

Abstract
To better understand the binding mechanism of TRIM5α to retrovirus capsid, we had previously selected N-tropic murine leukemia virus (N-MLV) mutants escaping from rhesus macaque TRIM5α (rhTRIM5α) by passaging the virus in rhTRIM5α-expressing cells and selecting for nonrestricted variants. To test the commonality of the findings from the rhTRIM5α study, we have now employed a similar genetic approach using human TRIM5α (huTRIM5α). Consistent with the rhTRIM5α study, the mapped huTRIM5α escape mutations were distributed across the capsid exterior, confirming the extended binding surface between virus and restriction factor. Compared to the results of the previous study, fewer escape mutations were identified, with particular mutants being repeatedly selected. Three out four huTRIM5α escape variants showed resistance to all primate TRIM5αs tested, but two of them sacrificed viral fitness, observations that were not made in the rhTRIM5α study. Moreover, differences in amino acid changes associated with escape from hu- and rhTRIM5αs suggested a charge dependence of the restriction by different TRIM5αs. Taken together, these results suggest that the recognition of the entire capsid surface is a general strategy for TRIM5α to restrict MLV but that significantly different specific interactions are involved in the binding of TRIM5α from different species to the MLV capsid core.
INTRODUCTION
Capsid (CA)-binding retrovirus restriction factors have arisen on at least five occasions in the course of evolution. Their independent evolution is testament to the utility of such factors, either to limit endogenous retrovirus amplification or to inhibit infection by exogenous viruses (1–3). This group of factors includes Fv1 (4, 5), TRIM5α (6), and three independently arising hybrid factors, TRIM5CypA1 (7, 8), TRIM5CypA2 (9–12), and TRIM5CypA3 (13) in which a retrotransposed copy of the cellular cyclophilin-A gene has been inserted in the 3′ end of the TRIM5 gene. At a minimum, these factors contain a virus CA-binding domain, as well as one or more multimerization domains (14–22). Restriction factor engagement appears to occur soon after retroviral core release into the cell cytoplasm, resulting either in proteasome-mediated CA degradation and an inhibition of reverse transcription (6) or an apparent sequestration of the reverse-transcribed preintegration complex en route to the cell nucleus (23).
One of the least-appreciated aspects of restriction by these factors is the ability to restrict multiple species of retrovirus, a property that presumably reflects the ability of these factors to bind several different CA molecules. For example, TRIM5α from the cotton-top tamarin can restrict one or more members of the lentivirus, gammaretrovirus, betaretrovirus, and foamy virus genera (24–26), despite extensive differences in the sequences of their CA molecules. Nevertheless, restriction can be sensitive to a single amino acid change, as best characterized by the single change at CA position 110 of murine leukemia virus (MLV) that distinguishes between N-tropic MLV (N-MLV) and B-tropic MLV (27).
As one way to understanding the nature of the binding interaction between virus and restriction factor, we have taken a genetic approach, by isolating escape mutants from virus grown under restrictive conditions. We had previously characterized a series of MLV variants that lack or show reduced sensitivity to rhesus macaque TRIM5α (rhTRIM5α), revealing that changes all over the surface of CA could affect restriction (28). We now report analogous experiments with human TRIM5α (huTRIM5α), asking whether we see the same spectrum of escape mutations and how the recognition by huTRIM5α and rhTRIM5α compares.
MATERIALS AND METHODS
Cells.
Tail fibroblast cells from Mus dunni (MDTF), MDTF cells that stably express restriction factors, and NIH-3T3 cells, as well as human 293T and TE671 cells, were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and antibiotics. MDTF cells expressing human and rhesus macaque TRIM5α, Fv1b, and Fv1n (abbreviated MDHu, MDRh, MDFv1b, and MDFv1n, respectively) were established by an endpoint dilution method. Stable, unselected expression of TRIM5α was observed in transduced cells for at least 1 month after cloning.
DNA.
The construction of chimeric human/primate TRIM5αs (human RBCC domain fused with primate B30.2 domain) has been described previously (25, 29). Such chimeric TRIM5αs were used throughout this study to generate, select, and test escape mutants of N-MLV, thereby allowing a direct comparison of interactions with the human and primate B30.2 domains. The generation of transduction vectors expressing TRIM5α or Fv1 plus enhanced yellow fluorescent protein (eYFP) using the Gateway system was also described previously (25, 30). Single-nucleotide mutations were introduced into the capsid genes of Gag-Pol vector plasmids (pCIG3N and pCIG3B) and an N-MLV provirus plasmid (pWN41) (31) by the QuikChange mutagenesis PCR protocol. Random mutations were introduced at position 92 on the MLV capsid by QuikChange mutagenesis PCR using the following primer pair: forward, 5′-CACCCAACTGCCCAACNN(G/T)GTTGACGCTGCTTTTC-3′, and reverse, 5′-GAAAAGCAGCGTCAAC(C/A)NNGTTGGGCAGTTGGGTG-3′, where N indicates any nucleotide and G/T and C/A indicate G or T and C or A, respectively. The sequences of the other primers used to prepare mutants are available upon request. All introduced mutations were verified by DNA sequencing.
Viruses.
Single-cycle vectors were produced by transfection of vesicular stomatitis virus (VSV) G envelope (pcz-VSV-G), retroviral Gag-Pol (pCIG3N and pCIG3B for N- and B-tropic viruses, respectively), and viral genomic plasmids (pczCFG2fEGFPf) into 293T cells by a conventional calcium phosphate method or using TurboFect (Fermentas) as described previously (28, 30). To prepare virus pseudotyped with an ecotropic MLV Env, the VSV-G Env plasmid was replaced with pHIT123, which encodes the Env gene from Moloney MLV (32).
Stocks of replication-competent N-MLV derived in three different ways were used (stocks A, B, and C) (Fig. 1). Unpassaged virus (A) was recovered from pWN41 DNA transfection of HEK293T cells with pWN41 as described previously (28). Stocks A1 to -6 were derived from independent transfections. As plasmid pWN41 encodes an ecotropic AKV strain of MLV that cannot infect human cells, stock A viruses will not have undergone reverse transcription. Stock B viruses are derived from stock A viruses following various numbers of viral passage on MDTF cells; stocks B1 to -6 were obtained in one experiment, while B7 to -11 were from one independently developed permanent producer cell line. Stock C viruses are viruses that were isolated following the growth of stock B in MDRh cells and are resistant to restriction by rhTRIM5α. Virus production in newly transfected/infected cells was monitored by protein blotting or by measurement of reverse transcriptase (RT) activity.
Fig 1.

Schematic view of the preparation of stock viruses. Stock As represent the supernatants of 293T cells transfected with repermuted pWN41. Stock Bs were obtained by further passage of stock A virus on MDTF cells. Stock Cs were derived from stock A by selection for resistance to rhTRIM5α restriction.
Mutant virus selection.
MDTFs expressing huTRIM5α were used for selection of escape viruses using methods analogous to those previously used for isolation of rhTRIM5α escape mutations (28).
Fluorescence-activated cell sorting (FACS).
Virus spread in infected cells was monitored by detection of an intracellular virus antigen, p12, as previously described (28).
RT assay.
Reverse transcriptase activity was measured in cell supernatants using a commercial RT assay kit (CavidiTec, Sweden).
Sequencing of the capsid gene.
Capsid-encoding sequences from recovered viruses were determined as previously described (28).
Abrogation assay.
Saturation of restriction factors was performed as described previously (33), with minor modifications. Virus-like particles (VLP) were prepared by transfection as described previously (28), with the green fluorescent protein (GFP)-encoding virus genome plasmid replaced by a plasmid bearing the LacZ gene (pHIT111). The titers of VLP and GFP-encoding N-MLV were standardized by RT activity as described above. Human TE671 cells were preplated in a 12-well plate at a density of 1 × 104 cells per well and incubated at 37°C overnight. For saturating huTRIM5α in TE671 cells, cells were infected by 2-fold dilutions of VLP with wild-type (wt) N, B, or mutant N capsids at 37°C for 2 h in the presence of 10 μg/ml Polybrene, followed by infection with 20 mU RT activity of GFP-encoding N-MLV at 37°C for 48 h. GFP expression was detected using a FACS LSR II.
Core stability.
The stability of viral cores was examined using the previously described fate-of-capsid assay (28) to compare extracts from cells infected in the absence of restriction factor with mutant and wild-type virus. MDTF cells were infected with freshly harvested virus from transfected 293T cultures (see above), and extracts were prepared for analysis at different times after infection.
RESULTS
Comparison of escape from huTRIM5α and rhTRIM5α.
