

Abstract
Activation of the mitogen-activated protein (MAP) pathway kinases p38 and MK2 induces phosphorylation of the chaperone Hsp27 and stabilization of mRNAs containing AU-rich elements (AREs) (ARE-mRNAs). Likewise, expression of phosphomimetic mutant forms of Hsp27 also stabilizes ARE-mRNAs. It appears to perform this function by promoting degradation of the ARE-mRNA decay factor AUF1 by proteasomes. In this study, we examined the molecular mechanism linking Hsp27 phosphorylation to AUF1 degradation by proteasomes. AUF1 is a target of β-TrCP, the substrate recognition subunit of the E3 ubiquitin ligase Skp1–cullin–F-box protein complex, SCFβ-TrCP. Depletion of β-TrCP stabilized AUF1. In contrast, overexpression of β-TrCP enhanced ubiquitination and degradation of AUF1 and led to stabilization of reporter mRNAs containing cytokine AREs. Enhanced AUF1 degradation required expression of phosphomimetic mutant forms of both Hsp27 and AUF1. Our results suggest that a signaling axis composed of p38 MAP kinase–MK2–Hsp27–β-TrCP may promote AUF1 degradation by proteasomes and stabilization of cytokine ARE-mRNAs.
INTRODUCTION
AU-rich elements (AREs) are perhaps the best-characterized mRNA-destabilizing elements in eukaryotes (1, 2). They are present in the 3′ untranslated regions (UTRs) of many mRNAs encoding cytokines, cell cycle regulators, signaling proteins, and oncoproteins. ARE-binding proteins associate with their target mRNAs to promote mRNA degradation or stabilization, depending upon the particular protein(s) bound. In a growing number of cases, ARE-binding proteins couple with microRNA target sites to provide coordinated control of mRNA decay (3). AUF1/hnRNP D is one family of ARE-binding proteins that controls degradation and/or translation of numerous ARE-containing mRNAs (ARE-mRNAs) (4). Alternative pre-mRNA splicing generates the four isoforms with apparent molecular masses of 37, 40, 42, and 45 kDa. These isoforms assemble with a complex of proteins, including the heat shock proteins Hsc70, Hsp70, and Hsp27, eukaryotic translation initiation factor 4G (eIF4G), poly(A)-binding protein, and additional unknown proteins (5–7). The AUF1 complex of proteins recruits messenger ribonucleases, such as exosomes and decapping factors, and in some cases the microRNA-induced silencing complex (miRISC), to degrade ARE-containing and other mRNAs (8–11).
AUF1 proteins control numerous biological processes, particularly in the immune system. For example, auf1−/− mice present severe endotoxemia due to failure to degrade TNF-α and IL-1β mRNAs (12). These animals also present pruritic inflammatory skin disease characterized by elevated production of interleukin 2 (IL-2), tumor necrosis factor alpha (TNF-α), and IL-1β (13), and they have reduced numbers of splenic follicular B cells (14). In monocytes exposed to endotoxin, p40AUF1 promotes translation of TAK1 mRNA (15). This ensures sufficient levels of TAK1 kinase to elicit NF-κB signaling for induction of IL-10 gene expression during the anti-inflammatory phase of an innate immune response (15, 16).
As noted above, AUF1 forms complexes with the heat shock proteins Hsc/Hsp70 and Hsp27, all of which are ARE-binding proteins as well (7, 17–20). Hsp27 is a ubiquitous protein that is induced in response to a wide variety of physiological and environmental stresses. Upon stimulation, Hsp27 is phosphorylated on three serine residues, Ser15, Ser78, and Ser82, by p38 mitogen-activated protein kinase (MAPK), MAPK-activated protein kinase 2 (MK2), Akt, and protein kinase C (21). Hsp27 promotes the catalytic activity of the proteasome machinery to increase degradation of ubiquitinated proteins in response to stress stimuli (22–24).
Hsp27 phosphorylation on Ser15, Ser78, and Ser82 also occurs by activation of p38 MAPK signaling upon binding of the IL-1 receptor by IL-1. This in turn promotes stabilization of ARE-mRNAs (25). Expression of phosphomimetically mutated Hsp27 in monocytes leads to destruction of AUF1 by proteasomes and stabilization of TNF-α and IL-1β ARE-mRNAs (17). However, the mechanisms by which Hsp27 phosphorylation promotes AUF1 degradation by proteasomes are unknown.
The ubiquitin-proteasome system is a major regulator of protein abundance in cells. Protein ubiquitination directs the modified proteins for proteolysis by the 26S proteasome. Protein ubiquitination consists of three sequential steps: activation of ubiquitin by a ubiquitin-activating enzyme (E1), transfer of ubiquitin from E1 to a ubiquitin-conjugating enzyme (E2), and conjugation of ubiquitin to target proteins by a ubiquitin ligase (E3). E3 ubiquitin ligases, which contain subunits responsible for recognizing specific substrate proteins, are thus vital components of the ubiquitination-proteasome machinery (26). The SCF (Skp1–cullin–F-box protein) ubiquitin ligase complex is one of the best-characterized mammalian cullin RING ubiquitin ligases (CRL). The F-box protein β-TrCP (β-transducin repeat-containing protein) is the substrate recognition subunit of the complex. β-TrCP-dependent ubiquitination often requires phosphorylation of its substrates on two serines separated by 2 to 4 amino acids (26–29).
Our previous study demonstrated that Hsp27 phosphorylation controls AUF1 abundance and ARE-mRNA degradation. Phosphomimetic mutation of Ser15, Ser78, and Ser82 to aspartic acid (single-letter code D) in Hsp27 (Hsp27DDD) promoted degradation of AUF1 by proteasomes and resulted in stabilization of ARE-mRNAs (7, 17). Here we identified β-TrCP as an F-box protein that promotes degradation of all AUF1 isoforms and stabilization of reporter ARE-mRNAs in cells expressing Hsp27DDD. Moreover, the p40 isoform has two phosphorylated serines, Ser83 and Ser87, that appear necessary for ubiquitination by β-TrCP and degradation by proteasomes. Thus, the p38 MAPK-MK2-Hsp27-SCFβ-TrCP signaling axis may induce AUF1 degradation to promote ARE-mRNA stabilization.
MATERIALS AND METHODS
Cell culture and stable transfections.
HeLa cells were maintained at 37°C and 5% CO2 in minimal essential medium (MEM) (MediaTech) supplemented with 10% fetal bovine serum (Gibco). Two plasmids in which known phosphorylation sites of Hsp27—Ser15, Ser78, and Ser82—had been mutated to either alanine or aspartic acid were used (17); each Hsp27 open reading frame was also linked to an N-terminal FLAG tag. Linearized plasmids were individually transfected into cells using Lipofectamine 2000 according to the manufacturer's instructions. Cells were selected in 500 μg/ml Geneticin (Gibco) for 3 to 4 weeks.
siRNA transfections.
The small interfering RNA (siRNA) 5′-GUGGAAUUUGUGGAACAUC-3′, which targets both βTrCP1 and βTrCP2 (30), and scrambled control siRNA were synthesized by Dharmacon. siRNA directed against mRNA encoding a control F-box protein, FBXW8, was from Santa Cruz Biotechnology. HeLa cells were seeded at 7.5 × 104 cells/well of a 6-well plate and incubated overnight. Two hundred forty picomoles of siRNA was diluted in 100 μl of Opti-MEM and mixed with Oligofectamine reagent (Invitrogen) according to the manufacturer's instructions. The mixture was added to cells. After 2 days, knockdowns of β-TrCP and FBXW8 were confirmed by Western blotting analyses as described below.
Protein decay assays.
Approximately 24 h prior to the assay, HeLa cells were seeded at a concentration of 1.5 × 105 cells/well in 6-well plates. Cells were cultured with 100 μg/ml cycloheximide (Sigma-Aldrich) to block translation and were harvested at various time points. Cells were lysed in SDS sample buffer, and 3 × 105 cell equivalents of proteins were resolved by 10% SDS-PAGE. AUF1 and internal control proteins were detected by Western blotting analyses as described below. Protein levels were quantified, normalized to internal control protein, and plotted as a percentage of the AUF1 levels at time zero. Data were analyzed by nonlinear regression, and the half-life was calculated from the first-order decay constant (k) obtained using the PRISM software program, version 3.03 (GraphPad, San Diego, CA): half-life = ln2/k.
Western blotting.
