

Abstract
The human adenovirus E3/19K protein is a type I transmembrane glycoprotein of the endoplasmic reticulum (ER) that abrogates cell surface transport of major histocompatibility complex class I (MHC-I) and MHC-I-related chain A and B (MICA/B) molecules. Previous data suggested that E3/19K comprises two functional modules: a luminal domain for interaction with MHC-I and MICA/B molecules and a dilysine motif in the cytoplasmic tail that confers retrieval from the Golgi apparatus back to the ER. This study was prompted by the unexpected phenotype of an E3/19K molecule that was largely retained intracellularly despite having a mutated ER retrieval motif. To identify additional structural determinants responsible for ER localization, chimeric molecules were generated containing the luminal E3/19K domain and the cytoplasmic and/or transmembrane domain (TMD) of the cell surface protein MHC-I Kd. These chimeras were analyzed for transport, cell surface expression, and impact on MHC-I and MICA/B downregulation. As with the retrieval mutant, replacement of the cytoplasmic tail of E3/19K allowed only limited transport of the chimera to the cell surface. Efficient cell surface expression was achieved only by additionally replacing the TMD of E3/19K with that of MHC-I, suggesting that the E3/19K TMD may confer static ER retention. This was verified by ER retention of an MHC-I Kd molecule with the TMD replaced by that of E3/19K. Thus, we have identified the E3/19K TMD as a novel functional element that mediates static ER retention, thereby increasing the concentration of E3/19K in the ER. Remarkably, the ER retrieval signal alone, without the E3/19K TMD, did not mediate efficient HLA downregulation, even in the context of infection. This suggests that the TMD is required together with the ER retrieval function to ensure efficient ER localization and transport inhibition of MHC-I and MICA/B molecules.
INTRODUCTION
Human adenoviruses (Ads) can cause a variety of acute diseases (1) but can also persist for variable lengths of time in a clinically inapparent state (2). More than 50 different Ad serotypes have been distinguished and classified into six different species, A to F (3). Ads devote a considerable part of their genome to immune evasion functions that facilitate infection and/or maintain a state of balance during persistence or latency (1, 4). If this balance is perturbed, as in immunosuppressed patients, serious or life-threatening disease may ensue (5). Ads are also widely used as vectors for vaccination and gene therapy (6). Thus, a better understanding of their interaction with the immune system has major medical implications.
Many of the Ad immune evasion genes are grouped together in the early transcription unit 3 (E3), which is nonessential for virus replication in vitro but is preserved in all human Ads (4, 7). This suggests an important role in vivo, which is supported by various in vivo models (1, 8, 9). E3 proteins counter a variety of immune responses (4, 7, 10), including antigen presentation and natural killer (NK) cells (11). In this context, the E3/19K protein has a dual function. It prevents the transport of newly synthesized major histocompatibility class I (MHC-I; HLA in humans) molecules to the cell surface, thereby interfering with peptide presentation to cytotoxic T lymphocytes (CTL) (12–15). E3/19K also suppresses recognition by natural killer cells via intracellular sequestration of the stress-induced MHC-I-related chain A and B (MICA/B) molecules (11), which serve as ligands for the major activating NK receptor, NKG2D (16).
E3/19K proteins are type I transmembrane glycoproteins expressed by Ads of species B to E. Despite their common function, their sequence homology is poor (4, 10, 17, 18). The mature Ad2 protein consists of 142 amino acids forming a luminal domain of ∼104 amino acids with two N-linked high-mannose carbohydrates, a transmembrane segment of ∼23 amino acids, and a 15-amino-acid cytoplasmic tail. E3/19K appears to combine two functional entities to block cell surface display of MHC-I and MICA/B molecules: (i) the luminal domain, which binds newly synthesized HLA-I molecules, and (ii) a dilysine motif in the cytoplasmic tail, which mediates the retrieval of E3/19K–MHC-I complexes from the cis-Golgi back to the endoplasmic reticulum (ER) (12, 13, 19–21).
The structural requirements of Ad2 E3/19K for MHC-I and MICA/B binding are well characterized; they include two crucial intramolecular disulfide bonds and a number of other conserved amino acids within the luminal domain (22–26). Recently, detailed insight into the interaction between the luminal E3/19K domain and a soluble form of HLA-A2 was provided by crystallography (26). However, the roles of the transmembrane domain (TMD) and cytoplasmic tail in this process remain controversial. While complex formation can clearly be demonstrated in vitro in their absence (22, 27, 28), in vivo studies within cells suggested that the interaction is reduced or even abrogated upon deletion of the cytoplasmic tail or the TMD, respectively (13, 28–30). Due to the lack of appropriate antibodies, it remained unclear whether this was caused by secondary structural alterations in the luminal domain induced by the deletions or whether it reflected merely a need for membrane anchoring mediated by the TMD (27, 28, 31). Also, the exact mechanism of ER localization is not completely understood. While deletion analysis of the cytoplasmic tail and the analysis of an E3/19K reporter chimera clearly identified a dilysine motif important for ER retrieval (4, 13, 19–21, 29, 32), these studies are complicated by the fact that the efficacy of ER retrieval by dilysine motifs depends on the sequence context (33). When the structural requirements for ER localization were studied in the context of E3/19K itself, conflicting data were obtained (29, 30), suggesting that other structural elements within the E3/19K protein may be responsible for ER retrieval.
The present study was prompted by the phenotype of an E3/19K molecule with a mutated dilysine retrieval motif. As expected, this mutant reached the cell surface, yet the great majority remained in the ER. Thus, elements in the mutated protein other than the ER retrieval signal clearly contributed to ER retention. By analyzing a series of chimeric molecules in which the cytoplasmic tail and TMD of E3/19K were replaced individually or in combination with corresponding domains of the bona fide murine cell surface molecule MHC-I Kd, we provide evidence that the E3/19K TMD acts as a signal for static ER retention that significantly enhances HLA complex formation. Thus, we have discovered a new functional element in E3/19K, and we propose that efficient transport inhibition of HLA and MICA/B molecules requires the combined activity of an ER retention signal in the E3/19K TMD and the ER retrieval signal in its cytoplasmic tail.
MATERIALS AND METHODS
Construction of E3/19K and Kd mutant proteins.
The Ad2 EcoRI D fragment (2,674 bp; nucleotides 27372 to 30046 of the Ad2 reference sequence), containing the E3/19K gene, was ligated into pBluescript II KS(−) (Stratagene), yielding pBS-EcoRI D. All mutants were introduced using PCR-mediated mutagenesis essentially as described previously (24). Generally, each construct was designed using mutant oligonucleotide pairs encompassing the desired sequence alteration in combination with respective flanking primers. For construction of the E3/19K-Kd chimeras EEK and EKK, a three-step protocol was used, separately amplifying E3/19K sequences with Kd-compatible ends by using pBS-EcoRI D as the target DNA and the corresponding Kd domains by using the Kd cDNA vector pSV-Kd (34) as the template. The last step consisted of the combination of the two PCR products, followed by PCR with flanking oligonucleotides. Constructs EKE and EKE* were constructed similarly, but EKK was used as the template DNA to amplify a 5′ fragment. In two independent reactions, the corresponding 3′ fragments were generated by using either the wild-type or the mutated (EEE*) pBS-EcoRI D fragment as template DNA. These PCR fragments were cleaved with BsrGI and BclI and were ligated into wild-type pBS-EcoRI D cleaved with the same enzymes.
The Kd gene containing the TMD of E3/19K (KEK) was generated by subcloning an 810-bp ApaI/NsiI fragment of the genomic clone p191.6 of murine Kd (35) into appropriately cleaved pGEM7+ (Promega), resulting in pKKK-AN. The sequence corresponding to the transmembrane domain of Ad2 E3/19K (nucleotides 29175 to 29243 of the GenBank Ad2 reference sequence) was amplified with flanking primers using pBS-EcoRI D as the template. The E3/19K-specific primers were flanked with 5′ sequences homologous to the upstream (nucleotides 3388 to 3397) and downstream (nucleotides 3466 to 3472) Kd coding sequences (numbered according to the published Kd gene sequence [35]). This E3/19K amplicon was first fused to an upstream Kd amplicon by using appropriate primers and p161.6 as the template. The Kd-E3/19K hybrid TMD amplicon was then fused by another assembly PCR to a downstream Kd-specific amplicon generated by using relevant primers with p191.6 as the template. The final fusion amplicon produced by the flanking Kd primers was restricted with SacI and was inserted into SacI-digested pKKK-AN, replacing its original small SacI fragment to yield pKEK-AN. pKEK-AN was then inserted into appropriately cleaved p191.6. All intermediates, as well as the final construct, were verified by sequencing. The sequences of the oligonucleotides used for the construction of the various chimeric molecules are available upon request. The generation of KKE has been described previously (36).
Cell lines, culture conditions, and transfections.