In our previous study (28), we took a genetic approach to investigate the interaction between rhTRIM5α and N-MLV CA protein by selecting and characterizing a series of escape mutants. That study revealed that the entire outer surface of the CA molecule in the viral core was involved in the binding interaction with rhTRIM5α. To investigate the generality of this conclusion, we took a similar approach to study the interaction of huTRIM5α with N-MLV. Three different protocols (A, B, and C) for virus stock preparation (see Materials and Methods) were employed that vary in the number of replication cycles following recovery from cloned DNA and presumably carry increasing numbers of mutations. In addition, the stock C viruses comprised several rhTRIM5α escape viruses that did not show altered growth on huTRIM5α (28), since we reasoned that such mutations might facilitate virus escape from huTRIM5α even if, by themselves, they did not alter restriction by huTRIM5α.
Since our previous study had shown efficient generation and selection of the N-MLV escape mutant from rhTRIM5α in mixed cell cultures (cells expressing the restriction factor cocultured with 10% nonexpressing cells to allow virus replication), we passaged N-MLV in such mixed cell cultures in our attempts to select escape mutants from huTRIM5α. Virus growth was monitored either by FACS detection of p12-positive cells among the eYFP-positive (i.e., huTRIM5α) fraction or by measuring RT activity in the culture supernatant. The results of these experiments are summarized in Table 1, with kinetics illustrated in Fig. 2. Comparison of the three types of virus preparation showed that stock C was the best source of escape virus, whereas stock A was the least efficient. Using stock A virus (A1 to A3), an escape mutant emerged in only one (stock A1) of three independent experiments (Table 1); in this single positive experiment, virus was first seen more than 50 days after culture initiation (Fig. 2A). Five of eight experiments with stock B gave escape virus on mixed cell cultures (Table 1, B1 to B8), with virus appearing after around 5 weeks in each case (Fig. 2B). Interestingly, it appears that multiple virus passages prior to selection facilitate the isolation of escape mutants (compare B1 to B3 with B4 to B7 in Table 1), presumably by allowing some diversification to occur. This effect of diversification may also have facilitated the escape mutant selection when stock B (B9 to B11) was passed directly in 100% huTRIM5α-expressing cells without rescue (Table 1). An escape mutant emerged in only one (B9) of three independent experiments under this experimental setting, with the escape variant appearing about 40 days after infection, followed by a steep decline in RT activity to an undetectable level (Fig. 2D). This was most likely due to Env-mediated cytopathic effects, resulting in cell death and considerably diminished virus production. In contrast, several rhTRIM5α escape mutations (stocks C) gave rise to viruses yielding very rapid and reproducible escape (Fig. 2C).
Table 1.
Isolation of escape mutants from human TRIM5αa
| Virus stock | Passage history of stock | Preexisting mutation | Passage schedule (% MDHu cells) | Mutation(s) recovered | Time of escape (days)b |
|---|---|---|---|---|---|
| A | |||||
| 1 | Unpassaged | 90 →100 | E92K | 55 | |
| 2 | Unpassaged | 90 →100 | No escape | ||
| 3 | Unpassaged | 90 →100 | No escape | ||
| 4 | Unpassaged | L10W | 90 →100 | L10W/E100K | 25 |
| 5 | Unpassaged | L10W | 90 →100 | L10W/E100K | 25 |
| 6 | Unpassaged | L10W | 90 →100 | L10W/E100K | 25 |
| B | |||||
| 1 | Passaged in MDTF 1 time | 90 →100 | No escape | ||
| 2 | Passaged in MDTF 2 times | 90 →100 | No escape | ||
| 3 | Passaged in MDTF 3 times | 90 →100 | No escape | ||
| 4 | Passaged in MDTF 4 times | 90 →100 | E100K | 35 | |
| 5 | Passaged in MDTF 5 times | 90 →100 | E100K | 35 | |
| 6 | Passaged in MDTF 6 times | 90 →100 | E100K | 35 | |
| 7 | Passaged in MDTF >10 times | 90 →100 | N7K/E211K | 35 | |
| 8 | Passaged in MDTF >10 times | 90 →100 | E100K | 35 | |
| 9 | Passaged in MDTF >10 times | 100 | E92K | 40 | |
| 10 | Passaged in MDTF >10 times | 100 | No escape | ||
| 11 | Passaged in MDTF >10 times | 100 | No Escape | ||
| C | |||||
| 1 | Selected by rhTRIM5α | L10W | 90 →100 | L10W/E100K | 15 |
| 2 | Selected by rhTRIM5α | L10W | 90 →100 | L10W/E100K | 15 |
| 3 | Selected by rhTRIM5α | G8D | 90 →100 | G8D/E92K | 25 |
| 4 | Selected by rhTRIM5α | N82D | 90 →100 | N82D/E100K | 15 |
| 5 | Selected by rhTRIM5α | N82D | 90 →100 | N82D/N113K | 15 |
| 6 | Selected by rhTRIM5α | H114D | 90 →100 | No escape |
Summary of data. Each line represents a different culture.
Time until the first increase of either the percentage of virus-infected cells or RT activity in the culture supernatant was observed.
Fig 2.
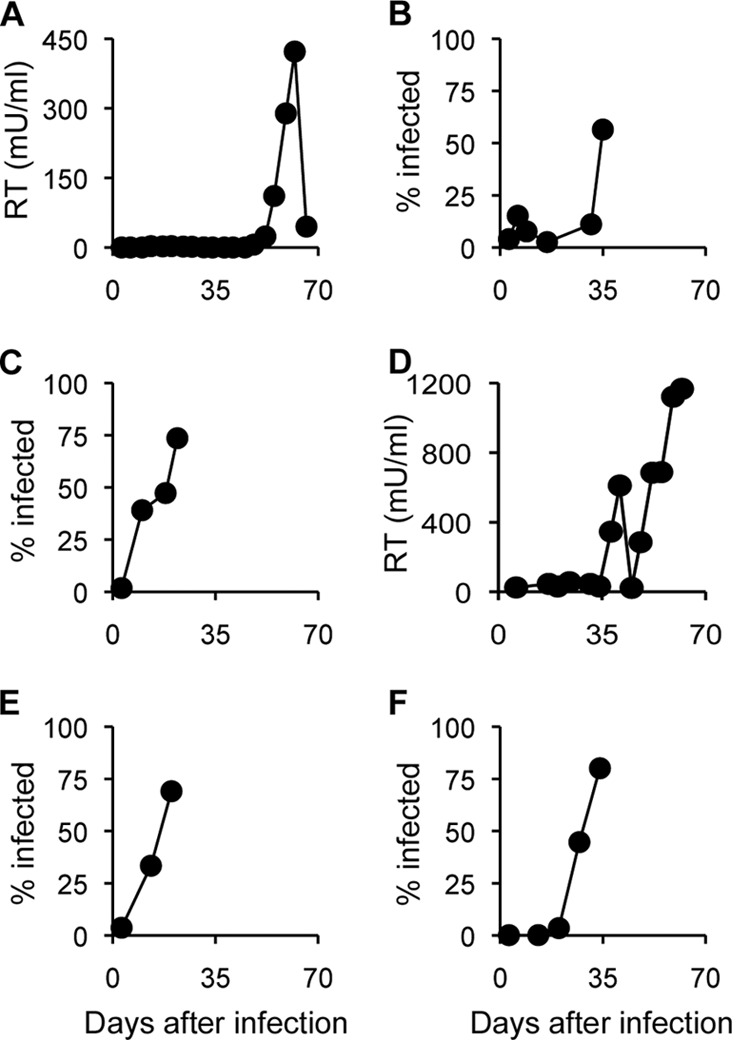
Selection of N-MLV escape mutants from human TRIM5α. Various cell populations were infected with three different preparations of virus and maintained for up to 10 weeks, with passaging every 3 days and virus production monitored by FACS or RT activity. (A) Passage of stock A1 virus in 90% MDHu cells. (B) Passage of stock B7 virus in 90% MDHu cells. (C) Passage of stock C1 virus in 90% MDHu cells. (D) Passage of stock B9 virus in 100% MDHu cells. (E) Passage of stock B7 virus in 90% MDRh cells. (F) Passage of stock B7 virus in 100% MDRh cells. Similar results were obtained from at least three independent experiments, each of which started with a stock virus prepared independently, and one representative result is shown.