Cells were lysed in SDS sample buffer. Proteins were fractionated by 10% SDS-PAGE and transferred to nitrocellulose membranes by wet transfer. Membranes were blocked with phosphate-buffered saline (PBS) containing 5% low-fat dry milk (Blotto). Membranes were incubated with primary antibody and then washed with PBS–0.2% Tween 20 or with PBS alone in some cases. Membranes were incubated with goat anti-mouse or anti-rabbit IgG conjugated to horseradish peroxidase (Promega). Reactions were developed using an enhanced chemiluminescence (ECL) kit (Thermo Scientific) and detected with X-ray film or the Kodak Image Station 4000R imaging system (Carestream). Primary antibodies were used at the following dilutions or concentrations: anti-AUF1 rabbit polyclonal, 1:15,000; anti-Hsp70 rabbit polyclonal (Stressgen), 1:30,000; anti-Hsp27 rabbit polyclonal (Enzo Life Sciences), 1:3,000; anti-Myc mouse monoclonal (clone 9E10; BD Pharmingen), 2 μg/ml; anti-FLAG mouse monoclonal (Sigma), 10 μg/ml; anti-FBXW8 rabbit polyclonal (Abcam), 1 μg/ml; anti-β-TrCP mouse monoclonal (Invitrogen), 1 μg/ml. To permit sequential detection of different proteins, antibodies were removed from membranes by washing them with OneMinute stripping buffer (GM Biosciences).
Plasmid constructions and transient transfections.
Both known phosphorylation sites of p40AUF1, Ser83 and Ser87, were mutated to either alanine or aspartic acid using the QuikChange kit (Stratagene) according to the manufacturer's directions. To confer resistance to short hairpin RNA (shRNA) against AUF1, each p40AUF1 cDNA was mutated using the QuikChange kit (Stratagene) to disrupt base pairing in the siRNA-annealing site while maintaining amino acid sequence (16). These were each cloned into plasmid pcDNA3 (Invitrogen) at the 5′ Acc65I site and 3′ XbaI or 3′ ApaI site. All mutations were verified by DNA sequencing.
Transfection of plasmids into HeLa cells was performed using the SuperFect transfection reagent (Qiagen) according to the manufacturer's instructions. Briefly, 4 × 105 cells were seeded with MEM–10% fetal bovine serum in each well of a 6-well plate and incubated overnight. Various amounts (0.1 to 0.8 μg) of plasmid expressing Myc-tagged β-TrCP1 (wild type or ΔF-box mutant) (28) in 100 μl serum-free MEM were mixed with 10 μl SuperFect reagent and incubated at room temperature for 10 min. Cells were refed 600 μl fresh MEM plus 10% FBS, and then the plasmid-SuperFect mixture was added to the culture dropwise. Cells were incubated at 37°C for 2 days. Cells were lysed with 400 μl SDS-PAGE sample buffer. Twenty microliters of cell extract (6 × 104 cell equivalents of protein) was fractionated by 10% SDS-PAGE, and Western blotting was performed to detect relevant proteins. For cotransfections of multiple plasmids, 2 μg of plasmid expressing shRNA-refractory p40AUF1, 2 μg of plasmid expressing control or AUF1 shRNAs (pSilencer-U6-hygro/shCTRL and pSilencer-U6-hygro/shAUF1) (7), 1 μg of plasmid expressing hemagglutinin (HA)-tagged ubiquitin (pcDNA-HA-Ubiquitin; Addgene), and 0.8 μg of plasmid expressing Myc-tagged wild-type or ΔF-box β-TrCP1 were mixed in 150 μl serum-free MEM and combined with 30 μl SuperFect reagent; this mixture was incubated at room temperature for 10 min before adding to cells. To inhibit proteasome activity in selected experiments, lactacystin (final concentration, 10 μM; Sigma-Aldrich) was added to cells 24 h after transfection.
Ubiquitination assay of p40AUF1 and coimmunoprecipitation assays.
HeLa cells (1.2 × 106) were transfected with various plasmids. Lactacystin was added to cultures to a final concentration of 10 μM 24 h later. Twenty-four hours later, cells were lysed with 200 μl PEB buffer (20 mM Tris-HCl, pH 7.4, 100 mM KCl, 5 mM MgCl2, and 0.5% Nonidet P-40) and clarified by centrifugation at 12,000 × g for 10 min. Cell extracts were immunoprecipitated with 2 μl anti-AUF1 antibody (final dilution in reaction, 1:100) and then subject to Western blot analysis with anti-HA antibody (clone 12CA5; Roche) to detect HA-ubiquitin-conjugated p40AUF1. In some experiments, anti-AUF1 immunoprecipitates were analyzed by Western blotting with anti-AUF1 antibody. In this case, the secondary antibody employed was TrueBlot (horseradish peroxidase [HRP]-conjugated IgG that recognizes native IgG only; eBiosciences); this prevents detection of heavy and light chains on immunoblots of gel-fractionated immunoprecipitates. For coimmunoprecipitation assays, pellets from immunoprecipitations using anti-AUF1 antibody were fractionated by SDS-PAGE and transferred to membranes. The presence of test proteins was determined by immunodetection with antibodies described above.
Reporter mRNA half-life determinations.
Half-life assays were carried out as described previously (31). Plasmids pTRE/Rβ-wt (wild type, lacking AREs), pTRE/Rβ-TNF-α ARE, and pTRE/Rβ-COX2 ARE were described previously (7, 25). Briefly, a mixture of Rβ reporter plasmids, pTet-Off (encoding the tetracycline (Tet)-responsive transcriptional activator tTA) (32), pEGFP-C2 (encoding internal control mRNA), and wild-type/mutant Myc–β-TrCP1, were cotransfected into the HeLa-Hsp27 cell lines using the SuperFect reagent (Qiagen). Some experiments included transfection of plasmids expressing control shRNA or shRNA directed against AUF1. Two days posttransfection, doxycycline (Sigma-Aldrich) was added to culture medium to a final concentration of 2 μg/ml in order to block transcription of reporter genes. Cells were harvested at various time points, and total RNA was extracted from cells using an RNeasy minikit (Qiagen). Total RNA (500 ng) was reverse transcribed into cDNA with a TaqMan reverse transcription (RT) kit (Applied Biosystems) according to the manufacturer's instructions. The resulting cDNA was analyzed by quantitative RT-PCR (qRT-PCR) using Power SYBR green PCR master mix (Applied Biosystems) and 15 pmol of each primer (for Rβ, 5′-GTGAACTGCACTGTGACAAGC-3′ and 5′-ATGATGAGACAGCACAATAACCAG-3′; for EGFP, 5′-GCGACACCCTGGTGAACC-3′ and 5′-GATGTTGTGGCGGATCTTGAAG-3′). Reactions were carried out using the Stratagene MX3005P thermocycler. Relative mRNA levels were calculated based on standard curves. Reporter mRNA levels were normalized with EGFP mRNA and plotted as a percentage of the levels at time zero. Data were analyzed by nonlinear regression, and the half-life was calculated from the first-order decay constant (k) obtained with the PRISM software program, version 3.03 (GraphPad, San Diego, CA): half-life = ln2/k. Comparisons between half-lives were performed with the unpaired two-tailed t test with three independent experiments. Differences yielding P values of <0.05 were considered significant.
RESULTS
Phosphomimetic mutant form of Hsp27 promotes AUF1 degradation.
Our previous studies showed that posttranslational modifications of AUF1 and Hsp27 within the AUF1 complex of proteins occur in response to activation of protein kinase C in the human monocyte cell line THP-1 (7). We also showed that phosphomimetic replacement of the three critical serine residues in Hsp27, Ser15, Ser78, and Ser82, promoted AUF1 protein degradation in a proteasome-dependent manner in THP-1 cells and resulted in mRNA stabilization; Hsp27 containing alanine substitutions for these serines had no effect on AUF1 abundance (17). Similarly, activation of p38 MAPK signaling in HeLa cells promotes Hsp27 phosphorylation and stabilization of ARE reporter mRNA (25). However, the underlying mechanisms by which Hsp27 phosphorylation promotes AUF1 degradation and ARE-mRNA stabilization remain unknown.