Transfection of 293 cells (ATCC CRL 1573) with the wild-type and mutant Ad2 EcoRI D and EcoRV C fragments (encoding E3A and the entire E3 region, respectively) led to the establishment of the 293.12, 293E22.7, and 293E3-45 cell lines, constitutively expressing wild-type E3/19K (12, 37). 293 cells expressing the Ad5 E1A and E1B genes were specifically used to provide the E1A transactivating function for the Ad E3 promoter present in the transfected EcoRV C and EcoRI D fragments. This system has been shown previously to yield good expression levels of E3 genes (23, 37). The mutant E3/19K cell lines were established by transfection of 293 cells with mutated EcoRI D fragments and the neomycin phosphotransferase gene essentially as described previously (23, 24). 293Kd2 and 293.12Kd8 were generated by transfection of 293 and 293.12 cells, respectively, with a genomic clone of the murine H-2Kd gene (14). 293KKE, previously termed 293KdE3, expresses a Kd molecule with its cytoplasmic tail replaced by that of E3/19K (36). Similarly, cells expressing Kd molecules with the TMD of E3/19K (KEK) were established. 293 cells were grown in complete Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS), 2 mM glutamine, and antibiotics. For routine culture, the medium of the transfectants was supplemented with 200 μg/ml of active G418 (Calbiochem). K562 is an erythroleukemia cell line lacking MHC class I expression (ATCC CCL243) that was grown in complete RPMI medium. The K562-EEE and K562-EEK cell lines, derived from K562 by transfection of constructs EEE and EEK, respectively, were cultured in complete RPMI medium supplemented with 600 μg/ml of G418.
Construction of recombinant adenoviruses expressing mutant E3/19K proteins.
In order to study mutant E3/19K proteins EEE*, EKE, and EKE* in the virus context, the wild-type Ad2 genome, cloned as a bacterial artificial chromosome (BAC) (38), was modified by a two-step mutagenesis procedure, essentially as described previously (39). First, the modified E3/19K sequences from the corresponding pBS-EcoRI D plasmids were transferred to pBS-EcoRV C (37), containing the Ad2-EcoRV C fragment (encompassing the entire E3 region) that had been labeled previously at its unique SwaI site by a PCR-amplified kanamycin resistance (Kmr) gene derived from pGPS1.1 (New England BioLabs). Next, the EcoRI fragments of these constructs containing the E3/19K mutation and the SwaI-flanked Kmr cassette were transferred to the Ad2-BAC by homologous recombination as described previously (38, 39). Likewise, the wild-type E3 sequence was replaced by an EcoRI fragment of an unmodified Kmr cassette-labeled construct as a control (EEE). After the selection of recombinant BACs with kanamycin, the resistance cassettes were removed from each construct by SwaI treatment, followed by recircularization with T4 DNA ligase (New England BioLabs). The repaired mutant BACs were retransformed into Escherichia coli and were screened for the loss of the Kmr cassette, and sequences were verified across the mutation and recombination site. Modified viruses (Ad2-EEE, Ad2-EEE*, Ad2-EKE, Ad2-EKE*) were reconstituted from the corresponding BACs upon transfection of 293 cells with purified BAC DNA as described previously (38).
MAbs and antisera.
The following monoclonal antibodies (MAbs) were used in this study: W6/32 against HLA-A, -B, and -C (ATCC HB95); BB7.2 against HLA-A2 and -Aw69 (40); BAMO3 against MICA/B (41); Tw1.3 (13) (kindly provided by J. Yewdell, NIH), 3A9, and 3F4 (42) against E3/19K. The C-tail (23) and Kc-tail (36) polyclonal antisera recognize the cytoplasmic tails of E3/19K and Kd, respectively.
Cell labeling, immunoprecipitation, SDS-PAGE, and endo H treatment.
Labeling of cells with [35S]methionine, immunoprecipitation of NP-40 extracts, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and endo-β-N-acetylglucosaminidase H (endo H) treatment were carried out essentially as described previously (12, 23, 43).
Flow cytometry analysis.
For the detection of antigens, unfixed cells were incubated with the MAbs listed above, followed by goat anti-mouse IgG conjugated with fluorescein isothiocyanate (Sigma) or Alexa Fluor 488 (Invitrogen), as described previously (23, 24), except that for internal staining of antigens, 0.075% saponin (Sigma) was used. For the last two washes and for analysis, saponin was omitted from the buffer. As demonstrated previously (23, 24), staining in the presence of the mild detergent saponin allows the Abs access to the ER lumen, preserving ER membrane proteins while concomitantly extracting a large fraction of cell surface proteins. This treatment therefore detects primarily intracellular proteins, and the values obtained with the saponin treatment were used as the denominators in the ratios (cell surface/internal staining) shown in Fig. 2B, 8B, and 9.
Fig 2.

Differential cell surface and intracellular distributions of mutant constructs in 293 cells stably expressing Kd, wild-type E3/19K, and E3/19K variants. Internal and cell surface expression of constructs was determined by quantitative flow cytometry in the presence and absence of saponin, respectively. MHC-I Kd was detected using MAb 34-1-2, while wild-type E3/19K and E3/19K mutants were detected using MAb Tw1.3. (A) Filled histograms represent the background staining obtained with the secondary antibody alone; open histograms denote specific staining. The MFI is given in the upper right corner of each histogram. (B) Subcellular distribution is expressed as the ratio of the MFI obtained upon cell surface staining to that obtained upon internal staining. At least three independent experiments were performed for at least three different clones of EEE*, EEK, EKK, EKE*, and EKE, two cell lines for EEE, and one cell line for KKK. Data are expressed as means and standard errors of the means.
Fig 8.

The E3/19K TMD confers ER retention on the bona fide cell surface molecule MHC-I Kd. The TMD of the MHC-I Kd molecule (KKK) was replaced by that of E3/19K, yielding KEK. (A) The identity of the KEK domain structure was verified by successful immunoprecipitation (IP) with antibodies against the luminal domain and the respective cytoplasmic tail. Detergent extracts of various control cell lines and 293 cells stably expressing KEK were immunoprecipitated using either a MAb to the luminal α1/α2 domains of Kd (34-1-2) (left) or an antiserum against the cytoplasmic tail (αKd C-tail) (right). 293 cells and K1, a G418-resistant, KEK-negative clone, served as negative controls. In addition, cell lines stably expressing wild-type Kd alone (KKK) (293Kd2 cells) or both wild-type Kd and E3/19K (KKK+EEE) (293.12Kd8 cells) were analyzed. The positions of Kd, KEK, β2-microglobulin (β2m), E3/19K, APLP2, and an unidentified protein (arrow) are indicated on the right, and those of molecular size markers (kDa) are given on the left. (B) The expression of KEK on the cell surface is similar to that of MHC-I Kd in the presence of wild-type E3/19K. 293 cells transfected with KKK+EEE, KKK, KEK, or KKE were analyzed as described above by flow cytometry using MAb 34-1-2 in the presence and absence of saponin. The ratio of cell surface expression to internal expression was calculated. Bars represent means derived from at least 3 independent experiments; error bars, standard errors of the means. Similar data were obtained using MAb SF1.1.1, directed against the α3 domain of Kd (data not shown). (C) The processing pattern and enhanced coprecipitation of APLP2 (100 and 110 kDa) with KEK indicate ER retention. 293 cells stably expressing KEK, KKE, or wild-type Kd in the absence (KKK) (293Kd2 cells) or presence (KKK+EEE) (293.12Kd8 cells) of E3/19K were pulse-labeled for 20 min (0) and then chased for 40 min and 120 min as indicated. The asterisk in lane 6 indicates a Kd species with complex-type carbohydrates. Note the enhanced coprecipitation of the APLP2 p100/p110 bands in cells expressing KKK+EEE, KEK, and KKE compared to that in cells expressing wild-type Kd alone (KKK), which is indicative of ER retention (36).
Fig 9.
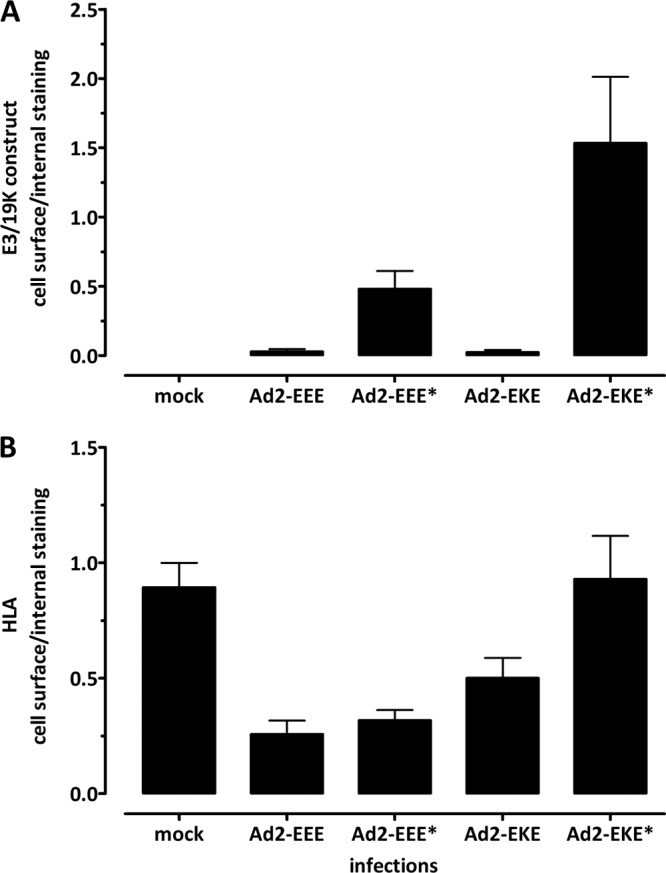
The cell surface and intracellular distributions of E3/19K variants and HLA are confirmed in the context of virus infection. The EEE*, EKE, and EKE* E3/19K mutants were inserted into the Ad2 genome by recombineering, and the reconstituted viruses were used to infect 293 cells. Cells were analyzed 18 h postinfection by flow cytometry for cell surface and internal expression of E3/19K constructs and of HLA by using MAbs Tw1.3 and W6/32, respectively. The ratios of cell surface staining to internal staining of E3/19K constructs (A) and HLA (B) are given as means and standard errors of the means derived from three independent experiments. Similar results were obtained upon infection of HeLa cells (data not shown).