Comparison of the growth of stock B virus in the presence of huTRIM5α (Fig. 2B and D) and rhTRIM5α (Fig. 2E and F) in selection cultures conducted in parallel showed a significant delay of the spread of the virus, by 2 to 3 weeks, in the presence of huTRIM5α. This occurred both in the mixed cell cultures (compare Fig. 2B and E) and in 100% resistant culture (Fig. 2D and F). In addition, in the mixed cell culture, virus escape was only observed in five of eight passage experiments in the presence of huTRIM5α (Table 1), while escape mutants emerged in all rhTRIM5α experiments (28). Taken together, these results imply that escape of N-MLV from huTRIM5α is, in some fashion, more difficult than from rhTRIM5α and suggest that the time required by the virus to spread in the first culture, which reflects the difficulty of escape, correlates with the strength of the restriction. In addition, they provide further evidence for the utility of mixed cell cultures in generating and selecting escape mutants even in the presence of a restriction factor with a strong restriction activity.
Characterization of huTRIM5α escape mutations.
To identify the sites in CA responsible for changes in restriction by huTRIM5α, the filtered virus-containing supernatants from the initial virus-positive cultures were passaged once more onto MDTF cells, all of which expressed huTRIM5α. Following the establishment of virus growth in the culture, viral genomic RNA was extracted from cell-free culture supernatant, viral cDNA was generated from viral RNA, and the whole capsid gene was sequenced. From the passage of stock C virus, in five independent experiments starting with three different rhTRIM5α escape mutations (G8D, L10W, and N82D, stocks C1 to -5), escape variants were isolated; the identified escape variants retained the preexisting mutations but additionally acquired an E100K, E92K, or N113K mutation (Table 1). In contrast, in the one experiment starting with virus carrying the H114D mutation (C6), no escape mutant was recovered. The growth of stock B virus in the mixed cell culture led to acquisition of the E100K mutant in four independent cultures (B4, -5, -6, and −8) (Table 1). On two further occasions, stock B virus acquired N7K/E211K or E92K changes. The E92K mutant also emerged once following passage of stock A virus (Table 1). As was the case with the escape mutations from rhTRIM5α, all of the escape mutations identified in this study resulted from single-nucleotide changes. These data suggest that only a limited number of mutations (N7K, E92K, E100K, and N113K) in N-MLV CA will lead to escape from huTRIM5α, with two mutants (E92K and E100K) being repeatedly selected. This contrasts with the selection of more than 10 different mutations by rhTRIM5α in our previous experiments (28), with fewer cases of the repeated selection of a particular mutant.
Whereas two independent passages of the N82D variant resulted in the selection of two different mutations (N82D/N113K and N82D/E100K), two independent passages of the L10W variant resulted in the selection of the same changes, L10W/E100K. This result implies a possibility of preferential acquisition of the E100K mutation by preexisting L10W CA. To test this possibility, the L10W mutation was introduced into pWN41 by PCR, and replication-competent N-MLV with the L10W mutation was prepared by a transient transfection of 293T cells. This virus was passaged in the mixed cell culture as described above. Strikingly, in three separate experiments (with independent preparations of the starting material, A4 to -6), the same mutant virus, L10W/E100K (Table 1), resulting from a single-nucleotide change (a G-to-A mutation at the first nucleotide of the codon that encodes E at amino acid 100 of CA), was isolated, with kinetics resembling those seen with stock C. These results suggest that the E100K mutation is a preferred means for N-MLV carrying the L10W change to escape huTRIM5α. Furthermore, the L10W mutation, even though it does not by itself give resistance to huTRIM5α (28), appears to act as an enabling mutation allowing rapid isolation of E100K.
Loss of fitness can accompany acquisition of escape.
To investigate the effect of these newly identified changes on virus growth and restriction, they were introduced into the N-MLV vector virus plasmid pCIG3N for further analysis. By comparison with wild-type N-MLV and using RT activity for normalization, viruses with either the N7K or E92K mutation showed greatly reduced infectivity on nonrestricting cells (Fig. 3, first column). However, on cells expressing huTRIM5α, these viruses, unlike the wild type, showed low but significant infectivity, indicating that these changes are responsible for resistance and reduced titers. Furthermore, virus derived from pWN41 by introduction of the N7K or E92K changes replicated in the presence of huTRIM5α (data not shown), confirming that for at least these two variants, escape mutations in CA conferred on the virus the ability to escape the restriction factor. The addition of the E211K change to N7K did not relieve the growth defect in MDTF cells or change the restriction properties (data not shown). The lower infectivity of N7K and E92K mutants does not seem to be caused by lower levels of Gag incorporation into virus released from the transfected cells, since comparable amounts of p30 viral proteins were detected by Western blot analyses of virus pellets (see Fig. 5A) and similar (N7K virus) or slightly reduced (E92K virus) levels of RT activity compared to that of wild-type virus were detected in the transfection culture supernatant measured (data not shown). In contrast, the E100K mutation showed only small reductions in infectivity of nonrestricting cells and gave substantial infection of huTRIM5α-expressing cells (Fig. 3). Virus carrying the N113K change showed an intermediate phenotype, with significant infection of MDTF cells and escape from huTRIM5α to a level comparable to that seen in virus with the E92K mutation. These data provide a good explanation for the frequency with which E100K was isolated in experiments with stocks A and B. However, the observation that viruses with infectivity levels significantly lower than those of E100K mutants (e.g., E92K mutants) were isolated implies (i) that the mutation rate is low (otherwise E100K would always grow out) and (ii) that escape mutations without loss of fitness are infrequent (otherwise E92K would not have appeared).
Fig 3.

Comparison of the ability of newly isolated mutants to grow with and without human TRIM5α. Enhanced-GFP-encoding vector viruses with or without escape mutations and normalized by RT activity were titrated on parental MDTF cells (open diamonds) or MDHu cells (filled squares). Similar results were obtained in two independent experiments, and the results for one are shown.
Fig 5.
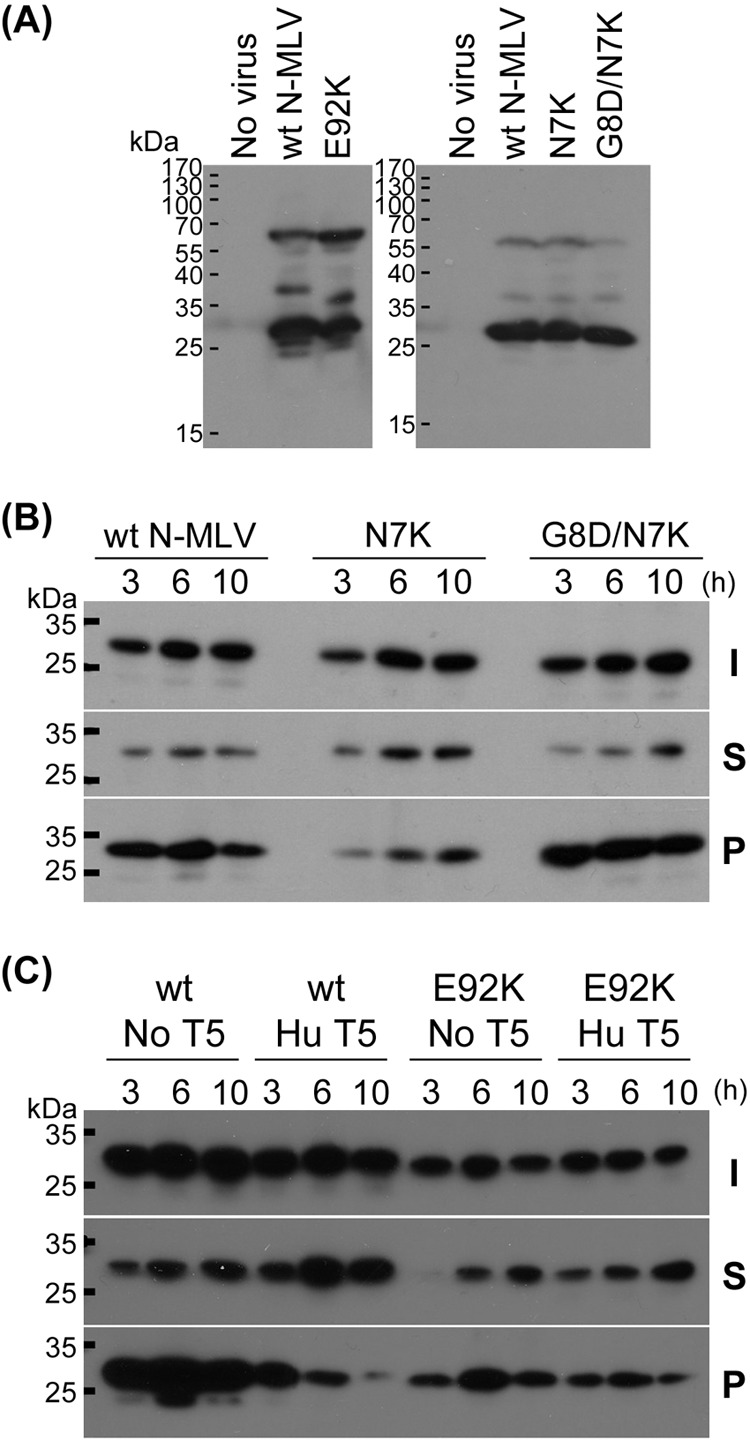
Staging N7K and E92K mutations. (A) Virus production was not significantly affected by N7K and E92K mutations. HEK293T cells were transfected with Gag-Pol-, Env-, and vector-encoding plasmids to produce the viruses, and the virus-containing supernatant was centrifuged to pellet the virus particles. p30CA was detected by Western blotting. (B and C) Fate-of-capsid assay was performed for wild-type N-MLV, N7K, G8D/N7K, and E92K viruses in the presence or absence of the restriction factor at three different time points (3, 6, and 10 h) after infection. T5, TRIM5α I, input; S, soluble fraction; P, pellet fraction.