To elucidate the mechanism, we established three HeLa cell lines: one stably expressing FLAG-tagged Hsp27 with phosphomimetic Ser-to-Asp substitutions at Ser15, Ser78, and Ser82 (Hsp27DDD), one expressing FLAG-tagged, phosphorylation-resistant Ser-to-Ala substitutions (Hsp27AAA), and one expressing FLAG-tagged, wild-type Hsp27 (Hsp27wt). Western blot analysis with an antibody against Hsp27 confirmed expression of the FLAG-tagged proteins, which migrate more slowly than endogenous Hsp27 (Fig. 1C). Cells were cultured with cycloheximide to block translation, cell extracts were prepared at various time points, and stability of AUF1 was examined by Western blotting with anti-AUF1 antibody. While HeLa cells express all four AUF1 isoforms, the p37AUF1 isoform is least abundant and is only observed with prolonged exposures; p40AUF1/p42AUF1 comigrate on gels but clearly separate from p45AUF1. While AUF1 proteins in HeLa stably expressing either wild-type or Hsp27AAA decayed with half-lives greater than 7 h (the limit of the time course), they collectively degraded with a half-life of 3.0 ± 0.2 h in HeLa/Hsp27DDD cells (Fig. 1A). Concurrent culturing of HeLa/Hsp27DDD cells with cycloheximide and the proteasome inhibitor lactacystin stabilized the AUF1 proteins (Fig. 1B) (t1/2 > 7 h). These results indicated that phosphomimetic mutation of Hsp27 promotes AUF1 degradation by proteasomes, consistent with our observations of THP-1 monocytes (17).
Fig 1.

Expression of phosphomimetic Hsp27 destabilizes AUF1 in HeLa cells. (A) Cells stably expressing the indicated wild-type (wt) and serine-replaced mutants of FLAG-Hsp27 were cultured with cycloheximide for the indicated times to block translation. Western blots were performed to detect AUF1 (top panels) and Hsp70 (bottom panels; loading control) to assess AUF1 protein stability. Each lane contains 3 × 105 cell equivalents of protein. The identification of AUF1 isoforms is noted on the rightmost panel. The p40/p42 AUF1 isoforms do not resolve in minigels; the p37 AUF1 isoform is the least abundant and is not observed in these exposures. (B) HeLa/Hsp27DDD cells were cultured with cycloheximide and 10 μM lactacystin (to block proteasome activity) to assess AUF1 protein stability. (C) Extracts were prepared from untransfected HeLa cells and cells stably expressing the indicated FLAG-Hsp27 proteins. Proteins were fractionated by SDS-PAGE and analyzed by Western blotting with antibody to Hsp27 to verify expression of FLAG-Hsp27 proteins. Positions of endogenous and FLAG-tagged Hsp27 proteins are noted on the right.
The F-box protein β-TrCP promotes Hsp27DDD-dependent degradation of AUF1.
Both the p40AUF1 and p45AUF1 isoforms possess a 19-amino-acid insert encoded by alternatively spliced exon 2 (4). Figure 2A shows a portion of the sequence including Ser83 and Ser87, both of which are phosphorylated in cellular p40AUF1 (33, 34). Ser83 lies within a consensus sequence for glycogen synthase kinase 3β (GSK-3β) and Ser87 for protein kinase A, and these kinases phosphorylate p40AUF1 at the respective serines in vitro (34). The F-box protein β-TrCP is the substrate recognition subunit of the SCFβ-TrCP ubiquitin E3 ligase. The canonical substrate sequence is DSpGX2–5Sp, in which the serines are phosphorylated (26). However, subsequent examination of numerous substrates has revealed broader consensus sequences (29) that suggested that phosphorylated p40AUF1 may be a target of β-TrCP (Fig. 2A).
Fig 2.

Phosphomimetic Hsp27 promotes AUF1 degradation via the F-box protein β-TrCP. (A) Alignment of AUF1 amino acid sequence with β-TrCP binding motifs. Shown on top, in single-letter codes, is a portion of the 19-amino-acid sequence encoded by exon 2 that distinguishes the p40/p45 AUF1 isoforms from the p37/p42 isoforms. The two indicated serines, Ser83 and Ser87, are phosphorylated in cellular p40AUF1 (33, 34). Shown below the AUF1 sequence are consensus binding motifs within β-TrCP substrates, with the phosphorylated serines in the canonical motif, DSGX2–5S, indicated as Sp (29). (B) HeLa/Hsp27DDD cells were transfected with control siRNA (siCTRL) or siRNA targeted to βTrCP1/2 (siTrCP). After 2 days, extracts were prepared for Western blot analyses of β-TrCP1/2 and β-actin (loading control). A 2-fold dilution series of extract from siCTRL-transfected cells (lanes 1 to 4) permitted estimates of β-TrCP knockdown efficiency (lane 5). (C) Knockdown of β-TrCP1/2 stabilizes AUF1. HeLa/Hsp27DDD cells were transfected with siCTRL (left panel) or siTrCP (right panel). After 2 days, cells were cultured with cycloheximide for protein stability assays by Western blotting analyses with antibodies to AUF1 and Hsp70 (as a control). (D) HeLa/Hsp27DDD cells were transfected with control siRNA (siCTRL) or siRNA targeted to FBXW8 (siFBXW8). After 2 days, extracts were prepared for Western blot analyses of FBXW8 and β-actin (loading control). A 2-fold dilution series of extract from siCTRL-transfected cells (lanes 1 to 4) permitted estimates of FBXW8 knockdown efficiency (lane 5). (E) Knockdown of FBXW8 has no effect on AUF1 protein stability. HeLa/Hsp27DDD cells were transfected with siCTRL (left panel) or siFBXW8 (right panel). After 2 days, cells were cultured with cycloheximide for protein stability assays by Western blotting analyses with antibodies to AUF1 and Hsp70 (as a control).
To determine whether β-TrCP plays a role in Hsp27-regulated turnover of AUF1, the effects of β-TrCP knockdown on AUF1 degradation were assessed. Control siRNA or siRNA targeting β-TrCP1 and β-TrCP2 was transfected into HeLa/Hsp27DDD cells, and knockdown efficiency was measured by Western blotting 2 days later. The level of β-TrCP was reduced at least 85% in cells transfected with siRNA targeting β-TrCP1/2 (Fig. 2B, compare lane 5 to a control, serial dilution series in lanes 1 to 4). Cycloheximide assays with these cells showed that while AUF1 decayed with a half-life of 2.5 ± 0.2 h in control siRNA-transfected cells (Fig. 2C, left panel), it was stabilized upon knockdown of β-TrCP (Fig. 2C, right panel; t1/2 > 7 h).
As a control, the effects of FBXW8 (F-box and WD40 domain protein 8) on AUF1 protein degradation were examined. Like β-TrCP, FBXW8 is an F-box protein that is a subunit of one of the SCF E3 ligase complexes involved in phosphorylation-dependent ubiquitination (35–37). While β-TrCP promotes CUL1-SKP1 association, FBXW7 promotes CUL7-SKP1 association. Thus, β-TrCP and FBXW8 are subunits of distinct SCFs. Knockdown of FBXW8 by siRNA transfection was confirmed by Western blotting to be at least 75% (Fig. 2D, compare lane 5 to a control, dilution series in lanes 1 to 4). AUF1 decayed with comparable half-lives of 3.2 ± 0.2 h and 3.5 ± 0.4 h in control and FBXW8 siRNA-transfected HeLa/Hsp27DDD cells (Fig. 2E, compare left and right panels). Thus, FBXW8 knockdown had no discernible effect on degradation of AUF1. Taken together, the knockdown experiments suggested that β-TrCP is specifically required for AUF1 degradation in cells expressing Hsp27DDD.
To substantiate this conclusion, the converse experiment—overexpression of β-TrCP—was performed to assess the effects on AUF1 degradation. Various amounts of plasmid expressing Myc-tagged β-TrCP1 were transfected into HeLa/Hsp27DDD and HeLa/Hsp27AAA cells, and abundances of Myc–β-TrCP1 and AUF1 were examined by Western blotting with anti-Myc and anti-AUF1 antibodies, respectively. As expected, levels of Myc–β-TrCP1 increased with increasing amounts of transfected expression plasmid (Fig. 3A, upper panel; the anti-Myc antibody also detected the endogenous c-Myc protein). While overexpression of Myc–β-TrCP1 had no effect on the levels of AUF1 in Hsp27/Hsp27AAA cells, AUF1 levels declined in HeLa/Hsp27DDD cells with increasing amounts of transfected Myc–β-TrCP1 (Fig. 3A, middle panel). These results indicated that overexpression of β-TrCP1 reduces AUF1 abundance, which also requires Hsp27DDD.
Fig 3.