Immunofluorescence.
Immunofluorescence was performed principally as described previously (44), with the modifications indicated below. Cells were fixed for 10 min in 3% paraformaldehyde (Sigma) in phosphate-buffered saline (PBS) and were washed twice with PBS. Internal staining was achieved by permeabilizing the cells for 5 min with 0.5% Triton X-100–PBS. Nonspecific binding sites were blocked for 30 min with 1% bovine serum albumin (BSA)-PBS. This buffer composition was kept throughout the subsequent incubation and washing steps. The primary antibody 3A9 (42) was applied for 1 h as a 1:5 dilution of MAb cell culture supernatant. As a secondary antibody, Cy3-conjugated goat anti-mouse IgG (Dianova) was used in a 1:200 dilution. Preparations were embedded with Vectashield (Serva) and were viewed with a confocal laser scanning microscope (CLSM) (TCS4D; Leica) equipped with a krypton-argon laser.
RESULTS
Constructs and cell lines utilized to identify the domains that contribute to the intracellular localization of E3/19K.
To identify the structural elements that contribute to the intracellular sequestration of E3/19K and HLA molecules, site-directed mutagenesis and domain swapping were employed to systematically manipulate the TMD and/or cytoplasmic tail of the Ad2 E3/19K protein (Fig. 1, EEE). Replacement of the dilysine motif at positions 139 and 140 of E3/19K by serine is denoted by an asterisk. In addition, the TMD (EKE, EKE*) and/or the cytoplasmic tail (EKK, EEK) of E3/19K was replaced by that of the murine MHC-I molecule Kd (Fig. 1, KKK). This MHC-I allele was chosen as a control for the distribution of a typical cell surface protein, since it allowed us to monitor the presence and correct folding of individual domains in the mutant constructs due to the availability of antibodies to various portions of E3/19K and Kd. All mutations were introduced into the EcoRI D fragment of Ad2. By this means, the known Ad-specific control elements, such as the promoter, donor, and acceptor splice sites, translation regulatory elements, and poly(A) signals, were retained. The mutagenized Ad2 fragments were transfected into 293 cells together with the neomycin resistance gene. A total of 25 to 30 cell clones derived from each transfection were screened initially for expression of the E3/19K constructs by quantitative fluorescence-activated cell sorting (FACS) analysis in the presence of the detergent saponin. Clones with E3/19K expression similar to that of cells expressing wild-type E3/19K, as verified by immunoprecipitation after 30 to 60 min of metabolic labeling, were chosen for further analysis. In addition, the relative distributions of mutant proteins on the cell surface and intracellularly were compared with those seen in a cell line constitutively expressing wild-type E3/19K (EEE).
Fig 1.

Schematic representation of wild-type E3/19K (EEE), murine MHC-I Kd (KKK), and various chimeric proteins. The composition of the chimera is indicated on the left by three capital letters representing the ER luminal, transmembrane, and cytoplasmic domains, respectively. An “E” refers to E3/19K-derived sequences (open rectangles) and a “K” to Kd-specific sequences (shaded rectangles). Asterisks denote mutation of the dilysine motif. All constructs were stably transfected into the human embryonic kidney cell line 293.
Efficient expression of E3/19K on the cell surface requires replacement of its transmembrane domain.
In line with the previous suggestion that the dilysine motif is the only structural determinant in the E3/19K protein required for inhibition of transport to the cell surface (20, 29, 34), mutation of this motif resulted in expression of the mutant protein on the cell surface (Fig. 2A, EEE*, bottom), whereas wild-type E3/19K (EEE) did not reach the cell surface. Remarkably, though, internal staining in the presence of the detergent saponin revealed that the majority of the EEE* protein remained intracellular (Fig. 2A; compare the mean fluorescence intensities [MFIs] of 98.5 and 31.9). An inverse distribution was seen for the bona fide plasma membrane protein MHC Kd (KKK), where the majority of the protein was found on the cell surface (MFIs, 55.0 versus 93.2 [Fig. 2A]). The histograms in Fig. 2A show typical results for one cell clone for each transfection. To control for clonal variation, three or more independent clones from each transfection were analyzed in at least three different experiments. The relative subcellular distribution of constructs was expressed as the ratio of the MFI obtained in the absence of saponin to the MFI in the presence of saponin (primarily reflecting cell surface versus internal expression; see Materials and Methods) (Fig. 2B). The collective data clearly confirm that EEE* is predominantly localized intracellularly. Therefore, amino acids in the E3/19K protein other than the dilysine motif in the cytoplasmic tail seem to contribute to intracellular localization. Since replacement of the entire cytoplasmic tail of E3/19K protein by that of Kd (Fig. 1, EEK) resulted in a distribution similar to that for EEE* (Fig. 2A [MFIs, 89.7 versus 36.0] and B), other elements within the C-terminal portion of E3/19K apart from the dilysine motif appear to play a minor role in ER retention. Thus, although EEE* and EEK could be detected on the cell surface, the majority of each of these mutant proteins was localized intracellularly. To assess the impact of additional structural features on intracellular localization, a chimeric E3/19K protein was generated in which both the TMD and the cytoplasmic tail were replaced by the respective Kd sequences (EKK [Fig. 1]). Interestingly, the distribution of EKK differed drastically from those of EEE* and EEK and was comparable to the staining ratio of the plasma membrane protein KKK (Fig. 2A [MFIs, 25.2 versus 88.4] and B). These results suggest that the TMD of Kd promotes cell surface expression and that, conversely, the E3/19K TMD contributes to its efficient intracellular retention. This view was confirmed by the phenotype of a chimeric protein containing the TMD of Kd together with an E3/19K tail where the dilysine motif was mutated (EKE* [Fig. 1]), thus eliminating the impact of ER retrieval. Indeed, EKE* was expressed on the cell surface to an extent similar to those of the EKK and KKK proteins (Fig. 2A and B). Finally, to more precisely delineate the relative importance of the two structural elements, a chimeric protein bearing the TMD of Kd together with the native E3/19K tail (EKE [Fig. 1]) was analyzed. As predicted for a protein with a functional dilysine motif, EKE shows a distribution similar to that of the wild-type E3/19K protein (Fig. 2A and B, compare EEE with EKE). Together, these results demonstrate that both the dilysine motif and the TMD of the E3/19K protein contribute significantly to its efficient intracellular localization, whereas other amino acids in the cytoplasmic tail are of minor importance for subcellular distribution.
The subcellular distribution of the chimeric proteins is consistent with their flow cytometry phenotype.
To confirm the distributions of the constructs between cell surface and intracellular compartments and to obtain more information as to their intracellular localization, immunofluorescence microscopy in the absence and presence of a detergent was performed with MAb 3A9 (42), directed to a formaldehyde-resistant, exposed linear epitope (residues 15 to 21) of the E3/19K protein (Fig. 3). In line with previous observations, EEE was not detected on the cell surface (Fig. 3A). The same holds true for EKE (Fig. 3F). In agreement with the flow cytometry analysis, EEE* and EEK were detected on the cell surface (Fig. 3B and C); however, their expression was generally weaker than that of EKK or EKE* (Fig. 3D and E). When the same cell lines were fixed and treated with detergent to additionally detect E3/19K in cytoplasmic compartments (Fig. 3G to L), cells expressing EKK and EKE* exhibited primarily a cell surface pattern and little internal staining (Fig. 3J, K). This contrasts dramatically with cell lines expressing wild-type E3/19K, which showed bright reticular staining of the perinuclear region, typical for the ER (Fig. 3G). Likewise, this perinuclear rim of fluorescence was present in cell lines expressing EEE*, EEK, and EKE (Fig. 3H, I, and L), with EEE* and EEK exhibiting somewhat higher numbers of vesicular structures. Thus, the chimeras may be classified into three groups. The proteins of the first group, with intact dilysine motifs, are efficiently localized in the ER (EEE, EKE). Also, proteins in the second group, containing the native TMD without a functional dilysine motif (EEE*, EEK), are localized predominantly in the ER at steady state, although the presence of vesicular structures and a low level of cell surface expression indicate transport to other compartments. Finally, the third group comprises proteins with the capacity to be transported efficiently to the cell surface (EKK, EKE*, and KKK).
Fig 3.

Subcellular distribution of wild-type E3/19K and E3/19K mutants as analyzed by immunofluorescence. Transfected 293 cells stably expressing wild-type E3/19K (EEE), EEE*, EEK, EKK, EKE*, or EKE were stained for immunofluorescence microscopy using MAb 3A9 followed by Cy3-conjugated goat anti-mouse IgG. Cells were either left intact for the assessment of expression on the cell surface (A to F) or permeabilized with Triton X-100 for internal staining (G to L).
Manipulations of the cytoplasmic tail and TMD of E3/19K do not significantly affect the conformation of the luminal region.