The potential enabling effects of rhTRIM5α escape mutations (G8D, L10W, and N82D) upon huTRIM5α escape mutations (N7K, E92K, E100K, and N113K) suggested by the rapid isolation of escape mutants when starting with stock C viruses led us to test the effects of combinations of these changes upon growth in restricted and unrestricted cells in a systematic fashion. We prepared a series of N-MLV mutants with the three rhTRIM5α escape mutations plus one of the four huTRIM5α escape mutations (Fig. 3). The infectivity of G8D mutants in MDTF (Fig. 3, second column) was little affected by any of the huTRIM5α escape mutations. However, the G8D mutation improved the ability of escape resulting from the E92K and N113K mutations, whereas the G8D/E100K mutant showed a level of escape from huTRIM5α similar to that of the E100K mutant. In contrast, the G8D/N7K mutant was strongly restricted by huTRIM5α, showing the same phenotype as the G8D mutant. The L10W change (Fig. 3, third column) had little effect on the phenotypes of the single huTRIM5α escape mutants, with only a modest influence on restriction. However, the L10W/E100K mutant showed the highest infectivity among the L10W mutants in the presence of huTRIM5α. The N82D variants (Fig. 3, fourth column) display two different phenotypes; the N82D/N7K and N82D/E92K mutants showed low infectivity in the presence or absence of huTRIM5α, whereas the N82D/E100K and N82D/N113K mutants showed high infectivity in both MDTF and MDHu cells. The latter two mutants showed comparable infectivity in MDHu cells and in MDTF cells. These data confirm that rhTRIM5α-resistant mutants can facilitate the outgrowth of some but not all huTRIM5α escape mutants. One might predict that further escape mutants from huTRIM5α derived from the G8D stock would contain E100K and N113K.
Loss of binding to the newly identified escape mutations by huTRIM5α.
To examine whether the escape mutations affect the binding interaction between TRIM5α and capsid core, we employed an abrogation assay in which cells expressing a restriction factor endogenously are treated with increasing amounts of viruslike particles (VLP) containing the CA protein of interest, followed by the infection of a fixed amount of the virus with restricted CA encoding a reporter gene. If the restriction is abrogated by the VLP treatment, it implies a direct interaction of the VLP CA with the restriction factor. As expected, wt N-MLV VLP abrogated TRIM5α restriction in human cells, whereas nonrestricting B-MLV VLP did not abrogate the restriction (Fig. 4). Similar amounts (based on RT activity) of VLPs comprising CA with E92K, N7K, N82D/N113K, and L10W/E100K mutations also failed to abrogate the restriction of huTRIM5α (Fig. 4). If the loss of fitness by the N7K and E92K mutations observed above is an indication of poor core stability, loss of abrogation by these mutations may simply be due to the inability of TRIM5α to recognize a core with partially disassembled CA lattice. This may be the case for the N7K mutant, because extracts of cells made at three different time points (3, 6, and 10 h) after infection with similar amounts of viruses (13.1 U RT activity for wild-type virus compared with 12.6 U RT activity for the N7K virus) contained less high-molecular-mass CA that can be pelleted through a sucrose cushion than extracts of cells infected with wild-type virus (Fig. 5B). The amount of pelletable N7K CA was restored by the further introduction of the G8D mutation (Fig. 5B), most likely explaining the observation that the G8D mutation restored the infectivity of the N7K virus (Fig. 3). In contrast, E92K CA was readily detected in the pellet fraction at a level similar to that of the wild-type N-MLV CA (the input virus titers were 34.3 U RT activity for wild-type virus and 14.2 U RT activity for the E92K virus) at three different time points (Fig. 5C), arguing against significant differences in core stability. Whereas the amount of pelleted wild-type CA was dramatically reduced in the presence of human TRIM5α at 6 and 10 h after infection, a similar level of E92K CA was pelleted through a sucrose cushion in the presence or absence of human TRIM5α at all of the three time points after infection (Fig. 5C), suggesting that the E92K mutation, instead of affecting the stability of the core, allows the virus to escape human TRIM5α-induced core degradation. Furthermore, viruses carrying the double mutations (N82D/N113K and L10W/E100K) can be restricted by Fv1b (see below), implying that their cores are stable enough for factor binding. Therefore, the results from the abrogation and fate-of-capsid assays, at least for E92K, N82D/N113K, and L10W/E100K viruses, strongly suggest that the effect of the escape mutations is to prevent the recognition and binding of N-MLV by huTRIM5α, whereas for the N7K virus, the capsid instability may contribute to its inability to saturate the restriction factor.
Fig 4.
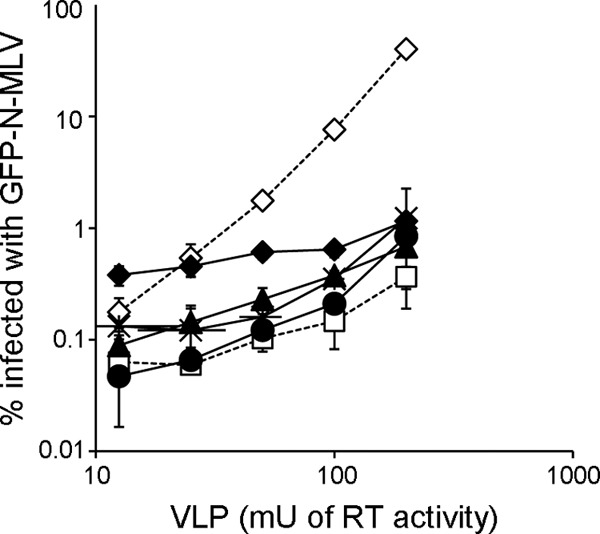
Abrogation assay. Human TE671 cells were treated for 2 h with increasing amounts of VLPs, followed by infection with a fixed amount of GFP-encoding N-MLV vector virus. GFP-expressing cells were enumerated by FACS 48 h after the infection. Symbols show different CAs on the VLPs: open diamond, wt N-MLV; open square, B-MLV; filled triangle, N7K; filled circle, E92K; asterisk, N82D/N113K; filled diamond, L10W/E100K. Each measurement was performed in triplicate, and mean values ± standard deviations from one representative experiment (of at least two) are plotted.
Effects of the huTRIM5α escape mutations on other restriction factors.
Given the observation that the escape mutations from rhTRIM5α that we had isolated previously (28) had various effects on restriction by different factors, it seemed plausible that the impact of new escape mutations from huTRIM5α might vary according to the restriction factors. We therefore examined the restriction of the new mutants by rhTRIM5α, Fv1b and Fv1n (the primary data are shown in Fig. 6 and summarized in Table 2), yielding some surprising results. E100K and N113K mutants that showed only partial escape from huTRIM5α restriction were sensitive to Fv1b but differed in their response to rhTRIM5α with E100K but not N113K, showing unchanged sensitivity to rhTRIM5α restriction. In contrast, both N7K and E92K mutations resulted in escape from Fv1b and the two TRIMs despite their poor growth properties. In general, but with several interesting exceptions, the addition of the rhTRIM5α escape mutations to the initial huTRIM5α escape mutations had more-pronounced effects on growth rates, with little effect on the restriction profile. For example, the addition of a second change to N113K had little effect on the restriction profile but could either increase or decrease growth fitness. The addition of a second mutation to E92K mutant virus also had little effect on restriction, although the G8D/E92K mutant showed somewhat-improved growth properties. E100K is sensitive to rhTRIM5α restriction and remains so even after the addition of the L10W or N82D changes, which individually give partial resistance. However, the L10W/E100K virus is fully resistant to rhTRIM5α. No restriction by either huTRIM5α or rhTRIM5α was seen with L10W/E100K or N82D/N113K. Unexpectedly, the N82D/E100K mutant was sensitive to rhTRIM5α but not huTRIM5α restriction, despite escape by the N82D single mutation from rhTRIM5α. Finally, the addition of G8D to N7K gave a virus that was no longer resistant to huTRIM5α or Fv1b but showed a significant gain in fitness.