β-TrCP overexpression accelerates AUF1 degradation. (A) The indicated amounts of plasmid expressing Myc–β-TrCP1 were transfected into HeLa/Hsp27DDD and HeLa/Hsp27AAA cells. Western blots were performed 2 days after transfection. Anti-Myc, anti-AUF1, and anti-Hsp27 were used to detect Myc–β-TrCP1, AUF1, and endogenous Hsp27/FLAG-Hsp27, respectively. Note: the anti-Myc antibody also detects the endogenous c-Myc protein, which migrates faster than Myc-tagged β-TrCP. (B) The indicated amounts of plasmids expressing wild-type or ΔF-box mutant (mut) Myc–β-TrCP1 were transfected into HeLa/Hsp27DDD cells. Western blotting was performed 2 days after transfection. Anti-Myc, anti-β-TrCP, anti-AUF1, and anti-Hsp27 were used to detect Myc–β-TrCP1, endogenous β-TrCP/Myc–β-TrCP1, AUF1, and endogenous Hsp27/FLAG-Hsp27, respectively. (C) Overexpression of β-TrCP1 accelerates degradation of AUF1. HeLa/Hsp27DDD cells were transfected with a plasmid expressing wild-type or mutant Myc–β-TrCP1 (0.8 μg, the maximum amounts used for panel A), as indicated at the top. After 2 days, cells were cultured with cycloheximide for protein stability assays by Western blotting analyses with antibodies to AUF1 and Hsp70 (as a control). (D) Hsp27DDD and β-TrCP1 promote AUF1 degradation by proteasomes. HeLa/Hsp27DDD cells were transfected with a plasmid expressing wild-type Myc–β-TrCP1 (0.8 μg, the maximum amount used for panel A). Lactacystin was either omitted or added to the indicated culture 24 h after transfection. Western blotting was performed as described for panel A.
The F box is the substrate recognition motif of β-TrCP. To determine whether this motif is critical for regulation of AUF1 stability, an F-box deletion mutant was tested. Various amounts of plasmids expressing Myc-tagged wild-type or mutant ΔF-box β-TrCP1 were transfected into HeLa/Hsp27DDD cells. Again, the levels of Myc–β-TrCP1 proteins reflected the amounts of transfected expression plasmids (Fig. 3B, top panel; ΔF-box β-TrCP migrates faster than the wild-type protein). Western blot analysis with antibody against β-TrCP allowed comparisons between the abundance of the wild-type/mutant Myc–β-TrCP1 proteins and the endogenous β-TrCP protein (Fig. 3B, second panel). While wild-type Myc–β-TrCP1 reduced abundance of AUF1, the deletion mutant did not (Fig. 3B, third panel). Thus, the F-box motif of β-TrCP1 is essential to reduce AUF1 abundance in cells expressing Hsp27DDD. That the effect is on protein degradation rates was confirmed by performing cycloheximide decay assays of AUF1 in HeLa/Hsp27DDD cells transfected with the plasmids expressing wild-type versus mutant Myc–β-TrCP1. In this case, cells were transfected with the maximum amount of plasmid (0.8 μg) tested for Fig. 3B. As expected, the abundance of AUF1 at time zero was lower (∼4-fold) in cells expressing wild-type Myc–β-TrCP1 than in the mutant-expressing cells (Fig. 3C). While the AUF1 half-life was 3.9 ± 0.2 h in cells expressing mutant Myc–β-TrCP1 (Fig. 3C, right panel), its half-life was reduced ∼5-fold, to 0.70 ± 0.06 h, in cells expressing wild-type Myc–β-TrCP1 (Fig. 3C, left panel). The 5-fold decrease in the AUF1 half-life is comparable to the 4-fold decrease in AUF1 abundance upon overexpression of wild-type Myc–β-TrCP1. Thus, accelerated degradation of AUF1 is sufficient to account for the decrease in its abundance upon overexpression of wild-type β-TrCP1.
To assess whether reduced abundance of AUF1 in cells expressing Hsp27DDD and β-TrCP1 required proteasomes, HeLa/Hsp27DDD cells were transfected with the maximal amount of expression plasmid tested (0.8 μg Myc–β-TrCP1, per Fig. 3B), and cells were cultured with or without the proteasome inhibitor lactacystin. Abundance of AUF1 was examined by Western blotting. Compared to results for cells not cultured with lactacystin, abundance of AUF1 increased in cells cultured with lactacystin (Fig. 3D, compare lane 2 with lane 1). Taken together, the results shown in Fig. 3 indicated that Hsp27DDD and β-TrCP1 promote AUF1 degradation by proteasomes.
Effects of β-TrCP on a phosphomimetic mutant form of AUF1.
Our previous studies showed that Ser83 and Ser87 in p40AUF1 are phosphorylated in cells (33, 34). As noted above, β-TrCP requires two properly spaced, phosphorylated serines within the recognition/binding sequence of its target proteins. We thus assessed whether Hsp27DDD- and β-TrCP1-induced degradation of p40AUF1 requires Ser83 and Ser87 phosphorylation of p40AUF1. Both serines were replaced with aspartic acid or alanine in a p40AUF1 cDNA that was mutated to make them refractory to an shRNA that targets all AUF1 isoforms. The expression plasmids thus encode shRNA-resistant wild-type (p40R), phosphomimetic [p40R(D)], and phosphor-resistant [p40R(A)] forms of p40AUF1. Depletion of endogenous AUF1 was necessary to permit Western blot detection of ectopically expressed p40AUF1 proteins lacking an epitope tag. (Pilot experiments revealed that an N-terminal FLAG tag on p40AUF1 blocked its degradation for reasons that are unknown.) Thus, shRNA plasmids were transfected in all experiments involving ectopic expression of p40AUF1 proteins.
To assess p40AUF1 protein stability, HeLa/Hsp27DDD cells were transfected with three sets of plasmids: (i) plasmid expressing control (shCTRL) or AUF1-directed shRNA (shAUF1); (ii) cells transfected with the shAUF1 plasmid were cotransfected with either p40R (wild-type), p40R(D) (phosphomimetic), or p40R(A) (phosphor-resistant) expression plasmids; and (iii) either a wild-type or mutant Myc–β-TrCP1 plasmid. Levels of the p40AUF1 proteins were determined by Western blotting with anti-AUF1 antibody. The wild-type and phosphomimetic p40AUF1 proteins were near background levels in cells expressing wild-type β-TrCP1 (Fig. 4A, upper panel, compare lanes 5 and 7 with lane 4) but were elevated in cells expressing mutant β-TrCP1 (Fig. 4A, upper panel, compare lane 6 with lane 5 and lane 8 with lane 7). In contrast, expression of wild-type β-TrCP1 did not reduce the level of phosphor-resistant p40AUF1 (Fig. 4A, upper panel, compare lane 9 with lanes 5 and 7). Reduced levels of wild-type and phosphomimetic p40AUF1 were not observed when cells were cultured with lactacystin (Fig. 4B, upper panel, compare lane 6 with lane 5 and lane 8 with lane 7). Collectively, these results suggested that degradation of p40AUF1 by proteasomes requires Ser83 and Ser87 phosphorylation.
Fig 4.

Degradation of p40AUF1 requires its phosphorylation. (A) HeLa/Hsp27DDD cells were transfected with plasmids encoding shAUF1, shAUF1-refractory wild-type p40 (p40R), phosphomimetic p40 [p40R(D)], or phosphor-resistant p40 [p40R(A)], and either wild-type or mutant Myc–β-TrCP1, as indicated. Two days after transfection, Western blotting was performed using antibodies against AUF1 and the Myc tag. To assess the efficiency of endogenous AUF1 knockdown, two cultures were transfected solely with plasmids expressing either shCTRL or shAUF1. AUF1 signal in an undiluted extract of shAUF1-expressing cells (lane 4) was compared to those from serial, 2-fold-diluted extract of cells expressing shCTRL (lanes 1 to 3). (B) HeLa/Hsp27DDD cells were transfected with plasmids encoding shAUF1, shAUF1-refractory wild-type p40 (p40R), phosphomimetic p40 [p40R(D)], or phosphor-resistant p40 [p40R(A)], and wild-type Myc–β-TrCP1, as indicated. Cells were cultured with or without lactacystin 24 h posttransfection, as noted. Western blotting was performed with antibodies against AUF1 and the Myc tag.
β-TrCP promotes p40AUF1 ubiquitination.
Ubiquitination of p40AUF1 by β-TrCP1 was next assessed. HeLa/Hsp27DDD and HeLa/Hsp27AAA cells were cotransfected with plasmids expressing shAUF1, p40R (wild-type) or p40R(D) (phosphomimetic), HA-tagged ubiquitin, and wild-type or mutant β-TrCP1. To observe ubiquitination, cells were cultured with lactacystin to block protein degradation. Cell extracts were prepared for immunoprecipitation with anti-AUF1 antibody. Precipitates were fractionated by SDS-PAGE, and ubiquitinated p40AUF1 was detected by Western blotting with anti-HA antibody. In HeLa/Hsp27DDD cells, overexpression of wild-type β-TrCP1 led to ubiquitination of both wild-type and phosphomimetic p40AUF1, while the ΔF-box mutant did not (Fig. 5A, upper panel, compare lane 2 with lane 3 and lane 4 with lane 5). (Compared to the size markers, the lowest band is likely monoubiquitinated p40AUF1.) No ubiquitination of p40AUF1 was observed in HeLa/Hsp27AAA cells (Fig. 5A, upper panel, lanes 6 to 9).