ER retention of EEE* and EEK might be due to misfolding and activation of the unfolded protein response by quality control systems residing in the ER (45). We therefore sought to find potential evidence for misfolding of the E3/19K mutants and chimeras by analyzing their abilities to be recognized by a panel of conformation-sensitive MAbs against the E3/19K protein (42). However, when the various E3/19K constructs were immunoprecipitated from [35S]methionine-labeled cells using MAbs 3A9, 3F4 (data not shown), and Tw1.3, no significant change in reactivity from that of wild-type E3/19K was observed, indicating that the conformation of the N-terminal luminal domain of E3/19K, present in all constructs, did not seem to be grossly altered by changes in the TMD or the cytoplasmic tail. As an example, immunoprecipitation data are shown for the conformation-sensitive MAb Tw1.3, which recognized all mutant proteins of the expected size with similar efficiencies (Fig. 4). Wild-type E3/19K contains two carbohydrates of the high-mannose type and migrates at approximately 25 kDa, with some faster-migrating species indicative of mannose processing. Accordingly, the predominant ∼25-kDa protein species of EEE, EEE*, EKE*, and EKE represent the fully glycosylated form, whereas the faster-migrating minor species (Fig. 4A, filled arrowheads) represent the respective monoglycosylated forms. Due to the different sizes of the cytoplasmic tails of Kd and E3/19K (40 versus 15 amino acids), the chimeric proteins EEK and EKK exhibit a 2.25-kDa increase in apparent molecular mass (Fig. 4A, lanes 3 and 4). The same results were obtained by using antisera against the respective cytoplasmic tails (data not shown). As expected, an antiserum against the C-terminal 11 amino acids of Kd recognized constructs EEK and EKK, while a serum against the C terminus of E3/19K (C-tail) recognized all other constructs. Thus, the correct apparent molecular weight and the ability of the proteins to be recognized by the corresponding antibodies suggest that all mutants contain the desired sequence alterations. Moreover, the similar efficiencies with which Tw1.3 immunoprecipitates the various constructs show that the alterations introduced into the TMD and cytoplasmic domain do not seem to significantly affect the conformation of the luminal part of E3/19K. Therefore, it is highly unlikely that the predominantly intracellular localization of EEE* and EEK is mediated by a quality control system that recognizes and retains unfolded proteins.
Fig 4.
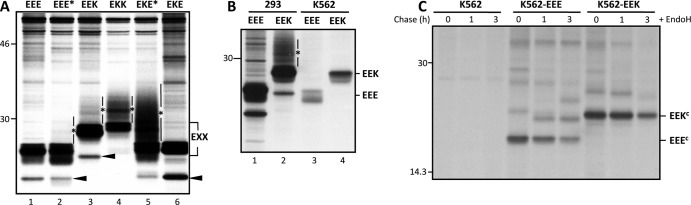
Size and carbohydrate modification of wild-type E3/19K and E3/19K variants as analyzed by immunoprecipitation. (A) E3/19K constructs were immunoprecipitated from lysates of stably transfected cell lines after 1 h of metabolic labeling with [35S]methionine by using MAb Tw1.3, directed to a conformational epitope in the luminal domain of E3/19K. Similar results were obtained after immunoprecipitation with the conformation-dependent MAbs 3A9 and 3F4 (data not shown). Filled arrowheads indicate monoglycosylated forms of E3/19K. The acquisition of complex-type glycans is visualized as slower-migrating protein species above the main labeled band (indicated by a line and an asterisk to the right of the respective lane). The positions of molecular size marker proteins are given on the left. (B) Wild-type E3/19K (EEE) and EEK constructs were immunoprecipitated from lysates of stably transfected 293 and K562 cell lines after 2.5 h of metabolic labeling with [35S]methionine by using MAb Tw1.3. (C) K562 cells and transfectants with wild-type E3/19K and EEK were pulse-labeled for 20 min and were chased for 1 and 3 h. The precipitated material was treated with endo H in order to identify constructs that still contain high-mannose carbohydrates that are cleaved by endo H (c) and therefore have not been transported through the medial Golgi complex.
Carbohydrate modifications reveal the importance of the TMD and cytoplasmic tail for transport inhibition.
The wild-type Ad2 E3/19K contains two N-linked carbohydrates of the high-mannose type characteristic of ER-resident proteins (46), whereas glycoproteins that exit the ER may undergo further modifications in post-ER compartments and incorporate complex-type carbohydrates. Because these processed proteins show slower electrophoretic mobility in SDS-PAGE, the appearance of these forms represents another means of assessing the abilities of the mutant proteins to exit the ER. Such slower-migrating protein species are visualized to some extent for EEK and EEE* but are much more prominent for EKK and EKE*, both containing the TMD of Kd (Fig. 4A, lanes 2 to 5). This conversion to complex-type sugars is indicative of transport through the medial and trans-Golgi apparatus, which was not seen for E3/19K proteins with intact dilysine motifs (EEE and EKE) (Fig. 4A, lanes 1 and 6). Quantitation of band intensities revealed that 44% of EKK, 47% of EKE*, 11% of EEK, and ∼5% of EEE* were processed within the labeling period of 1 h (data not shown). Therefore, the mutation of the dilysine motif principally allowed the exit of the E3/19K protein from the ER to the medial Golgi apparatus, where it was shown to acquire endoglycosidase H-resistant complex carbohydrates (data not shown), in line with previous data for a secreted E3/19K variant (28). The extent of this processing, and thus the degree of transport through the medial Golgi apparatus, is drastically enhanced by the TMD of Kd (Fig. 4A, compare the processing of EEE* and EEK with that of EKK and EKE*). This was verified by the acquisition of endo H-resistant carbohydrates in pulse-chase analysis (data not shown). The limited acquisition of complex-type sugars for EEE* and EEK confirmed the immunofluorescence data and strongly suggests that the constructs lacking dilysine motifs (EEE* and EEK) remain largely in the ER. Because this could be caused by complex formation with HLA, we tested whether transport to the cell surface was increased in the absence of HLA molecules. To this end, wild-type E3/19K and EEK were stably expressed in K562 cells, which lack HLA. While the expression levels of these E3/19K constructs were lower in K562 than in 293 cells due to the lack of E1A, which transactivates the E3 promoter (Fig. 4B), the absence of HLA molecules did not result in enhanced cell surface transport: neither construct showed evidence for processing to a higher apparent molecular weight (Fig. 4B, lane 2, asterisk; compare EEK in 293 and K562 cells) or for acquisition of endo H resistance in pulse-chase analyses (Fig. 4C). This is consistent with the lack of expression of both types of transfectants on the cell surface (data not shown). Therefore, we exclude the possibility that HLA complex formation causes ER retention of constructs with mutated dilysine motifs. Rather, the lack of transport of EEK in K562 cells suggests that the presence of HLA molecules contributes to the limited EEK transport in 293 cells.
The capacity of the E3/19K chimera to bind HLA depends on the nature of the TMD.
To assess whether the manipulations of the E3/19K protein would affect its ability to associate with HLA molecules, complex formation was measured by coimmunoprecipitation. HLA antigens were precipitated using MAb W6/32, directed to HLA-A, -B, and –C (Fig. 5A). In cells expressing wild-type E3/19K (EEE), E3/19K was coprecipitated as an additional band of 25 kDa that was not present in nontransfected cells (Fig. 5A, lanes 1 and 2). E3/19K-related protein species of the expected molecular weight were also seen in cells expressing the mutant E3/19K molecules (Fig. 5A, lanes 3 to 7). Although the visualization of EKK and EKE* was compromised by the heterogenous nature of the coprecipitated band (Fig. 4A), both proteins were clearly visible in longer exposures (data not shown). Thus, all mutants retain the ability to form complexes with HLA molecules. Quantitative phosphorimager analysis of the coprecipitated mutants relative to wild-type E3/19K revealed that the levels of complex formation with HLA by EEE, EEE*, and EEK were comparable within the 1-h labeling period (Fig. 5A, lanes 2 to 4, and B), whereas the amounts of coprecipitating protein were substantially lower in cell lines expressing EKK, EKE*, or EKE (Fig. 5A, lanes 5 to 7, and B). This indicates that the TMD of E3/19K contributes significantly to the efficiency of complex formation with HLA molecules.
Fig 5.
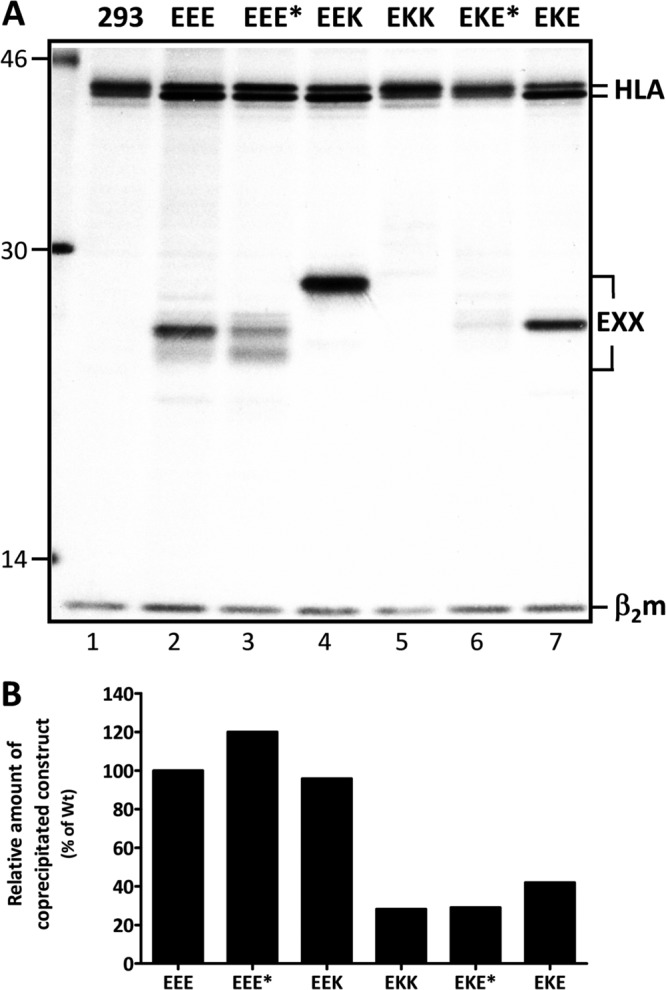
E3/19K constructs exhibit differential complex formation with HLA. 293 cells and cell clones stably expressing E3/19K constructs were metabolically labeled with [35S]methionine for 1 h, and lysates were subjected to immunoprecipitation with MAb W6/32. (A) The positions of HLA-I heavy chains (HLA), the various E3/19K forms (EXX), and β2-microglobulin (β2m) are marked on the right; those of molecular size markers (in kilodaltons) are given on the left. (B) The association with HLA-I of wild-type (Wt) E3/19K and the mutants indicated in panel A was quantitatively assessed by measuring the amount of radioactive E3/19K coprecipitated with HLA and relating it to the total amount expressed as detected by direct precipitation with Tw1.3 using a phosphorimager (data not shown). The ability of each mutant to bind to HLA-I is expressed as the ratio of the radioactivity detected in the coprecipitated band to that detected in the directly precipitated E3/19K band. The ratio obtained for wild-type E3/19K was set to 100%. The data are from a single coprecipitation experiment.