Fig 6.
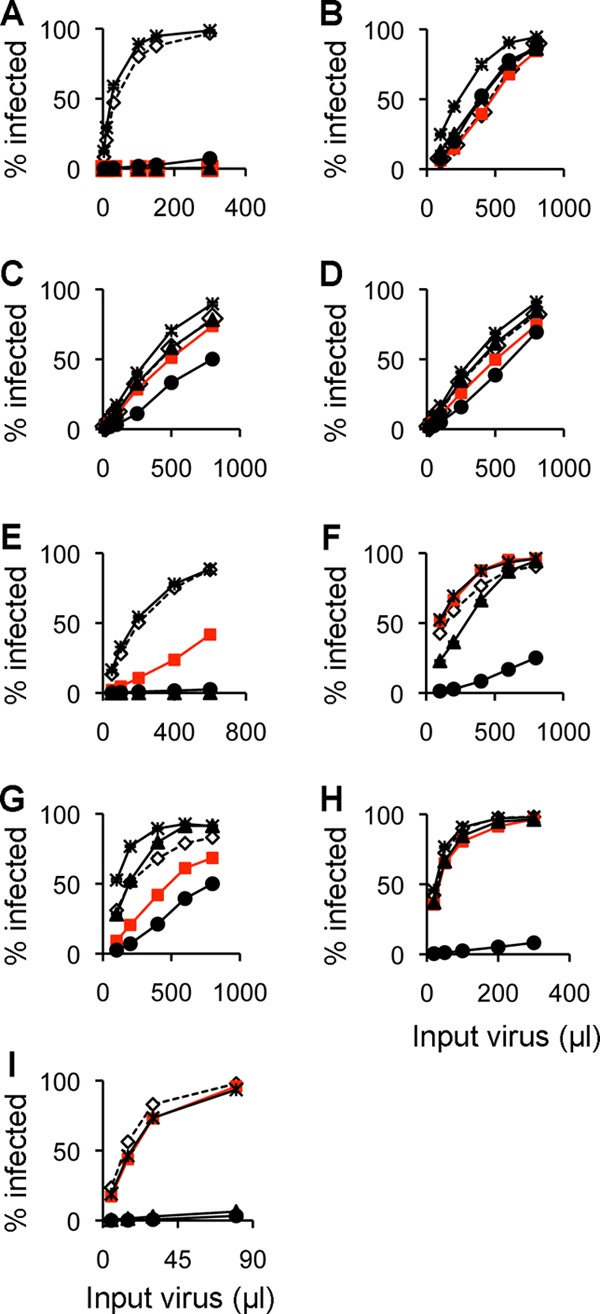
Restriction of N-MLV vectors carrying various escape mutations. eGFP-encoding vector viruses [wt (A); E92K (B); N7K/E211K (C); N7K (D); E100K (E); L10W/E100K (F); N113K (G); N82D/N113K (H); N82D/E100K (I)] were titrated on MDTF cells stably expressing restriction factors (open diamond, TRIM5-negative; filled red square, human TRIM5α; filled triangle, rhesus TRIM5α; filled circle, Fv1b; asterisk, Fv1n). Experiments were performed in triplicate; mean values from one representative experiment of three are plotted. These data are summarized in Table 2.
Table 2.
Restriction profiles of human TRIM5α escape mutants
| Virus or mutation(s) | Restriction factora |
Relative infectivity (%)b | |||
|---|---|---|---|---|---|
| HuTRIM5α | RhTRIM5α | Fv1b | Fv1n | ||
| wt N-MLV | ++ | ++ | ++ | − | 100 |
| G8D | ++ | + | ++ | ++ | >80 |
| L10W | ++ | − | ++ | − | >80 |
| N82D | ++ | + | ++ | − | >80 |
| N7K | − | − | − | − | <10 |
| G8D/N7K | ++ | − | ++ | − | >80 |
| L10W/N7K | + | − | + | − | <10 |
| N82D/N7K | − | − | + | − | 10–20 |
| E92K | − | − | − | − | 10–20 |
| G8D/E92K | − | − | − | − | 20–50 |
| L10W/E92K | + | − | − | − | <10 |
| N82D/E92K | + | − | − | − | <10 |
| E100K | + | ++ | ++ | − | >80 |
| G8D/E100K | + | ++ | ++ | − | >80 |
| L10W/E100K | − | − | ++ | − | 20–50 |
| N82D/E100K | − | ++ | ++ | − | >80 |
| N113K | + | − | + | − | 20–50 |
| G8D/N113K | + | − | ++ | − | >80 |
| L10W/N113K | + | − | + | − | 10–20 |
| N82D/N113K | − | − | ++ | − | >80 |
Restriction data are summarized as follows: ++, more than 10-fold difference in titer with and without restriction factor; +, 2- to 10-fold difference in titer with and without restriction factor; −, less than 2-fold reduction of infectivity. For primary restriction data from these mutants, see Figure 6 or reference 2. Similar results were obtained from three independent experiments.
Relative infectivity is in comparison to the infectivity of the wild-type N-MLV in MDTF cells.
The observation that the N7K and E92K mutants are resistant to the four restriction factors tested prompted us to further characterize the two mutants to see if they escape other primate TRIM5αs. To show the effects of CA mutations that are directly attributable to the binding of the B30.2 domain, we utilized a series of chimeric TRIM5αs that contain the RBCC domain of human origin and the B30.2 domain derived from various primates. MDTF cells were infected with delivery vector virus encoding the various TRIM5αs at a multiplicity of infection of approximately one and tested for infectivity by the mutant viruses using a two-color FACS assay. As summarized in Table 3 (Fig. 7 presents the primary titration data), the N7K and E92K N-MLV mutants were not restricted by any TRIM5αs tested, including three ape, three Old World monkey and three New World monkey TRIM5αs, indicating the very efficient escape of the N7K and E92K mutants from both TRIM5α and Fv1 restriction. We also examined the E100K and N113K mutants because the E100K mutant was the only variant that retained the susceptibility of the wild-type virus to restriction factors other than huTRIM5α (Table 2). The E100K variant moderately affected restriction by human, African green monkey, and tamarin TRIM5α but had a much stronger effect on capuchin and marmoset TRIM5α. In contrast, the N113K mutant was resistant to all primate TRIM5αs tested. As the N82D mutation enhanced the ability of E100K and N113K variants to resist human TRIM5α, the effects of these two double mutations on restriction by other primate TRIM5αs were also investigated. Whereas the N82D mutation potentiated the ability of the E100K mutant to escape African green monkey and tamarin TRIM5αs, it rendered the N113K mutant susceptible only to tamarin TRIM5α and did not affect restriction by other TRIM5αs. These results suggest that mutations at the two positions 7 and 92 affect recognition by all of the CA-recognizing restriction factors but impart significant fitness cost, whereas position 100 is instead a species-specific site for restriction. Position 113 seems specific to TRIM5α but not Fv1.
Table 3.
Restriction of human TRIM5α escape mutants by primate TRIM5α
| Origin of B30.2a | Restriction of indicated virusb |
||||||
|---|---|---|---|---|---|---|---|
| wt N-MLV | N7K | E92K | E100K | N113K | N82D/E100K | N82D/N113K | |
| Human | ++ | − | − | + | + | − | − |
| Gorilla | ++ | − | − | ++ | ND | ++ | − |
| Orangutan | ++ | − | − | ++ | − | ++ | − |
| Rhesus | ++ | − | − | ++ | − | ++ | − |
| AGM | ++ | − | − | + | − | − | − |
| S. mangabey | ++ | − | − | ++ | − | ++ | − |
| Squirrel m. | − | − | − | ND | ND | ND | ND |
| Capuchin | ++ | − | − | − | − | − | − |
| Tamarin | ++ | − | − | + | − | − | ++ |
| Marmoset | − | ND | ND | − | ND | ND | ND |
Rhesus, Rhesus macaque; AGM, African green monkey; S. mangabey, Sooty mangabey; Squirrel m., Squirrel monkey.
Restriction data are summarized as follows: ++, more than 10-fold difference in titer with and without restriction factor; +, 2- to 10-fold difference in titer with and without restriction factor; −, less than 2-fold reduction of infectivity; ND, not done. For the complete profile of titration curves, see Figure 7. Representative results from at least three independent experiments are shown.
