta phosphorylates Dok1 serines in response to TNF, IL-1, or gamma radiation. Proc. Natl. Acad. Sci. U. S. A. 101:17416–17421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. 2004. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117:225–237 [DOI] [PubMed] [Google Scholar]
- 36. Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. 2007. IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130:440–455 [DOI] [PubMed] [Google Scholar]