Replacement of the E3/19K TMD and cytoplasmic tail dramatically affects the efficiency of HLA and MICA/B retention.
To determine whether the ability of HLA molecules to interact with the mutant E3/19K constructs correlates with their transport, the carbohydrate processing of HLA molecules was analyzed in a pulse-chase experiment (Fig. 6). Stable transfectants and nontransfected control cells were pulse-labeled for 20 min, washed, and further incubated for 150 min in the presence of an excess of unlabeled methionine. Subsequently, HLA-A2 was precipitated first (Fig. 6A), and thereafter the remaining HLA-A, -B, and -C alleles (Fig. 6B). The processing of HLA-A2 was analyzed separately, since it has been shown to have a high affinity for Ad2 E3/19K (22, 47). Furthermore, by analyzing only one HLA allele, small changes in apparent molecular weight accompanying the conversion of the single HLA glycan to complex-type sugars in the medial and trans-Golgi apparatus become more apparent. Precipitated HLA antigens were digested with endo H, which cleaves high-mannose sugars, resulting in species of HLA that migrate faster (HLA-A2c, HLAc) than the HLA species that acquired endo H-resistant, complex carbohydrates in the medial and trans-Golgi apparatus (HLA-A2R, HLAR). Quantitation of the amounts of endo H-resistant protein in the individual cell lines (Fig. 6, bottom) revealed that processing of HLA antigens in the presence of EKK and EKE* was indistinguishable from that seen in the absence of E3/19K in 293 cells. In contrast, in the presence of wild-type E3/19K (EEE), EEE*, or EKE, essentially all HLA-A2 molecules remained endo H sensitive and thus contained high-mannose-type sugars. Similar results were obtained for the processing of the remaining, lower-affinity HLA alleles (Fig. 6B), except for EEK- and particularly EKE-expressing cells. In cells expressing EEK, a considerably larger proportion of HLA molecules acquired endo H resistance than in the EEE* transfectant. This is not reflected in the distribution of the two E3/19K constructs themselves (Fig. 2B), indicating that amino acids in the cytoplasmic tail of E3/19K other than the dilysine motif may not affect their transport but may have an influence on the interaction with MHC class I molecules. The phenotype of EKE is surprising considering the presence of the native E3/19K retrieval signal but is consistent with its reduced level of complex formation with HLA-A, -B, and -C alleles observed in coprecipitation experiments (Fig. 5) and with the generally lower binding affinity of HLA-B and -C alleles for E3/19K (25, 47).
Fig 6.

Differential transport of HLA molecules in cells expressing E3/19K chimeras. Transport of HLA molecules was analyzed in a pulse-chase experiment (20-min pulse, 2.5-h chase). First, HLA-A2 was immunoprecipitated with MAb BB7.2 (A). The remaining HLA-A, -B, and -C molecules were precipitated with MAb W6/32 (B). Precipitated material was treated with endo H to enable differentiation between molecules transported through the medial Golgi apparatus, where they acquire endo H-resistant (R) complex-type carbohydrates, and those that still contain high-mannose carbohydrates that are cleaved by endo H (c) and therefore have not reached this compartment. The percentage of HLA molecules that have acquired endo H resistance (% endo H resistant) relative to the total amount of HLA precipitated is shown separately in corresponding bar diagrams for HLA-A2 (A) and the remaining HLA molecules (B).
Finally, the steady-state expression of HLA and MICA/B on the cell surface was quantitatively determined using flow cytometry. At least three different cell clones derived from each transfection were analyzed using MAb W6/32 (Fig. 7A). The HLA expression level in 293 cells and three G418-resistant, E3/19K-negative cell lines was set at 100%. The results confirm the data obtained from coprecipitation and pulse-chase analysis. As shown previously, HLA expression on the cell surface is drastically reduced in the presence of wild-type E3/19K (EEE) (12–14). In line with the pulse-chase experiments, the removal of the ER retrieval signal in EEE* and EEK alleviates the transport block, whereby HLA cell surface expression reached considerably higher levels in EEK- than in EEE*-expressing cell lines (reduced by 34% versus 66%). Some reduction in the level of HLA expression (by approximately 25%) was visible in EKK transfectants, whereas cell lines expressing EKE* exhibited normal HLA levels. Remarkably, despite the presence of the ER retrieval signal in EKE, HLA cell surface expression at steady state is only slightly suppressed in EKE-expressing cell lines, consistent with the reduced ability to interact with HLA molecules and the ability to acquire endo H resistance. Expression of HLA-A2 on the plasma membrane correlated with the pulse-chase results (data not shown). Of note, the cell surface expression pattern for MICA/B, the other known target molecule of E3/19K, was largely similar in these transfectants (Fig. 7B). This indicates that the downregulation of both MICA/B and MHC-I molecules similarly depends on the TMD and cytoplasmic tail of E3/19K.
Fig 7.
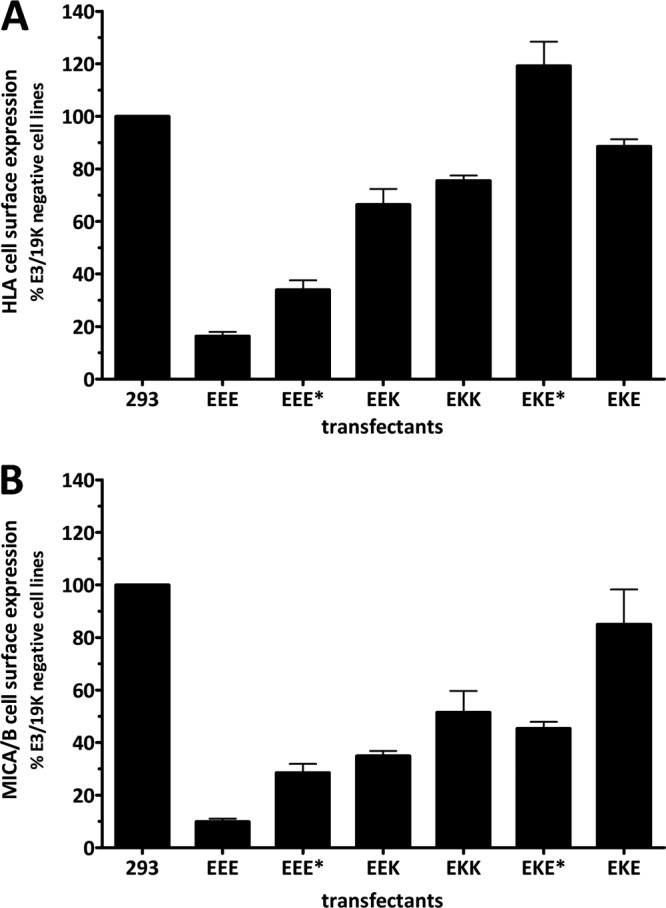
Relative cell surface expression of the E3/19K target molecules HLA and MICA/B in cell clones expressing mutant and wild-type E3/19K. Steady-state expression of HLA (A) and MICA/B (B) molecules on the cell surface was determined by flow cytometry using MAbs W6/32 and BAMO3, respectively, followed by incubation with Alexa Fluor 488-coupled goat anti-mouse IgG. At least three clones from each transfection were analyzed in at least two independent experiments. The background staining obtained with the secondary antibody alone was subtracted, and the MFI was related to that for 293 cells and three E3/19K-negative transfectant clones (293), which was set to 100%. Bars represent mean values; error bars, standard errors of the means.
The E3/19K TMD actively suppresses the expression of a bona fide plasma membrane protein on the cell surface.
The data presented above show that the MHC-I TMD promotes cell surface expression of E3/19K and, conversely, that the E3/19K TMD has features that contribute to the retention of E3/19K and HLA molecules in the ER. To directly investigate whether the TMD alone is sufficient to promote ER retention and suppress cell surface expression of a heterologous bona fide plasma membrane protein, the Kd TMD was replaced by that of E3/19K to yield KEK (Fig. 1). Stable 293 transfectants were screened for expression of KEK by flow cytometry in the presence and absence of saponin using MAbs SF1.1.1 and 34-1-2, directed to the α3 and α1/α2 domains of Kd, respectively. Successful immunoprecipitations with 34-1-2 and antibodies directed to the cytoplasmic tail of Kd (C-tail) confirmed the presence of the respective domains in KEK and the expected molecular weight (Fig. 8A). Nontransfected 293 cells and KEK-negative, G418-resistant cell clones, such as K1, exhibited only background bands. As shown in Fig. 8B, KEK-expressing cell clones exhibited drastically reduced cell surface expression and, concomitantly, increased intracellular staining relative to wild-type Kd (KKK), a pattern very similar to that of Kd in cells coexpressing wild-type E3/19K (KKK+EEE). As expected, a hybrid Kd molecule with its cytoplasmic tail replaced by that of E3/19K (KKE) (Fig. 1) showed an exclusively intracellular localization (Fig. 8B). The surface expression of other typical cell surface molecules, such as CD46 or HLA, was not significantly affected by KEK expression (data not shown).