Fig 7.

Titration curves of selected N-MLV escape mutants in the presence of various primate TRIM5αs. Chimeric TRIM5α constructs expressing the human RBCC domain fused with different primate B30.2 domains (25) were used to test restriction of N7K, E92K, E100K, N113K, and N82D/N113K escape mutant viruses. Titration curves in the presence of various ape, Old World monkey (OWM), and New World monkey (NWM) TRIM5αs are shown. One representative result from three independent experiments is shown. These data are summarized in Table 3.
Charge-dependent effects on restriction and infectivity from mutation at position 92.
The mutation at position 92 is particularly interesting, both because it was repeatedly selected for by passage against rhTRIM5α (28), huTRIM5α (this study), and Fv1b (unpublished data), despite compromised infectivity, and because it escaped all restriction factors tested. There are data (34) suggesting that the sensitivity to CA mutation can be affected by the nature of the Env gene used. Since the escape mutants were isolated in the context of virus with ecotropic Env and the fitness testing was done in single-cycle experiments with VSV-G, we sought to investigate whether the nature of the Env gene had any effect on the infectivity of viral vectors carrying CA N7K and E92K. Viruses were pseudotyped with an ecotropic MLV envelope, and after normalization for RT activity (150 mU), their infectivity was compared with that of the viruses pseudotyped with VSV-G envelope in N-3T3 cells. MLV-Env pseudotypes showed 2 to 3 times more infected cells than VSV-G pseudotypes but did not rescue N7K or E92K (data not shown).
To see if there is an amino acid preference for infectivity and escape at position 92, 16 amino acids were introduced at this position (Fig. 8 and Table 4). They can be assigned into four groups based on infectivity and restriction data. The first group includes the negatively charged amino acids D and E. Viruses with these two amino acids show both full infectivity and complete restriction by huTRIM5α (Table 4) and rhTRIM5α (data not shown). This result is consistent with the fact that Rauscher and Friend strains of MLV have D at the corresponding position. The second pair of amino acids are positively charged K and R. Substitution with these amino acids gave rise to virus that was resistant to both hu- and rhTRIM5α (Table 4 and data not shown) but poorly infectious. The third group includes many of the amino acids introduced (C, F, L, M, Q, S, T, W, and Y). Viruses with these amino acids showed complete restriction by huTRIM5α and reduced infectivity. The last group includes G, N, and P. Variants containing these amino acids are not infectious. These data indicate that negatively charged amino acids are preferred for full infectivity and positively charged amino acids are preferred for efficient escape from huTRIM5α.
Fig 8.
Restriction by human TRIM5α of N-MLV carrying different amino acids at CA position 92. A variety of amino acids were introduced at position 92 of CA by random mutagenesis. Unique isolates were tested for growth in the presence (filled red squares) or absence (open diamonds) of human TRIM5α. Polybrene was added to the virus stocks to increase the infectivity, except for E92 (wt) and E92D viruses. One representative result from three independent experiments is shown. These data are summarized in Table 4.
Table 4.
Restriction sensitivity of CA 92 mutantsa
| Amino acid at position 92 | Infectivity | Escape from huTRIM5α |
|---|---|---|
| D or E | Fully infectious | No escape |
| K or R | Reduced infectivity | Escape |
| C, F, L, M, Q, S, T, W, or Y | Reduced infectivity | No escape |
| G, N, or P | Not infectious |
For primary restriction data for these mutants, see Figure 8. Similar results were obtained from three independent experiments.
Opposite charge preference for escape from huTRIM5α and rhTRIM5α.
The restriction profiles of the rhTRIM5α escape mutants (28) and the huTRIM5α escape mutants (Table 2) show different tendencies regarding interaction with factors that were not involved in the selection process; many of the rhTRIM5α escape mutations do not affect restriction by other factors (huTRIM5α and Fv1b), whereas the huTRIM5α escape mutations affect restriction by factors that were not involved in the mutant selection (rhTRIM5α and Fv1b). In addition, site-directed mutagenesis revealed a charge-related, inverse correlation between restriction and full infectivity for mutations at CA position 92. These observations collectively lead us to hypothesize that a change in the charge distribution on the capsid surface causes the differences in susceptibility to the different restriction factors. To examine this hypothesis, huTRIM5α escape mutations were plotted on the capsid structure and the sites involved in rh- and huTRIM5α restriction were compared (Fig. 9). This comparison revealed that changes over the entire span of the capsid molecule can affect restriction by both huTRIM5α and rhTRIM5α. However, opposite charge preferences for escape from rh- and huTRIM5α-mediated restriction are observed; rhTRIM5α escape tends to result from mutations generating negatively charged amino acids, whereas huTRIM5α escape results from mutation to positively charged residues. Indeed, many of the rhTRIM5α escape mutations resulted in replacement of the wild-type amino acids by aspartic acid, whereas all of the huTRIM5α escape mutations were changes to lysine.
Fig 9.

Comparison of the CA molecular surfaces of rhTRIM5α and huTRIM5α escape mutants. Residues of wt N-MLV capsid (PDB 1U7K) were replaced with corresponding escape mutations computationally, using UCSF Chimera software, and hexamers of the wt N-MLV (A), rhTRIM5α escape mutant (L4S, G8D, L10W, N82D, A95D, E92K, H114D, V116D) (B), and huTRIM5α escape mutant (N7K, E92K, E100K, N113K) (C) capsid were visualized using CCP4 molecular graphics software. Top views of the N-MLV CA N-terminal domain structure are displayed, and the molecular surface is represented. Positive charges are highlighted in blue, and negative charges are highlighted in red. (D) The residues of huTRIM5α escape mutations are shown as sticks in the protein backbone of the N-MLV CA N-terminal domain structure displayed in cartoon representation of side (left) and top (right) views. Each helix is numbered in black. Residues involved in restriction factor specificity are shown. Lys7 is shown in red; Leu10 and Glu100 are shown in green; Lys92 is shown in orange; Asp82 and Lys113 are shown in blue.
In this context, it is noteworthy that the V1 region of the human B30.2 domain contains two additional Arg residues compared with that of the corresponding rhesus domain (25), at positions 332 and 335. This prompted us to further investigate the restriction of L10W and N82D/E100K viruses by V1 region mutants with mutations at these positions, since the former is restricted by huTRIM5α but not by rhTRIM5α and vice versa. As expected, wild-type N-MLV was sensitive to all of the V1 region mutants. While the L10W virus was restricted by all of the V1 region mutations of human TRIM5α, the loss of the Arg residues at either position in the V1 region enabled TRIM5α to restrict the N82D/E100K virus (Fig. 10A and Table 5). Conversely, the acquisition of Arg at the corresponding positions (P334R and LFTFPSLT337RYQTFV) enabled rhTRIM5 to restrict the N82D/E100K virus (Fig. 7B) at a level similar to the restriction by huTRIM5α, confirming the involvement of these Arg residues in the recognition of this virus. In contrast to huTRIM5α V1 variants, Arg acquisition in the V1 region conferred on rhTRIM5α the ability to restrict L10W virus, although its effect was only modest for the P334R variant (Fig. 10B and Table 5).
Fig 10.

Effects of the charged amino acids in variable regions of the B30.2 domain on restriction. Restriction of wt N-MLV, L10W, and N82D/E100K viruses was tested against human (A) or rhesus (B) TRIM5αs with amino acid substitutions that add or deplete charge in V1 (R332P and RYQTFV335LFTFPSLT for huTIRM5α and P334R or LFTFPSLT337RYQTFV for rhTRIM5α), V2 (K389Q for huTRIM5α and Q393K for rhTRIM5α), or V3 (E405Q for huTRIM5α and Q409E for rhTRIM5α) regions of the B30.2 domain. In these experiments, intact rhesus TRIM5α (29) was used instead of the human-rhesus chimeric TRIM5α construct. Similar results were obtained in two independent experiments; values for one example are shown.
Table 5.