Fig 5.

β-TrCP promotes ubiquitination of p40AUF1. HeLa/Hsp27DDD and HeLa/Hsp27AAA cells were transfected with plasmids expressing shAUF1, HA-ubiquitin (HA-Ub), wild-type or ΔF-box mutant Myc–β-TrCP1, and wild-type p40R or phosphomimetic p40R(D), as indicated. Lactacystin was added to cultures 24 h posttransfection. (A) Cell extracts were immunoprecipitated with anti-AUF1 antibody and analyzed by Western blotting with anti-HA antibody to detect ubiquitinated p40AUF1 (upper panel). The positions of molecular mass markers are noted on the left. In the lower panel, aliquots of the same cell extracts were analyzed by Western blotting with anti-AUF1 antibody. (B) Cells were transfected as described for panel A and cultured with lactacystin. Cell extracts were immunoprecipitated with anti-AUF1 antibody and then analyzed by Western blotting with anti-AUF1 antibody. (C) As a control, cells were transfected as described for lane 2 of panel A. Cell extracts were immunoprecipitated with either nonimmune or AUF1 antiserum and then analyzed by Western blotting with AUF1 antibody.
Three controls were performed. (i) Western blot analysis of ectopically expressed p40AUF1 proteins in extracts from the above-described experiment indicated roughly equivalent levels (Fig. 5A, bottom panel, lanes 2 to 9). Thus, failure to detect ubiquitinated forms (in Fig. 5A, lanes 3 and 5 to 9) was not due to an absence of the p40AUF1 proteins. (ii) The transfection assay of ubiquitination described above was repeated, except that Western blots of anti-AUF1 immunoprecipitates were probed with anti-AUF1 antibody rather than anti-HA antibody. Similar to the results shown in Fig. 5A, higher-molecular-mass forms of wild-type and phosphomimetic p40AUF1 were detected in transfections with wild-type but not mutant β-TrCP1 (Fig. 5B, compare lane 3 with lane 4 and lane 5 with lane 6), consistent with their ubiquitination. Even in lanes without apparent ubiquitination, nonubiquitinated p40AUF1 proteins were observed (Fig. 5B, lanes 4 and 6 to 10). Thus, the failure to detect ubiquitinated forms was not due to an absence of p40AUF1 proteins in immunoprecipitates. We note that the pattern of high-molecular-mass bands (i.e., ubiquitinated proteins) is somewhat different for those detected with the HA antibody versus detection with the AUF1 antibody (compare Fig. 5A and B). We believe the most likely explanation is that for reasons that are unclear, the AUF1 antibody (a rabbit polyclonal) recognizes only certain forms of ubiquitinated p40AUF1. (iii) As a final control, to confirm specificity of the anti-AUF1 antibody, immunoprecipitations were performed with nonimmune serum and AUF1 antiserum. HeLa/Hsp27DDD cells were transfected with plasmids expressing p40R, shAUF1, HA-Ub, and wild-type β-TrCP1 and then cultured with lactacystin. Extracts were prepared, and immunoprecipitations were performed in parallel with either nonimmune rabbit serum or AUF1 antiserum. Immune precipitates were analyzed by Western blotting with anti-AUF1 antibody. Antibody to AUF1 precipitated ubiquitinated p40AUF1, while nonimmune serum did not, as expected (Fig. 5C, compare lane 3 with lane 2). Thus, the immunoprecipitation conditions employed are highly specific for AUF1 and its ubiquitinated forms.
To substantiate the hypothesis that phosphorylation of p40AUF1 is required for its ubiquitination and degradation, the experiments of Fig. 5A and B were repeated with HeLa/Hsp27DDD cells and transfections that included phosphor-resistant p40R(A). Again, immunoprecipitations with anti-AUF1 and Western blot analyses with either anti-HA antibody or anti-AUF1 antibody revealed that wild-type β-TrCP1 increased ubiquitination of wild-type and phosphomimetic p40AUF1 but not phosphor-resistant p40AUF1 (Fig. 6A, upper panel, compare lane 7 with lanes 3 and 5; Fig. 6B, compare lane 8 with lanes 4 and 6). Taken together, the results shown in Fig. 5 and 6 indicated that ubiquitination of p40AUF1 requires Hsp27DDD, β-TrCP1, and phosphorylation/phosphomimetic replacement of Ser83 and Ser87.
Fig 6.

Phosphorylation-deficient p40AUF1 is not ubiquitinated by β-TrCP. HeLa/Hsp27DDD cells were transfected with plasmids expressing shAUF1, HA-ubiquitin, wild-type or ΔF-box mutant Myc–β-TrCP1, and wild-type p40R, phosphomimetic p40R(D), or phosphor-resistant p40R(A), as indicated. Lactacystin was added to cultures 24 h posttransfection. (A) Cell extracts were immunoprecipitated with anti-AUF1 antibody and analyzed by Western blotting with anti-HA antibody to detect ubiquitinated p40AUF1 (upper panel). The positions of molecular mass markers are noted on the left. In the lower panel, the same cell extracts were analyzed by Western blotting with anti-AUF1 antibody. (B) Cells were transfected as described for panel A and cultured with lactacystin. Cell extracts were immunoprecipitated with anti-AUF1 antibody and then analyzed by Western blotting with anti-AUF1 antibody.
Interactions between p40AUF1, Hsp27, and β-TrCP1 require phosphomimetic substitutions.
The results so far indicate that β-TrCP and phosphomimetic mutant forms of both Hsp27 and p40AUF1 promote ubiquitination and degradation of p40AUF1. The question is, are those the same requirements for the proteins to form complexes in cells? HeLa/Hsp27DDD and HeLa/Hsp27AAA cells were transfected with plasmids expressing wild-type Myc–β-TrCP1 and shAUF1, together with a wild-type (p40R), phosphomimetic [p40R(D)], or phosphor-resistant [p40R(A)] form of p40AUF1. After 24 h, cells were cultured with lactacystin to block AUF1 degradation. For all experiments, cell extracts were prepared and aliquots were examined by Western blotting to confirm expression of all proteins (Fig. 7A to C, top three panels); extracts were immunoprecipitated with AUF1 antibody, and coprecipitated proteins were identified by Western blotting with appropriate antibodies (Fig. 7A to C, bottom three panels). Coimmunoprecipitation was observed for Myc–β-TrCP1 and Hsp27DDD with both wild-type and phosphomimetic p40AUF1 but not with phosphor-refractory p40AUF1 (Fig. 7A, bottom three panels, compare lanes 1 and 2 with lane 3). The observed interactions were likely not due to RNA bridging, since RNase A digestions of extracts prior to immunoprecipitations had no effects on interactions (Fig. 7B, lower three panels). In cells expressing Hsp27AAA, there was no coimmunoprecipitation of any p40AUF1 proteins with either Hsp27AAA or Myc–β-TrCP1 (Fig. 7A, bottom panels, lanes 4 to 6). Finally, immunoprecipitation of AUF1 with extracts from HeLa/Hsp27DDD cells expressing mutant β-TrCP1 did not coprecipitate either Hsp27DDD or the β-TrCP1 mutant (Fig. 7C, bottom three panels; compare lanes 1 and 3 with lanes 2 and 4). Taken together, these results indicated that association of p40AUF1, Hsp27, and β-TrCP1 requires the F box of β-TrCP and phosphomimetic substitution forms of both p40AUF1 and Hsp27. Since these are the same requirements for ubiquitination of p40AUF1 demonstrated earlier, ubiquitination likely requires association of phosphorylated p40AUF1 with multiprotein complexes composed of the E3 ubiquitin ligase SCFβ-TrCP and phosphorylated Hsp27.
Fig 7.