Taken together, these results demonstrate that the TMD of E3/19K can dominantly convert a plasma membrane protein into an intracellular protein; thus, it contains features that favor intracellular localization/ER retention. Comparative pulse-chase analyses of KEK and KKE with wild-type Kd in the presence and absence of E3/19K (KKK+EEE and KKK, respectively) clearly indicated that both constructs are retained in the ER. Unlike wild-type Kd (KKK), which shows the typical processing associated with the acquisition of complex-type sugars (Fig. 8C, lanes 4 to 6, asterisk), KEK and KKE failed to acquire complex-type sugars, like wild-type Kd in the presence of E3/19K (KKK+EEE) (Fig. 8C, compare lanes 7 to 12 with lanes 1 to 3). The progressively lower apparent molecular weights of the chased Kd molecule, reflecting mannose trimming in the ER, and the drastically enhanced complex formation with the 100- and 110-kDa species of the amyloid precursor-like protein 2 (APLP2) (Fig. 8C, compare lanes 7 to 12 with lanes 1 to 3), which is known to occur when transport of Kd out of the ER is inhibited, e.g., by coexpression of E3/19K (36), provide independent evidence for ER localization of KEK, KKE, and Kd in the presence of E3/19K. The close correlation between APLP2 coimmunoprecipitation and ER localization is confirmed by the loss of association of the two APLP2 species upon the transport of Kd out of the ER and the acquisition of complex-type sugars during the chase period (Fig. 8C, lanes 4 to 6).
ER retention by the E3/19K TMD also occurs during virus infection.
To assess the physiological relevance of our findings, the functional activity of the newly discovered ER retention signal in the E3/19K TMD was analyzed during viral infection. To this end, the E3/19K mutants EEE*, EKE*, and EKE were introduced into the Ad2 genome using recombineering (38, 39). HeLa cells (data not shown) and 293 cells were infected with wild-type Ad2, Ad2 viruses expressing the modified E3/19K molecules, and Ad2 dl810, an Ad2 lacking all E3 genes, as a negative control (48) (data not shown). As indicated by the ratios of cell surface expression to internal staining, the distributions of the mutant E3/19K molecules (Fig. 9A) and HLA target molecules (Fig. 9B) were essentially the same as those in the transfection system (Fig. 2 and 7). Wild-type E3/19K (EEE) and EKE were exclusively found intracellularly, whereas some cell surface expression and an increased ratio were seen for EEE*. Remarkably, Ad2-mediated expression of EKE* resulted in a ratio typical for a cell surface protein. The HLA expression profile was consistent with that of the transfected cell lines (Fig. 7) and correlated with that of the E3/19K mutant constructs except for EKE, where downregulation of HLA was only modest, even though EKE was completely intracellular. Thus, the results obtained by the transfection system used here for most experiments were essentially reproduced during virus infection, suggesting that the newly discovered functional activities of the TMD are physiologically relevant.
DISCUSSION
The adenovirus E3/19K protein subverts the recognition of T cells and NK cells by retaining MHC-I and MICA/B molecules in the ER (11, 12, 15, 24). To date, this capacity was thought to be based on the combined action of two functional entities: the ER luminal domain, which binds to MHC-I and MICA/B molecules, and an ER retrieval signal in the cytoplasmic tail that mediates the retrograde transport of E3/19K with its attached target molecules from the ER-Golgi intermediate compartment (ERGIC)/cis-Golgi to the ER (20, 21, 28, 29). In this study, we show that the TMD of E3/19K represents an additional functional element that crucially contributes both to efficient ER localization of E3/19K and to the downregulation of target molecules. This conclusion was reached by analyzing a series of chimeric constructs that contained the luminal domain of E3/19K together with the TMD and/or cytoplasmic tail of the bona fide plasma membrane protein MHC-I Kd. Analysis of these E3/19K–MHC-I chimeras revealed the importance of the E3/19K TMD in this process, since its replacement led to high cell surface expression. Conversely, replacement of the TMD of Kd by that of E3/19K was sufficient to cause ER retention, showing the dominant activity of the latter. Based on these data, we propose that the TMD of E3/19K mediates static ER retention. In agreement with this conclusion, the TMD contributes significantly to the binding and retention of HLA-I and MICA/B molecules, since its replacement severely compromised the downregulation of these molecules.
Figure 10 illustrates the transport characteristics of the various E3/19K molecules and the HLA molecule and summarizes the overall efficiency of HLA-I downregulation. Chimeras with similar phenotypes are grouped together. In wild-type E3/19K (EEE), the ER retrieval signal and the ER retention signal of the TMD act in concert to maintain a high concentration of E3/19K in the ER, where it efficiently associates with its target molecules (Fig. 10A). Without the ER retrieval signal, but in the presence of the cognate E3/19K TMD (Fig. 10B, EEE* and EEK), the bulk of these constructs remains in the ER through static ER retention, and only a small fraction of E3/19K-HLA complexes is further transported to the cell surface. The predominant intracellular retention of these constructs is not caused by complex formation with HLA in the ER, because there was no transport in HLA-negative cells. The higher HLA cell surface expression observed for EEK-expressing cells than for EEE*-expressing cells may suggest that residues in the cytoplasmic tail other than the ER retrieval signal impact on the efficiency of HLA downregulation, as previously suggested (29). In the absence of both the dilysine motif and the E3/19K TMD (Fig. 10C, EKK and EKE*), there is no static ER retention, and consequently, large amounts of chimeric proteins rapidly exit the ER. Due to the lack of an ER retrieval signal, these constructs are further transported through the Golgi apparatus to the cell surface. Thus, these constructs have a distribution and cell surface expression comparable to that of typical surface proteins, such as Kd. It is not clear to what extent EKE* and EKK reach the cell surface in complex with HLA molecules. Coimmunoprecipitation of these constructs is substantially lower than for wild-type E3/19K and the other mutants with a native E3/19K TMD. However, these constructs can still be coprecipitated after 1 h of chase (data not shown), indicating that considerable fractions of EKK and EKE* remain associated with HLA molecules during transport to, and possibly on, the cell surface. As expected, no cell surface expression was demonstrated for EKE, which, due to the absence of the E3/19K TMD, is rapidly transported from the ER to the ERGIC/cis Golgi but is also efficiently returned to the ER owing to the presence of the ER retrieval signal (Fig. 10D). Surprisingly, this was not the case for the majority of HLA-I and MICA/B molecules in those cells, which were transported further to the cell surface. This suggests that complexes with EKE that reach the ERGIC/cis-Golgi compartment are prone to dissociation, releasing the HLA and MICA/B molecules for transport to the cell surface (Fig. 10D). Conversely, the E3/19K TMD seems to prevent rapid ER export and hence dissociation and thus contributes to efficient HLA downregulation. Viral infection with corresponding Ad2 mutants confirmed the important functional contributions of the TMD and the cytoplasmic tail. Taking these findings together, this model can explain the transport characteristics of the chimeras and their effects on HLA cell surface expression. Accordingly, it is the combination of static ER retention provided by the polar TMD and ER retrieval provided by the dilysine motif that brings about efficient HLA and MICA/B downmodulation.
Fig 10.

Summary of transport characteristics of wild-type E3/19K and chimeras and their effects on HLA transport in transfected cells. The transport of E3/19K constructs and HLA molecules between the ER and the ERGIC/cis-Golgi complex, and from there to the plasma membrane, is indicated by filled and shaded arrows, respectively. The thickness of the arrows represents the intensity of transport. Mutants are grouped in panels A to D according to their transport behavior. The capacity for static ER retention and the extent of HLA downregulation are indicated semiquantitatively on the left and right, respectively.
Our findings with EEK and EEE* are in line with previous studies using C-terminally deleted E3/19K proteins, which also showed cell surface expression (13, 29). However, since the intracellular expression was not concomitantly quantified, those studies provided no information on the relative distribution of constructs to the cell surface versus intracellular compartments (13, 19, 29). Moreover, the previous studies did not specifically assess the role of the TMD. Deletion analyses have shown that E3/19K mutants truncated within the luminal domain (at residue 96) adjacent to the TMD or within the TMD exhibited no interaction with HLA molecules and were secreted. In contrast, constructs truncated at a point C-terminal of the TMD within the cytoplasmic tail associated with HLA molecules and were expressed on the surface (28, 31). This may suggest that in vivo, the luminal domain alone, without a TMD or a cytoplasmic tail, is insufficient for HLA interaction. However, in those studies, the possibility that misfolding, e.g., due to inappropriate disulfide bond formation, was the potential cause for the loss of complex formation could not be ruled out. Furthermore, the question of whether the TMD was required as a mere membrane anchor or had additional functions remained open. In contrast, the conformation of the luminal domains in the constructs generated here does not seem to be significantly altered, since all conformation-dependent MAbs tested showed similar efficiencies in precipitating the mutant and wild-type E3/19K molecules (Fig. 4). This panel of MAbs was shown to be highly sensitive to conformational changes induced by mutations of individual amino acids, small deletions, or disruption of intramolecular disulfide bonds (23, 24, 42, 49). Our data emphasize the crucial importance of the TMD not as a mere membrane-anchoring device but rather as a functional module that both mediates static ER retention and enhances the efficiency of complex formation with target molecules. In light of our data, the loss of interaction upon deletion of the TMD (28, 31) might be explained by the lack of static ER retention and ER retrieval, leading to an insufficient concentration of E3/19K in the ER. These in vivo data are not necessarily in conflict with the successful interaction of the purified soluble luminal E3/19K domain with HLA molecules in vitro, since this may have been forced by the high protein concentrations used in those systems (22, 25, 26, 28, 31).