Effects on restriction of the charged amino acids in variable regions
| Origin of TRIM5α, motifa | Amino acid(s) at indicated positionb |
Restriction of indicated virusc |
|||||
|---|---|---|---|---|---|---|---|
| V1 | V2 | V3 | wt N-MLV | L10W mutant | N82D/E100K mutant | ||
| Human | 332 | 335–340 | 389 | 405 | |||
| wt | R | RYQTFV | K | E | − | ++ | − |
| R332P | P | RYQTFV | K | E | ++ | ++ | ++ |
| RYQ | R | LFTFPSLT | K | E | ++ | ++ | ++ |
| PR double | P | LFTFPSLT | K | E | ++ | ++ | ++ |
| V2 | R | RYQTFV | Q | E | ++ | ++ | + |
| R332P V2 | P | RYQTFV | Q | E | ++ | ++ | ++ |
| RYQ V2 | R | LFTFPSLT | Q | E | ++ | ++ | ++ |
| V3 | R | RYQTFV | K | Q | ++ | ++ | − |
| R332P V3 | P | RYQTFV | K | Q | ++ | ++ | + |
| RYQ V3 | R | LFTFPSLT | K | Q | ++ | ++ | − |
| Rhesus | 334 | 337–344 | 393 | 409 | |||
| wt | P | LFTFPSLT | Q | Q | ++ | − | + |
| P334R | R | LFTFPSLT | Q | Q | ++ | + | − |
| LFT | P | RYQTFV | Q | Q | ++ | + | − |
| V2 | P | LFTFPSLT | K | Q | ++ | − | + |
| P334R V2 | R | LFTFPSLT | K | Q | ++ | − | − |
| LFT V2 | P | RYQTFV | K | Q | ++ | + | − |
| V3 | P | LFTFPSLT | Q | E | ++ | + | ++ |
| P334R V3 | R | LFTFPSLT | Q | E | ++ | ++ | ++ |
| LFT V3 | P | RYQTFV | Q | E | ++ | ++ | + |
Amino acids or regions changed in mutant TRIM5α. V1 to V3, variable regions 1 to 3.
Boldface with underlining, positively charged amino acid; italic with underlining, negatively charged amino acid.
Restriction data are summarized as follows: ++, more than 10-fold difference in titer with and without restriction factor; +, 2- to 10-fold difference in titer with and without restriction factor; −, less than 2-fold reduction of infectivity. For the complete profile of titration curves, see Figure 10.
However, the Arg residues in V1 are not the sole determinants of restriction. Amino acid comparisons reveal additional charge differences within the V2 and V3 regions; Lys at 389 in the V2 and Glu at 405 in the V3 regions of the human B30.2 domain, compared with Gln at the corresponding sites in the rhesus B30.2 domain. These amino acid residues were exchanged by themselves or in combination with the V1 region mutations described above. The restriction of the wild-type virus was not altered by any of the V2 and V3 region changes. The effect of the V2 region mutation on restriction was relatively subtle, with partial effects seen for only two constructs, huTRIM5α K389Q, which conferred partial restriction of N82D/E100K virus, and rhTRIM5α P334R/Q393K, which resulted in some loss in the ability to restrict L10W. In contrast, restriction by both hu- and rhTRIM5α was influenced significantly by the V3 region mutations. Whereas the V3 mutation counteracted the restrictive effect of huTRIM5α V1 variants on N82D/E100K virus, the corresponding V3 mutation enabled the restriction of both L10W and N82D/E100K viruses by rhTRIM5α. In addition, the V3 mutation exerted an additional effect on the restriction of L10W virus by rhTRIM5α V1 variants, while it determined the restrictive phenotype of rhTRIM5α variants to N82D/E100K virus regardless of the V1 region. These results confirm the proposed involvement of charged amino acids of the V3 region in restriction (35). Taken together, these results point to the importance of the four charged amino acids in the variable regions for restriction, with the specific consequences dependent on the surrounding amino acids (36).
DISCUSSION
Prompted by our interest in understanding the mechanism(s) by which cellular factors like TRIM5α bind and restrict incoming retroviruses and in the absence of informative structural data, we have set out to isolate viruses that escape restriction. Sequence changes in CA allowing escape might facilitate the identification of CA residues participating in the interaction with TRIM5α. Such changes might reflect direct contacts between CA and restriction factor or, perhaps less likely, identify residues that are indirectly important for multivalent binding (17, 37), such as those involved in core stability. Comparison of N-MLV escape variants from rhesus (28) and human TRIM5αs (this paper) reveals both similarities and differences in the changes allowing virus growth.
In both cases, escape mutants with amino acid changes distributed across the CA surface were isolated, thereby suggesting multiple contacts between CA monomers in the assembled core and the restriction factor. These would include the CA N-terminal beta hairpin loop, a region C-terminal to helix 4, and the surface-exposed N-terminal end of helix 6. A similar conclusion has been reached from studies of different lentiviruses (38–42). Such an extended interaction domain would be consistent with structural studies of the TRIM5α PRY-SPRY region that suggest a large, triangular surface region containing the specificity determinants of restriction (25, 43, 44). Studies of CA determinants of Fv1 tropism also suggest an extended interaction domain (45).
However, N-MLV escape from huTRIM5α restriction appears to be more difficult than escape from rhTRIM5α, as judged by the time taken for factor-resistant variants to emerge (Fig. 2) and the identification of fewer mutations giving resistance to huTRIM5α (Table 1), with three of four showing significantly reduced fitness even in the absence of restriction factor (Fig. 3). Moreover, all the huTRIM5α escapees were the products of nucleotide changes resulting in the incorporation of a positively charged lysine (Table 1), whereas many rhTRIM5α escape mutants were substitutions resulting in negatively charged Asp residues (28). Furthermore, most of the huTRIM5α escapees, although showing reduced fitness, also became resistant to TRIM5α from multiple species (Table 3). This was not generally true for the variants selected with rhTRIM5α. Taken together, these results suggest that the course of evolution resulted in a human TRIM5α developing a tighter binding interaction with MLV than did rhTRIM5α, perhaps involving the acquisition of two additional Arg residues in the V1 region, probably in combination with the other variable regions, of its B30.2 domain (25, 36). The virus(es) driving this selection have not been and may never be identified.
To escape from the more-extensive interaction with huTRIM5α likely requires substantial changes in CA, with consequent effects on viral fitness. It has been demonstrated that Moloney MLV is easily inactivated by the incorporation of alanine scanning mutations into CA (46). Perhaps gammaretroviruses are more sensitive to changes in CA than currently appreciated; possible constraints might include the need to form a hexameric lattice in immature or mature cores and/or interactions with cellular factors involved in nuclear transport. The reason(s) for the fitness loss in N7K and E92K viruses are not fully explained. Both virus production and maturation appear normal, but the stability of N7K virus appears to be compromised (Fig. 5). Reduced stability of the N7K core might allow more-rapid transit through a TRIM5α-sensitive state and apparent escape. However, this does not appear to be the case for E92K.
To begin to define the scope for possible capsid change, viruses with 16 different amino acids at CA position 92 were compared (Table 4). Consistent with the results of our selection experiments (Table 1), the only changes to give rise to escape variants were introduction of the basic amino acids Lys and Arg (His was not obtained in our mutagenesis protocol), though at considerable cost to fitness. All other changes away from acidic amino acids resulted in the production of either noninfectious virus or viruses that were fully restricted by huTRIM5α. Such data reinforce the idea that only a limited number of escape mutations are possible and emphasize the potential effectiveness of TRIM5α restriction. Interestingly, no viruses carrying an Arg residue were isolated in our experiments (Table 1), despite the similar growth properties of E92K and E92R (data not shown). We note that this would require two nucleotide changes as opposed to the one for a Glu-to-Lys change. Two changes would also be required for an Arg-to-Glu change at position 110, the position that determines Fv1 tropism (27) and allowed the original definition of MLV sensitivity to TRIM5 (47). We conclude that the concurrent change of two nucleotides is rare unless the first change provides an amino acid alteration providing a selective advantage.
In this light, the observation that the presence of the rhTRIM5α escape mutations L10W and N82D can facilitate the development of escape from huTRIM5α by E100K or N113K (Table 1) without individually affecting restriction by huTRIM5α appears somewhat surprising. It is unlikely that this is due to direct interaction between the first and second altered amino acid, as the distance between the two affected residues is at least 15 Å (Fig. 9D). Rather, we speculate that the two residues of the double mutations may affect the binding surfaces independently, with the first mutation acting to constrain the positioning of CA relative to TRIM5 at a second, distant site.
The foregoing discussion makes the implicit assumption that escape mutations affect only direct interaction between the N-terminal domain of CA and the B30.2 of TRIM5α. All the mutations we have isolated map to the CA N-terminal domain, but we cannot exclude the possibility that mutations in other parts of, say, gag might play such a role. Furthermore, it is likely that at least in some instances, the RBCC domain may influence the interaction. The strength of restriction may be affected by the nature of the RBCC domain (22, 48, 49); for example, N82D/E100K virus was restricted strongly by human RBCC-rhesus B30.2 chimeric TRIM5α (Table 3), whereas the same virus was only partially restricted by authentic rhesus TRIM5α (Fig. 10B). It may be that the coiled-coil domain or linker-2 region has an effect on the spatial arrangement of the B30.2 domain, thereby affecting the binding interaction with retrovirus CA core to some extent (19, 50). Whether the same region plays a role in determining the specificity of potential targets remains to be determined.