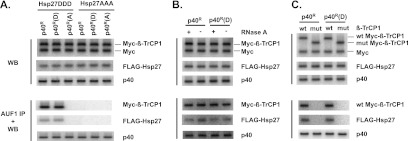
Phosphomimetic p40AUF1 and Hsp27 form complexes with β-TrCP1 in cells. (A) HeLa/Hsp27DDD and HeLa/Hsp27AAA cells were transfected with plasmids expressing shAUF1, wild-type Myc–β-TrCP1, and wild-type p40R, phosphomimetic p40R(D), or phosphor-resistant p40R(A), as indicated. Lactacystin was added to cultures 24 h posttransfection. Cell extracts were prepared. Western blots (WB) confirmed expression of each protein indicated on the right (top three panels). Extracts were immunoprecipitated with anti-AUF1 antibody, and the copurifying proteins were detected by Western blotting with anti-Myc, anti-FLAG, and anti-AUF1 antibodies, as indicated on the right (AUF1 IP + WB, bottom three panels). (B) Extracts of HeLa/Hsp27DDD cells expressing wild-type Myc–β-TrCP1 and either p40R or p40R(D) were treated or mock treated with RNase A prior to immunoprecipitation of AUF1. Proteins indicated to the right were detected by Western blotting of crude extracts (top three panels) and pellets following immunoprecipitation of AUF1 (bottom three panels). (C) Extracts from HeLa/Hsp27DDD cells expressing either wild-type or mutant Myc–β-TrCP1 and either p40R or p40R(D) were analyzed by Western blotting of crude extracts (top three panels) and pellets following immunoprecipitation of AUF1 (bottom three panels).
Overexpression of β-TrCP1 stabilizes reporter ARE-mRNAs.
The effects of Hsp27DDD and β-TrCP1 on ARE-mRNA decay were assessed. Reporter mRNAs containing the AREs from TNF-α and COX2 (cyclooxygenase 2 gene) mRNAs were examined, since mRNP immunoprecipitation assays with antibody to AUF1 showed that it binds these ARE-mRNAs in cells (11, 38). Tet-responsive reporter plasmids containing either an unmodified rabbit β-globin gene (Rβ-wt) or β-globin linked to the TNF-α or COX2 ARE in the 3′ UTR (Rβ–TNF-α or Rβ-COX2) were cotransfected into HeLa/Hsp27DDD cells expressing either wild-type or mutant β-TrCP1 together with a plasmid encoding a fusion protein of the Tet repressor DNA-binding domain and VP16 transactivation domain to permit reporter gene transcription (32). Two days after transfection, doxycycline was added to cells to inhibit reporter gene transcription, and RNA abundance was analyzed for each time point. In HeLa/Hsp27DDD cells expressing mutant β-TrCP1 (which does not affect AUF1 abundance), Rβ–TNF-α mRNA was relatively unstable, with a half-life of 0.46 ± 0.03 h; in contrast, the mRNA half-life increased 5-fold to 2.3 ± 0.2 h in cells expressing wild-type β-TrCP1 (Fig. 8A) (P = 0.0002). Likewise, Rβ-COX2 mRNA was relatively unstable, with a half-life of 0.50 ± 0.03 h in cells expressing mutant β-TrCP1, but the half-life increased almost 3-fold, to 1.4 ± 0.2 h, in cells expressing wild-type β-TrCP1 (Fig. 8C) (P = 0.0015). Thus, overexpression of β-TrCP1 in cells expressing Hsp27DDD is sufficient to promote stabilization of the reporter ARE-mRNAs tested.
Fig 8.

Effects of β-TrCP1 and Hsp27DDD on decay of reporter ARE-mRNAs. (A and C) Overexpression of β-TrCP1 in HeLa/Hsp27DDD cells stabilizes reporter ARE-mRNAs. Tetracycline-responsive plasmids expressing the rabbit β-globin gene (Rβ) containing the TNF-α (A) or COX2 (C) ARE in the 3′ UTR were cotransfected into HeLa/Hsp27DDD cells with the plasmid pTet-Off, expressing the Tet transactivator protein, plasmid pEGFP-C2 (an internal control), and a plasmid expressing either wild-type or mutant Myc–β-TrCP1. After 48 h, doxycycline (Dox) was added to block transcription of the reporter genes. Levels of Rβ-ARE reporter mRNAs were measured by qRT-PCR, normalized to that of enhanced green fluorescent protein (EGFP) mRNA, and plotted as percent reporter mRNA remaining versus time following doxycycline (Dox) treatment. Each time point represents the mean result ± SD from 3 independent experiments. Nonlinear regression analyses yielded first-order decay constants (k) and associated mRNA half-lives. Average half-lives are given in the text. (B and D) AUF1 knockdown stabilizes reporter ARE-mRNAs. HeLa/Hsp27DDD cells were cotransfected with Rβ–TNF-α (B) or Rβ-COX2 (D) reporter plasmids, pTet-Off, pEGFP-C2, mutant Myc–β-TrCP1, and a plasmid expressing either shCTRL or shAUF1. Dox time course assays were performed as described above to determine average half-life values, given in the text. (E) Overexpression of β-TrCP1 has no effect on reporter mRNA lacking AREs. HeLa/Hsp27DDD cells were cotransfected with the wild-type β-globin reporter gene (Rβ-wt), which lacks AREs, pTet-Off, pEGFP-E2, and either wild-type or mutant β-TrCP1. Dox time course assays were performed as described above to determine average half-life values, given in the text.
Since, as expected, cells expressing mutant Myc–β-TrCP1 (Fig. 8A and C) present rapid reporter mRNA decay kinetics, the effects of AUF1 knockdown on mRNA decay were examined next in these cells. HeLa/Hsp27DDD cells were cotransfected with a mutant β-TrCP1 plasmid, plasmids expressing shCTRL or shAUF1, and reporter ARE-mRNA plasmids. Rβ–TNF-α reporter mRNA decayed rapidly, with a half-life of 0.46 ± 0.02 h, in shCTRL-expressing cells, and AUF1 knockdown increased the mRNA half-life more than 4-fold, to 2.1 ± 0.1 h (Fig. 8B) (P = 0.0002). Likewise, Rβ-COX2 mRNA decayed with a half-life of 0.5 ± 0.1 h in shCTRL-expressing cells, and AUF1 knockdown increased the mRNA half-life 3-fold, to 1.5 ± 0.1 h (Fig. 8D) (P = 0.0003). Thus, AUF1 is necessary for rapid decay of reporter ARE-mRNAs. As a specificity control for the effects of β-TrCP1 overexpression on mRNA degradation, decay of Rβ-wt reporter mRNA (which lacks AREs) was examined in HeLa/Hsp27DDD cells expressing either wild-type or mutant β-TrCP1. The reporter mRNA half-lives of 3.3 ± 0.2 h and 3.0 ± 0.2 h for cells expressing wild-type and mutant β-TrCP1, respectively, were not statistically different (Fig. 8E) (P = 0.14). This control experiment suggests that the effect of β-TrCP1 overexpression on decay rates of ARE-mRNAs is not universal for all mRNAs. Collectively, the mRNA decay experiments suggested that AUF1 is necessary for decay of the reporter ARE-mRNAs tested and that β-TrCP may control ARE-mRNA degradation in part by determining AUF1 abundance (see Discussion).
DISCUSSION
As noted earlier, IL-1 activates p38 MAPK signaling and leads to a cascade of MK2 and Hsp27 phosphorylation that cause stabilization of ARE-mRNAs (25). Our recent studies demonstrated that phosphorylation of Hsp27 enhances AUF1 degradation by proteasomes to reduce its abundance and stabilize ARE-mRNAs in monocytes (7, 17). Also, replacement of Ser15, Ser78, and Ser82 in Hsp27 with aspartic acid (Hsp27DDD) but not alanines (Hsp27AAA) led to increased turnover of AUF1 and stabilization of ARE-mRNAs encoding the proinflammatory cytokines TNF-α and IL-1β. In this study, we investigated the mechanism by which phosphorylated Hsp27 reduces AUF1 abundance by protein degradation. Collectively, our results show that a substrate-binding subunit of the SCF E3 ubiquitin ligase, β-TrCP, controls ubiquitination and degradation of AUF1. Phosphorylation of Hsp27 is essential for β-TrCP-mediated degradation of AUF1, since the level of AUF1 declined significantly in HeLa cells overexpressing phosphomimetic Hsp27 and wild-type β-TrCP1 but not in cells expressing Hsp27AAA (Fig. 3). Furthermore, β-TrCP1-mediated ubiquitination and degradation required phosphomimetic replacement of Ser83 and Ser87 in p40AUF1 (Fig. 4 to 6). Consistent with these results, phosphomimetically mutated p40AUF1 and Hsp27 formed complexes in HeLa cells (by coimmunoprecipitation) with wild-type but not mutant β-TrCP1 (Fig. 7C). Phosphomimetic substitution in both Hsp27 and p40AUF1 was absolutely essential, since alanine substitution in either protein abolished ternary interactions between Hsp27, AUF1, and β-TrCP1 (Fig. 7A). Functionally, overexpression of β-TrCP1 reduced AUF1 abundance and stabilized reporter mRNAs harboring the TNF-α or COX2 AREs (Fig. 8). Thus, β-TrCP and Hsp27 together play key roles in regulating AUF1 abundance and stability of ARE-mRNAs.