Due to the common function of all E3/19K proteins examined to date (7, 12, 17, 25, 50), the specific feature of the TMD, identified here as promoting ER retention, should be preserved in E3/19K proteins. However, none of the 20 strictly conserved amino acids of E3/19K is localized in the TMD (24). Also, its length is not distinctly different from those of TMDs of plasma membrane proteins (23 amino acids in E3/19K versus 24 in Kd) (51). A well-conserved property of the TMDs of all E3/19K proteins, however, is the unusually high number of polar amino acids. Nine polar residues are present in species C (e.g., in the Ad2 E3/19K protein), 9 to 12 in species B, 9 in species E, and 4 in species D (4). Therefore, the high number of polar amino acids in the E3/19K TMD could contribute to static ER retention. In contrast, the TMD of Kd, used here as a bona fide plasma membrane protein to replace the corresponding domain of E3/19K, contains only two polar residues. While we cannot recognize major differences from the TMDs of other typical cell surface proteins, such as CD4, CD8, or the interleukin 2 (IL-2) receptor alpha chain (21, 32, 52), used in similar domain-swapping studies, we cannot completely rule out the possibility that those TMDs would give identical results. It will be interesting to mutate the various polar amino acids individually and in combination so as to elucidate their potential role in this process. Moreover, it is tempting to speculate as to whether lower numbers of polar amino acids in the TMDs of other E3/19K proteins may in fact be less potent at promoting ER retention of target molecules. In support of this, the Ad19a E3/19K protein, with four polar amino acids in the TMD, is less efficient than Ad2 E3/19K at downregulating HLA alleles (17). Apart from E3/19K, numerous other proteins have been described where uncharged polar or charged amino acids in the TMD contribute to efficient ER localization or are otherwise involved in regulating the transport of protein complexes to the cell surface (45). Examples include the B-cell immunoglobulin receptor (54, 55), cytochrome P450 (56, 57), and the ER-resident protein UDP-glucuronosyltransferase (GT) (58). Interestingly, like E3/19K, the latter contains a dilysine motif, the mutation of which also does not affect the predominant ER localization (59). In addition, various viral proteins have been reported where ER localization is mediated by TMDs, although in many cases ER retention is more complex, involving multiple TMDs, e.g., the E1 and E2 complexes of hepatitis C virus and rubella virus (59, 60). In both cases, these represent viral envelope proteins that have to assemble in the ER to mediate virus budding or are retained individually to avoid the export of unassembled protein complexes. In light of the striking polarity of the E3/19K TMD, this quality control mechanism may have been exploited for the immune evasion function of E3/19K, in that it guarantees a high local concentration in the ER membrane for efficient sequestration of HLA and MICA/B molecules.
We cannot rule out the possibility that the E3/19K TMD, apart from increasing the local concentration in the ER and increasing the time for interaction, may also directly enhance complex formation with HLA molecules. Since the proposed ER retention function impacts on complex formation, these two phenomena are interrelated and cannot easily be separated. Based on the fact that polar amino acids tend to interact with other polar TMDs (61), the polarity of the E3/19K TMD is unlikely to directly mediate interaction with the nonpolar TMD of HLA molecules. Rather, the E3/19K TMD may mediate self-association, and this may be required for efficient HLA complex formation. Accordingly, the E3/19K TMD may constitute the inner core of the complex, with the larger HLA molecule covering the exposed surface. In support of this, we consistently observe efficient coprecipitation of E3/19K by using MAbs against HLA molecules, yet very little if any coprecipitation of HLA molecules by use of E3/19K-specific MAbs. Oligomerization may also be mediated by cysteines, since all E3/19K molecules have at least 1 cysteine in the TMD, and those of Ad species B, C, and E have 2 to 5 cysteines. In support of this notion, we and others previously provided evidence for the existence of disulfide-linked E3/19K dimers (13, 23, 28), although no individual cysteine could be assigned to this phenomenon. For species D E3/19K, oligomerization may also involve a GxxxG motif in the TMD, which has been shown to serve as a module to support interactions between TMD helices (62–64). Thus, the E3/19K TMD may have an additional role in promoting oligomerization, thereby contributing to improved complex formation with target molecules. Further mutagenesis analysis will be necessary to clarify the roles of these amino acids in oligomerization and HLA or MICA/B downregulation.
It is not immediately obvious why two elements for ER localization should be employed for E3/19K function, since the dilysine motif is highly efficacious and does not require an additional element to ensure its ER localization. However, our findings clearly show that neither the ER retrieval signal on its own nor the TMD alone can provide complete suppression of HLA and MICA/B cell surface levels. Given the efficiency of ER localization by the ER retrieval signal, we suggest that the TMD of E3/19K has been evolutionarily selected to maximize its immunomodulatory function.
ACKNOWLEDGMENTS
We are grateful to A. Steinle and G. Kettner for the gifts of antibodies, plasmids, and Ad dl810. We thank C. Ebenau-Jehle and A. Osterlehner for excellent technical assistance. We thank Robert Spooner and Ann Dixon (University of Warwick) for critical readings of the manuscript and helpful comments.
Footnotes
Published ahead of print 20 March 2013
REFERENCES
- 1. Wold WSM, Horwitz MS. 2007. Adenoviruses, p 2395–2436 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. 2009. Latent species C adenoviruses in human tonsil tissues. J. Virol. 83:2417–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berk AJ. 2007. Adenoviridae: the viruses and their replication, p 2355–2394 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4. Burgert HG, Ruzsics Z, Obermeier S, Hilgendorf A, Windheim M, Elsing A. 2002. Subversion of host defense mechanisms by adenoviruses. Curr. Top. Microbiol. Immunol. 269:273–318 [DOI] [PubMed] [Google Scholar]
- 5. Echavarría M. 2008. Adenoviruses in immunocompromised hosts. Clin. Microbiol. Rev. 21:704–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto M, Curiel DT. 2010. Current issues and future directions of oncolytic adenoviruses. Mol. Ther. 18:243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Windheim M, Hilgendorf A, Burgert HG. 2004. Immune evasion by adenovirus E3 proteins: exploitation of intracellular trafficking pathways. Curr. Top. Microbiol. Immunol. 273:29–85 [DOI] [PubMed] [Google Scholar]
- 8. Ginsberg HS, Lundholm-Beauchamp U, Horswood RL, Pernis B, Wold WS, Chanock RM, Prince GA. 1989. Role of early region 3 (E3) in pathogenesis of adenovirus disease. Proc. Natl. Acad. Sci. U. S. A. 86:3823–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horwitz MS, Efrat S, Christen U, von Herrath MG, Oldstone MB. 2009. Adenovirus E3 MHC inhibitory genes but not TNF/Fas apoptotic inhibitory genes expressed in beta cells prevent autoimmune diabetes. Proc. Natl. Acad. Sci. U. S. A. 106:19450–19454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burgert HG, Blusch JH. 2000. Immunomodulatory functions encoded by the E3 transcription unit of adenoviruses. Virus Genes 21:13–25 [PubMed] [Google Scholar]
- 11. McSharry BP, Burgert HG, Owen DP, Stanton RJ, Prod'homme V, Sester M, Koebernick K, Groh V, Spies T, Cox S, Little AM, Wang EC, Tomasec P, Wilkinson GW. 2008. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 82:4585–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burgert HG, Kvist S. 1985. An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 41:987–997 [DOI] [PubMed] [Google Scholar]
- 13. Cox JH, Bennink JR, Yewdell JW. 1991. Retention of adenovirus E19 glycoprotein in the endoplasmic reticulum is essential to its ability to block antigen presentation. J. Exp. Med. 174:1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burgert HG, Maryanski JL, Kvist S. 1987. “E3/19K” protein of adenovirus type 2 inhibits lysis of cytolytic T lymphocytes by blocking cell-surface expression of histocompatibility class I antigens. Proc. Natl. Acad. Sci. U. S. A. 84:1356–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hansen TH, Bouvier M. 2009. MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 9:503–513 [DOI] [PubMed] [Google Scholar]
- 16. Jonjic S, Babic M, Polic B, Krmpotic A. 2008. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 20:30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deryckere F, Burgert HG. 1996. Early region 3 of adenovirus type 19 (subgroup D) encodes an HLA-binding protein distinct from that of subgroups B and C. J. Virol. 70:2832–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Flomenberg P, Szmulewicz J, Gutierrez E, Lupatkin H. 1992. Role of the adenovirus E3-19k conserved region in binding major histocompatibility complex class I molecules. J. Virol. 66:4778–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nilsson T, Jackson M, Peterson PA. 1989. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell 58:707–718 [DOI] [PubMed] [Google Scholar]
- 20. Jackson MR, Nilsson T, Peterson PA. 1990. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 9:3153–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jackson MR, Nilsson T, Peterson PA. 1993. Retrieval of transmembrane proteins to the endoplasmic reticulum. J. Cell Biol. 121:317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu H, Fu J, Bouvier M. 2007. Allele- and locus-specific recognition of class I MHC molecules by the immunomodulatory E3-19K protein from adenovirus. J. Immunol. 178:4567–4575 [DOI] [PubMed] [Google Scholar]