ACKNOWLEDGMENTS
We thank Melvyn Yap, Kate Bishop, and Ian Taylor for their helpful comments.
This work was supported by the UK Medical Research Council (file reference U117512710).
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 5:1109–1115 [DOI] [PubMed] [Google Scholar]
- 2. Feschotte C, Gilbert C. 2012. Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13:283–296 [DOI] [PubMed] [Google Scholar]
- 3. Stoye JP. 2012. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 10:395–406 [DOI] [PubMed] [Google Scholar]
- 4. Jolicoeur P. 1979. The Fv-1 gene of the mouse and its control of murine leukemia virus replication. Curr. Top. Microbiol. Immunol. 86:67–122 [DOI] [PubMed] [Google Scholar]
- 5. Lilly F. 1970. Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J. Natl. Cancer Inst. 45:163–169 [PubMed] [Google Scholar]
- 6. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 7. Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573 [DOI] [PubMed] [Google Scholar]
- 8. Nisole S, Lynch C, Stoye JP, Yap MW. 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. U. S. A. 101:13324–13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, Towers GJ. 2008. Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 105:3557–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T. 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. U. S. A. 105:3563–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brennan G, Kozyrev Y, Hu SL. 2008. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. U. S. A. 105:3569–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O'Neil SP, Johnson W. 2008. Evolution of a TRIM5-CypA splice isoform in Old World monkeys. PLoS Pathog. 4:e1000003 doi:10.1371/journal.ppat.1000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malfavon-Borja R, Wu LI, Emerman M, Malik HS. 2013. Birth, decay, and reconstruction of an ancient TRIMCyp gene fusion in primate genomes. Proc. Natl. Acad. Sci. U. S. A. 110:E583–E592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bishop KN, Bock M, Towers G, Stoye JP. 2001. Identification of the regions of Fv1 necessary for murine leukemia virus restriction. J. Virol. 75:5182–5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bishop KN, Mortuza GB, Howell S, Yap MW, Stoye JP, Taylor IA. 2006. Characterization of an amino-terminal dimerization domain from retroviral restriction factor Fv1. J. Virol. 80:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, Sodroski J. 2009. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 83:10737–10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li X, Sodroski J. 2008. The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J. Virol. 82:11495–11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD. 2005. Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J. Virol. 79:8969–8978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sastri J, O'Connor C, Danielson CM, McRaven M, Perez P, Diaz-Griffero F, Campbell EM. 2010. Identification of residues within the L2 region of rhesus TRIM5alpha that are required for retroviral restriction and cytoplasmic body localization. Virology 405:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sawyer SL, Wu LI, Emerman M, Malik HS. 2005. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. U. S. A. 102:2832–2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stremlau M, Perron M, Welikala S, Sodroski J. 2005. Species-specific variation in the B30.2(SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J. Virol. 79:3139–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yap MW, Nisole S, Stoye JP. 2005. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr. Biol. 15:73–78 [DOI] [PubMed] [Google Scholar]
- 23. Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 103:7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diehl WE, Stansell E, Kaiser SM, Emerman M, Hunter E. 2008. Identification of postentry restrictions to Mason-Pfizer monkey virus infection in New World monkey cells. J. Virol. 82:11140–11151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohkura S, Yap MW, Sheldon T, Stoye JP. 2006. All three variable regions of the TRIM5alpha B30.2 domain can contribute to the specificity of retrovirus restriction. J. Virol. 80:8554–8565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yap MW, Lindemann D, Stanke N, Reh J, Westphal D, Hanenberg H, Ohkura S, Stoye JP. 2008. Restriction of foamy viruses by primate Trim5α. J. Virol. 82:5429–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kozak CA, Chakraborti A. 1996. Single amino acid changes in the murine leukemia virus capsid protein gene define the target of Fv1 resistance. Virology 225:300–305 [DOI] [PubMed] [Google Scholar]
- 28. Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP. 2011. Novel escape mutants suggest an extensive TRIM5alpha binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog. 7:e1002011 doi:10.1371/journal.ppat.1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yap MW, Nisole S, Lynch C, Stoye JP. 2004. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. U. S. A. 101:10786–10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bock M, Bishop KN, Towers G, Stoye JP. 2000. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J. Virol. 74:7422–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boone LR, Myer FE, Yang DM, Ou CY, Koh CK, Roberson LE, Tennant RW, Yang WK. 1983. Reversal of Fv-1 host range by in vitro restriction endonuclease fragment exchange between molecular clones of N-tropic and B-tropic murine leukemia virus genomes. J. Virol. 48:110–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 23:628–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dodding MP, Bock M, Yap MW, Stoye JP. 2005. Capsid processing requirements for abrogation of Fv1 and Ref1 restriction. J. Virol. 79:10571–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jiang J, Ablan SD, Derebail S, Hercik K, Soheilian F, Thomas JA, Tang S, Hewlett I, Nagashima K, Gorelick RJ, Freed EO, Levin JG. 2011. The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology 421:253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perron M, Stremlau M, Sodroski J. 2006. Two surface-exposed elements of the B30.2/SPRY domain as potency determinants of N-tropic murine leukemia virus restriction by human TRIM5α. J. Virol. 80:5631–5636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maillard PV, Reynard S, Serhan F, Turelli P, Trono D. 2007. Interfering residues narrow the spectrum of MLV restriction by human TRIM5alpha. PLoS Pathog. 3:e200 doi:10.1371/journal.ppat.0030200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. 2011. Hexagonal assembly of a restricting TRIM5a protein. Proc. Natl. Acad. Sci. U. S. A. 108:534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miyamoto T, Yokoyama M, Kono K, Shioda T, Sato H, Nakayama EE. 2011. A single amino acid of human immunodeficiency virus type 2 capsid protein affects conformation of two external loops and viral sensitivity to TRIM5a. PLoS One 6:e22779 doi:10.1371/journal.pone.0022779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Song H, Nakayama EE, Yokoyama M, Sato H, Levy JA, Shioda T. 2007. A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5αs. J. Virol. 81:7280–7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuroishi A, Saito A, Shingai Y, Shioda T, Nomaguchi M, Adachi A, Akari H, Nakayama EE. 2009. Modification of a loop sequence between alpha-helices 6 and 7 of virus capsid (CA) protein in a human immunodeficiency virus type 1 (HIV-1) derivative that has simian immunodeficiency virus (SIVmac239) vif and CA α-helices 4 and 5 loop improves replication in cynomolgus monkey cells. Retrovirology 6:70 doi:10.1186/1742-4690-6-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pacheco B, Finzi A, Stremlau M, Sodroski J. 2010. Adaptation of HIV-1 to cells expressing rhesus monkey TRIM5a. Virology 408:204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kono K, Song H, Yokoyama M, Sato H, Shioda T, Nakayama EE. 2010. Multiple sites in the N-terminal half of simian immunodeficiency virus capsid protein contribute to evasion from rhesus monkey TRIM5alpha-mediated restriction. Retrovirology 7:72 doi:10.1186/1742-4690-7-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, Ivanov DN. 2012. Structure of the rhesus monkey TRIM5alpha PRYSPRY domain, the HIV capsid recognition module. Proc. Natl. Acad. Sci. U. S. A. 109:13278–13283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, Xiong Y. 2012. Structural insight into HIV-1 capsid recognition by rhesus TRIM5alpha. Proc. Natl. Acad. Sci. U. S. A. 109:18372–18377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stevens A, Bock M, Ellis S, LeTissier P, Bishop KN, Yap MW, Taylor W, Stoye JP. 2004. Retroviral capsid determinants of Fv1 NB- and NR-tropism. J. Virol. 78:9592–9598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Auerbach MR, Shu C, Kaplan A, Singh IR. 2003. Functional characterization of a portion of the Moloney murine leukemia gag gene by genetic footprinting. Proc. Natl. Acad. Sci. U. S. A. 100:11678–11683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O. 2000. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. U. S. A. 97:12295–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Newman RM, Hall L, Connole M, Chen G-L, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, Johnson WE. 2006. Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5a. Proc. Natl. Acad. Sci. U. S. A. 103:19134–19139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462 doi:10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li X, Yeung DF, Fiegen AM, Sodroski J. 2011. Determinants of the higher order association of the restriction factor TRIM5alpha and other tripartite motif (TRIM) proteins. J. Biol. Chem. 286:27959–27970 [DOI] [PMC free article] [PubMed] [Google Scholar]