Our cumulative findings lead us to propose a mechanism by which IL-1-induced p38 MAPK signaling controls ARE-mRNA decay in the systems we have employed. While AUF1 and Hsp27 normally act to promote degradation of ARE-mRNAs such as those of TNF-α and IL-1β (7), p38 activation phosphorylates Hsp27. (The effects of p38 signaling on AUF1 phosphorylation await further investigation.) Phospho-Hsp27 serves to facilitate interaction of phospho-p40AUF1, and perhaps the other AUF1 isoforms, with the β-TrCP subunit of the SCFβ-TrCP E3 ubiquitin ligase. The ligase complex ubiquitinates p40AUF1 and targets it, together with the other AUF1 isoforms, for codegradation by proteasomes. The resulting reduction in AUF1 abundance contributes to ARE-mRNA stabilization. We note that ectopic expression of alanine-substituted p40AUF1 in HeLa/Hsp27DDD cells overexpressing β-TrCP1 (which stabilizes reporter ARE-mRNAs) did not restore rapid decay of reporter ARE-mRNAs (data not shown). However, microRNAs and additional mRNP-associated proteins contribute to degradation of TNF-α and COX2 ARE-mRNAs as well. Proteins include CUGBP1/2 (CUG-binding proteins 1/2), HuR (human antigen R), RBM3 (RNA binding motif protein 3), TIA-1 (T cell intracellular antigen 1), and TTP (tristetraprolin) (39–46), and in some cases, AUF1 acts as a cofactor with additional ARE-binding proteins to control mRNA degradation (1, 11, 47). Nonetheless, AUF1 is essential for rapid decay of these ARE-mRNAs, since its knockdown with shRNA was sufficient to stabilize the mRNAs 3- to 5-fold (Fig. 8B and D). Future work will examine to what extent β-TrCP affects abundance of the aforementioned ARE-binding proteins and AUF1 cofactors.
The sequence encompassing Ser83 and Ser87 in p40AUF1 likely forms the β-TrCP recognition motif, although the sequence differs from the canonical β-TrCP recognition/binding motif, DSpGSX2–5Sp (48). Even so, numerous β-TrCP substrates all lack canonical consensus sequences (28, 29, 49, 50), suggesting that β-TrCP is capable of recognizing sequence variations (Fig. 2A). Notably, one or both serines in the canonical motif are correspondingly either aspartic acid or glutamic acid (single letter codes D and E, respectively) in the expanded binding motifs. These acidic amino acids can act as phosphomimetics for phosphoserine. However, proteins with aspartic acid and/or glutamic acid at the critical positions would not be subject to regulated phosphorylation within the β-TrCP binding motif. It is also possible that Hsp27DDD aids recognition/binding of β-TrCP to phosphorylated p40AUF1. Consistent with this hypothesis is the observation that Myc–β-TrCP1 coimmunoprecipitated with p40AUF1 from extracts of cells expressing Hsp27DDD but not those expressing Hsp27AAA (Fig. 7A). To our knowledge, there is no precedent for Hsp27 acting as a cofactor for β-TrCP recognition/binding of its substrates. (A search of PubMed for the dual search terms Hsp27 and TrCP did not yield any results.) In any event, how β-TrCP recognizes p40AUF1 (and perhaps p45AUF1) raises the question of how it acts together with Hsp27DDD to promote degradation of all the AUF1 isoforms, even though the p37/p42 isoforms lack the β-TrCP recognition motif (Fig. 1 to 3) (17). As we noted in earlier work (17), the most likely mechanism is by codegradation of AUF1 proteins associated together within a complex. The AUF1 isoforms can each interact with the others since they all possess an N-terminal dimerization domain (51). Indeed, they do interact with the others as heterocomplexes in cells during nucleocytoplasmic shuttling (52). There is a precedent for codegradation of all AUF1 isoforms: treatment of eosinophils with juglone, an immunosuppressive inhibitor of peptidyl-prolyl isomerases, induces AUF1 degradation (53). There is a precedent for codegradation of other proteins as well, such as the c-Fos oncoprotein during targeted degradation of c-Jun by proteasomes (17, 54).
β-TrCP recognition of p40AUF1 that is phosphorylated at Ser83 and Ser87 raises an intriguing conundrum: phosphorylation of these serines also affects disparate biochemical properties of p40AUF1 that appear unrelated to its degradation. For example, while phosphorylation has little effect on RNA-binding affinity, it abolishes the ability of p40AUF1 to form compact RNA structures; this may in turn affect local RNA presentation and accessibility of the mRNA to other proteins and microRNAs (11, 34, 55). Phosphorylation of these serines also induces cis-trans isomerization of the Ser83-Pro84 peptide bond by the peptidyl-prolyl isomerase Pin1 in T cells and eosinophils; isomerization promotes dissociation of AUF1 from the ARE-mRNA encoding granulocyte-macrophage colony-stimulating factor (GM-CSF) and stabilization of the mRNA (53, 56). Most likely, the pathway(s) taken to effect protein degradation versus RNA binding and/or RNA folding will be dictated by cell type, intracellular localization of AUF1, ancillary proteins that assemble with AUF1, or combinations of these.
In conclusion, we propose that activation of p38 MAPK-MK2 signaling phosphorylates Hsp27 to allow β-TrCP to bind and ubiquitinate phosphorylated p40AUF1 and perhaps other AUF1 isoforms. This leads to destruction of all AUF1 isoforms by proteasomes and stabilization of cytokine ARE-mRNAs. Future work will address how widespread the p38–MK2–Hsp27–β-TrCP axis acts to regulate abundance of AUF1 and other ARE-binding proteins to thereby control ARE-mRNA decay.
ACKNOWLEDGMENTS
We thank Tianyan Gao and Binhua Zhou for providing β-TrCP plasmids, Andy Clark for the β-globin/COX2-ARE plasmid, and Estela Jacinto for the FBXW8 siRNA and antibody. Very special thanks go to Alexei Kisselev for recommending β-TrCP during a visit by G.B. to Dartmouth Medical School.
This work was supported by NIH grant CA052443 to G.B.
Footnotes
Published ahead of print 25 March 2013
REFERENCES
- 1. Wu X, Brewer G. 2012. The regulation of mRNA stability in mammalian cells: 2.0. Gene 500:10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garneau NL, Wilusz J, Wilusz CJ. 2007. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8:113–126 [DOI] [PubMed] [Google Scholar]
- 3. Jacobsen A, Wen J, Marks DS, Krogh A. 2010. Signatures of RNA binding proteins globally coupled to effective microRNA target sites. Genome Res. 20:1010–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gratacos FM, Brewer G. 2010. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 1:457–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laroia G, Cuesta R, Brewer G, Schneider RJ. 1999. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science 284:499–502 [DOI] [PubMed] [Google Scholar]
- 6. Lu JY, Bergman N, Sadri N, Schneider RJ. 2006. Assembly of AUF1 with eIF4G-poly(A) binding protein complex suggests a translation function in AU-rich mRNA decay. RNA 12:883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sinsimer KS, Gratacos FM, Knapinska AM, Lu J, Krause CD, Wierzbowski AV, Maher LR, Scrudato S, Rivera YM, Gupta S, Turrin DK, De La Cruz MP, Pestka S, Brewer G. 2008. Chaperone Hsp27, a novel subunit of AUF1 protein complexes, functions in AU-rich element-mediated mRNA decay. Mol. Cell. Biol. 28:5223–5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. 2004. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 23:3092–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen CY, Gherzi R, Ong SE, Chan EL, Raijmakers R, Pruijn GJ, Stoecklin G, Moroni C, Mann M, Karin M. 2001. AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell 107:451–464 [DOI] [PubMed] [Google Scholar]
- 10. Lehner B, Sanderson CM. 2004. A protein interaction framework for human mRNA degradation. Genome Res. 14:1315–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu X, Chesoni S, Rondeau G, Tempesta C, Patel R, Charles S, Daginawala N, Zucconi BE, Kishor A, Xu G, Shi Y, Li ML, Irizarry-Barreto P, Welsh J, Wilson GM, Brewer G. 2013. Combinatorial mRNA binding by AUF1 and Argonaute 2 controls decay of selected target mRNAs. Nucleic Acids Res. 41:2644–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu JY, Sadri N, Schneider RJ. 2006. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 20:3174–3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sadri N, Schneider RJ. 2009. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J. Invest. Dermatol. 129:657–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sadri N, Lu JY, Badura ML, Schneider RJ. 2010. AUF1 is involved in splenic follicular B cell maintenance. BMC Immunol. 11:1 doi:10.1186/1471-2172-11-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sarkar S, Han J, Sinsimer KS, Liao B, Foster RL, Brewer G, Pestka S. 2011. RNA-binding protein AUF1 regulates lipopolysaccharide-induced IL10 expression by activating IkappaB kinase complex in monocytes. Mol. Cell. Biol. 31:602–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarkar S, Sinsimer KS, Foster RL, Brewer G, Pestka S. 2008. AUF1 isoform-specific regulation of anti-inflammatory IL10 expression in monocytes. J. Interferon Cytokine Res. 28:679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knapinska AM, Gratacos FM, Krause CD, Hernandez K, Jensen AG, Bradley JJ, Wu X, Pestka S, Brewer G. 2011. Chaperone Hsp27 modulates AUF1 proteolysis and AU-rich element-mediated mRNA degradation. Mol. Cell. Biol. 31:1419–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson GM, Sutphen K, Bolikal S, Chuang KY, Brewer G. 2001. Thermodynamics and kinetics of Hsp70 association with A + U-rich mRNA-destabilizing sequences. J. Biol. Chem. 276:44450–44456 [DOI] [PubMed] [Google Scholar]