- 23. Sester M, Burgert HG. 1994. Conserved cysteine residues within the E3/19K protein of adenovirus type 2 are essential for binding to major histocompatibility complex antigens. J. Virol. 68:5423–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sester M, Koebernick K, Owen D, Ao M, Bromberg Y, May E, Stock E, Andrews L, Groh V, Spies T, Steinle A, Menz B, Burgert HG. 2010. Conserved amino acids within the adenovirus 2 E3/19K protein differentially affect downregulation of MHC class I and MICA/B proteins. J. Immunol. 184:255–267 [DOI] [PubMed] [Google Scholar]
- 25. Fu J, Li L, Bouvier M. 2011. Adenovirus E3-19K proteins of different serotypes and subgroups have similar, yet distinct, immunomodulatory functions toward major histocompatibility class I molecules. J. Biol. Chem. 286:17631–17639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L, Muzahim Y, Bouvier M. 2012. Crystal structure of adenovirus E3-19K bound to HLA-A2 reveals mechanism for immunomodulation. Nat. Struct. Mol. Biol. 19:1176–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H, Stafford WF, Bouvier M. 2005. The endoplasmic reticulum lumenal domain of the adenovirus type 2 E3-19K protein binds to peptide-filled and peptide-deficient HLA-A*1101 molecules. J. Virol. 79:13317–13325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pääbo S, Weber F, Nilsson T, Schaffner W, Peterson PA. 1986. Structural and functional dissection of an MHC class I antigen-binding adenovirus glycoprotein. EMBO J. 5:1921–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pääbo S, Bhat BM, Wold WS, Peterson PA. 1987. A short sequence in the COOH-terminus makes an adenovirus membrane glycoprotein a resident of the endoplasmic reticulum. Cell 50:311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gabathuler R, Kvist S. 1990. The endoplasmic reticulum retention signal of the E3/19K protein of adenovirus type 2 consists of three separate amino acid segments at the carboxy terminus. J. Cell Biol. 111:1803–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gabathuler R, Levy F, Kvist S. 1990. Requirements for the association of adenovirus type 2 E3/19K wild-type and mutant proteins with HLA antigens. J. Virol. 64:3679–3685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Teasdale RD, Jackson MR. 1996. Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 12:27–54 [DOI] [PubMed] [Google Scholar]
- 33. Zerangue N, Malan MJ, Fried SR, Dazin PF, Jan YN, Jan LY, Schwappach B. 2001. Analysis of endoplasmic reticulum trafficking signals by combinatorial screening in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 98:2431–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lapham CK, Bacik I, Yewdell JW, Kane KP, Bennink JR. 1993. Class I molecules retained in the endoplasmic reticulum bind antigenic peptides. J. Exp. Med. 177:1633–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kvist S, Roberts L, Dobberstein B. 1983. Mouse histocompatibility genes: structure and organisation of a Kd gene. EMBO J. 2:245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sester M, Feuerbach D, Frank R, Preckel T, Gutermann A, Burgert HG. 2000. The amyloid precursor-like protein 2 associates with the major histocompatibility complex class I molecule Kd. J. Biol. Chem. 275:3645–3654 [DOI] [PubMed] [Google Scholar]
- 37. Körner H, Fritzsche U, Burgert HG. 1992. Tumor necrosis factor alpha stimulates expression of adenovirus early region 3 proteins: implications for viral persistence. Proc. Natl. Acad. Sci. U. S. A. 89:11857–11861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruzsics Z, Wagner M, Osterlehner A, Cook J, Koszinowski U, Burgert HG. 2006. Transposon-assisted cloning and traceless mutagenesis of adenoviruses: development of a novel vector based on species D. J. Virol. 80:8100–8113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hilgendorf A, Lindberg J, Ruzsics Z, Honing S, Elsing A, Lofqvist M, Engelmann H, Burgert HG. 2003. Two distinct transport motifs in the adenovirus E3/10.4–14.5 proteins act in concert to down-modulate apoptosis receptors and the epidermal growth factor receptor. J. Biol. Chem. 278:51872–51884 [DOI] [PubMed] [Google Scholar]
- 40. Parham P, Brodsky FM. 1981. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 3:277–299 [DOI] [PubMed] [Google Scholar]
- 41. Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, Rammensee HG, Steinle A. 2003. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur. J. Immunol. 33:194–203 [DOI] [PubMed] [Google Scholar]
- 42. Menz B, Sester M, Koebernick K, Schmid R, Burgert HG. 2008. Structural analysis of the adenovirus type 2 E3/19K protein using mutagenesis and a panel of conformation-sensitive monoclonal antibodies. Mol. Immunol. 46:16–26 [DOI] [PubMed] [Google Scholar]
- 43. Burgert HG, Kvist S. 1987. The E3/19K protein of adenovirus type 2 binds to the domains of histocompatibility antigens required for CTL recognition. EMBO J. 6:2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Windheim M, Burgert HG. 2002. Characterization of E3/49K, a novel, highly glycosylated E3 protein of the epidemic keratoconjunctivitis-causing adenovirus type 19a. J. Virol. 76:755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ellgaard L, Helenius A. 2003. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4:181–191 [DOI] [PubMed] [Google Scholar]
- 46. Kornfeld R, Wold WS. 1981. Structures of the oligosaccharides of the glycoprotein coded by early region E3 of adenovirus 2. J. Virol. 40:440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beier DC, Cox JH, Vining DR, Cresswell P, Engelhard VH. 1994. Association of human class I MHC alleles with the adenovirus E3/19K protein. J. Immunol. 152:3862–3872 [PubMed] [Google Scholar]
- 48. Berg M, Difatta J, Hoiczyk E, Schlegel R, Ketner G. 2005. Viable adenovirus vaccine prototypes: high-level production of a papillomavirus capsid antigen from the major late transcriptional unit. Proc. Natl. Acad. Sci. U. S. A. 102:4590–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hermiston TW, Tripp RA, Sparer T, Gooding LR, Wold WS. 1993. Deletion mutation analysis of the adenovirus type 2 E3-gp19K protein: identification of sequences within the endoplasmic reticulum lumenal domain that are required for class I antigen binding and protection from adenovirus-specific cytotoxic T lymphocytes. J. Virol. 67:5289–5298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pääbo S, Nilsson T, Peterson PA. 1986. Adenoviruses of subgenera B, C, D, and E modulate cell-surface expression of major histocompatibility complex class I antigens. Proc. Natl. Acad. Sci. U. S. A. 83:9665–9669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lankford SP, Cosson P, Bonifacino JS, Klausner RD. 1993. Transmembrane domain length affects charge-mediated retention and degradation of proteins within the endoplasmic reticulum. J. Biol. Chem. 268:4814–4820 [PubMed] [Google Scholar]
- 52. Hennecke S, Cosson P. 1993. Role of transmembrane domains in assembly and intracellular transport of the CD8 molecule. J. Biol. Chem. 268:26607–26612 [PubMed] [Google Scholar]
- 53. Reference deleted. [Google Scholar]
- 54. Cherayil BJ, MacDonald K, Waneck GL, Pillai S. 1993. Surface transport and internalization of the membrane IgM H chain in the absence of the Mb-1 and B29 proteins. J. Immunol. 151:11–19 [PubMed] [Google Scholar]
- 55. Williams GT, Venkitaraman AR, Gilmore DJ, Neuberger MS. 1990. The sequence of the mu transmembrane segment determines the tissue specificity of the transport of immunoglobulin M to the cell surface. J. Exp. Med. 171:947–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Murakami K, Mihara K, Omura T. 1994. The transmembrane region of microsomal cytochrome P450 identified as the endoplasmic reticulum retention signal. J. Biochem. 116:164–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Szczesna-Skorupa E, Ahn K, Chen CD, Doray B, Kemper B. 1995. The cytoplasmic and N-terminal transmembrane domains of cytochrome P450 contain independent signals for retention in the endoplasmic reticulum. J. Biol. Chem. 270:24327–24333 [DOI] [PubMed] [Google Scholar]
- 58. Barré L, Magdalou J, Netter P, Fournel-Gigleux S, Ouzzine M. 2005. The stop transfer sequence of the human UDP-glucuronosyltransferase 1A determines localization to the endoplasmic reticulum by both static retention and retrieval mechanisms. FEBS J. 272:1063–1071 [DOI] [PubMed] [Google Scholar]
- 59. Cocquerel L, Duvet S, Meunier JC, Pillez A, Cacan R, Wychowski C, Dubuisson J. 1999. The transmembrane domain of hepatitis C virus glycoprotein E1 is a signal for static retention in the endoplasmic reticulum. J. Virol. 73:2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hobman TC, Lemon HF, Jewell K. 1997. Characterization of an endoplasmic reticulum retention signal in the rubella virus E1 glycoprotein. J. Virol. 71:7670–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dixon AM, Stanley BJ, Matthews EE, Dawson JP, Engelman DM. 2006. Invariant chain transmembrane domain trimerization: a step in MHC class II assembly. Biochemistry 45:5228–5234 [DOI] [PubMed] [Google Scholar]
- 62. Moore DT, Berger BW, DeGrado WF. 2008. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure 16:991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Unterreitmeier S, Fuchs A, Schaffler T, Heym RG, Frishman D, Langosch D. 2007. Phenylalanine promotes interaction of transmembrane domains via GxxxG motifs. J. Mol. Biol. 374:705–718 [DOI] [PubMed] [Google Scholar]
- 64. Langosch D, Arkin IT. 2009. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 18:1343–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]