- 19. Duttagupta R, Vasudevan S, Wilusz CJ, Peltz SW. 2003. A yeast homologue of Hsp70, Ssa1p, regulates turnover of the MFA2 transcript through its AU-rich 3′ untranslated region. Mol. Cell. Biol. 23:2623–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Henics T, Nagy E, Oh HJ, Csermely P, von Gabain A, Subjeck JR. 1999. Mammalian Hsp70 and Hsp110 proteins bind to RNA motifs involved in mRNA stability. J. Biol. Chem. 274:17318–17324 [DOI] [PubMed] [Google Scholar]
- 21. Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. 2006. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J. Biol. Chem. 281:37215–37226 [DOI] [PubMed] [Google Scholar]
- 22. Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, Kroemer G. 2006. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 5:2592–2601 [DOI] [PubMed] [Google Scholar]
- 23. Parcellier A, Brunet M, Schmitt E, Col E, Didelot C, Hammann A, Nakayama K, Nakayama KI, Khochbin S, Solary E, Garrido C. 2006. HSP27 favors ubiquitination and proteasomal degradation of p27Kip1 and helps S-phase re-entry in stressed cells. FASEB J. 20:1179–1181 [DOI] [PubMed] [Google Scholar]
- 24. Parcellier A, Schmitt E, Gurbuxani S, Seigneurin-Berny D, Pance A, Chantome A, Plenchette S, Khochbin S, Solary E, Garrido C. 2003. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol. Cell. Biol. 23:5790–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. 2000. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol. Cell. Biol. 20:4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuchs SY, Spiegelman VS, Kumar KG. 2004. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene 23:2028–2036 [DOI] [PubMed] [Google Scholar]
- 27. Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. 2003. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol. Cell 11:1445–1456 [DOI] [PubMed] [Google Scholar]
- 28. Li X, Liu J, Gao T. 2009. beta-TrCP-mediated ubiquitination and degradation of PHLPP1 are negatively regulated by Akt. Mol. Cell. Biol. 29:6192–6205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. 2006. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol. Cell. Biol. 26:4316–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. 2006. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314:467–471 [DOI] [PubMed] [Google Scholar]
- 31. Ysla RM, Wilson GM, Brewer G. 2008. Chapter 3. Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods Enzymol. 449:47–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gossen M, Bujard H. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. U. S. A. 89:5547–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G. 2003. Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J. Biol. Chem. 278:33029–33038 [DOI] [PubMed] [Google Scholar]
- 34. Wilson GM, Lu J, Sutphen K, Suarez Y, Sinha S, Brewer B, Villanueva-Feliciano EC, Ysla RM, Charles S, Brewer G. 2003. Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J. Biol. Chem. 278:33039–33048 [DOI] [PubMed] [Google Scholar]
- 35. Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, Wang R, Pan ZQ. 2008. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol. Cell 30:403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim SJ, DeStefano MA, Oh WJ, Wu CC, Vega-Cotto NM, Finlan M, Liu D, Su B, Jacinto E. 2012. mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Mol. Cell 48:875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ponyeam W, Hagen T. 2012. Characterization of the Cullin7 E3 ubiquitin ligase—heterodimerization of cullin substrate receptors as a novel mechanism to regulate cullin E3 ligase activity. Cell. Signal. 24:290–295 [DOI] [PubMed] [Google Scholar]
- 38. Kishor A, Tandukar B, Ly YV, Toth EA, Suarez Y, Brewer G, Wilson GM. 2013. Hsp70 is a novel posttranscriptional regulator of gene expression that binds and stabilizes selected mRNAs containing AU-rich elements. Mol. Cell. Biol. 33:71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, Hla T. 2003. The RNA-binding protein HuR regulates the expression of cyclooxygenase-2. J. Biol. Chem. 278:25227–25233 [DOI] [PubMed] [Google Scholar]
- 40. Zhang L, Lee JE, Wilusz J, Wilusz CJ. 2008. The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. J. Biol. Chem. 283:22457–22463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, Sternjohn JR, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, Bohjanen PR. 2008. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol. Cell 29:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee JE, Lee JY, Wilusz J, Tian B, Wilusz CJ. 2010. Systematic analysis of cis-elements in unstable mRNAs demonstrates that CUGBP1 is a key regulator of mRNA decay in muscle cells. PLoS One 5:e11201 doi:10.1371/journal.pone.0011201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. 2004. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. U. S. A. 101:2011–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cok SJ, Morrison AR. 2001. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J. Biol. Chem. 276:23179–23185 [DOI] [PubMed] [Google Scholar]
- 45. Lopez de Silanes I, Galban S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. 2005. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol. Cell. Biol. 25:9520–9531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin SC, Gram H, Han J. 2005. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 120:623–634 [DOI] [PubMed] [Google Scholar]
- 47. Chang N, Yi J, Guo G, Liu X, Shang Y, Tong T, Cui Q, Zhan M, Gorospe M, Wang W. 2010. HuR uses AUF1 as a cofactor to promote p16INK4 mRNA decay. Mol. Cell. Biol. 30:3875–3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shaik S, Nucera C, Inuzuka H, Gao D, Garnaas M, Frechette G, Harris L, Wan L, Fukushima H, Husain A, Nose V, Fadda G, Sadow PM, Goessling W, North T, Lawler J, Wei W. 2012. SCFbeta-TRCP suppresses angiogenesis and thyroid cancer cell migration by promoting ubiquitination and destruction of VEGF receptor 2. J. Exp. Med. 209:1289–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soond SM, Townsend PA, Barry SP, Knight RA, Latchman DS, Stephanou A. 2008. ERK and the F-box protein betaTRCP target STAT1 for degradation. J. Biol. Chem. 283:16077–16083 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50. van Kerkhof P, Putters J, Strous GJ. 2007. The ubiquitin ligase SCF(betaTrCP) regulates the degradation of the growth hormone receptor. J. Biol. Chem. 282:20475–20483 [DOI] [PubMed] [Google Scholar]
- 51. DeMaria CT, Sun Y, Long L, Wagner BJ, Brewer G. 1997. Structural determinants in AUF1 required for high affinity binding to A + U-rich elements. J. Biol. Chem. 272:27635–27643 [DOI] [PubMed] [Google Scholar]
- 52. Sarkar B, Lu JY, Schneider RJ. 2003. Nuclear import and export functions in the different isoforms of the AUF1/heterogeneous nuclear ribonucleoprotein protein family. J. Biol. Chem. 278:20700–20707 [DOI] [PubMed] [Google Scholar]
- 53. Shen ZJ, Esnault S, Malter JS. 2005. The peptidyl-prolyl isomerase Pin1 regulates the stability of granulocyte-macrophage colony-stimulating factor mRNA in activated eosinophils. Nat. Immunol. 6:1280–1287 [DOI] [PubMed] [Google Scholar]
- 54. Papavassiliou AG, Treier M, Chavrier C, Bohmann D. 1992. Targeted degradation of c-Fos, but not v-Fos, by a phosphorylation-dependent signal on c-Jun. Science 258:1941–1944 [DOI] [PubMed] [Google Scholar]
- 55. Zucconi BE, Ballin JD, Brewer BY, Ross CR, Huang J, Toth EA, Wilson GM. 2010. Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. J. Biol. Chem. 285:39127–39139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Esnault S, Shen ZJ, Whitesel E, Malter JS. 2006. The peptidyl-prolyl isomerase Pin1 regulates granulocyte-macrophage colony-stimulating factor mRNA stability in T lymphocytes. J. Immunol. 177:6999–7006 [DOI] [PubMed] [Google Scholar]