

Abstract
In polarized T cells, HIV-1 Gag localizes to a rear-end protrusion known as the uropod in a multimerization-dependent manner. Gag-laden uropods participate in formation of virological synapses, intercellular contact structures that play a key role in cell-to-cell HIV-1 transmission. Our previous observations suggest that Gag associates with uropod-directed microdomains (UDMs) that eventually comigrate with Gag to the uropod over the cell surface. However, the nature of Gag multimerization required for this movement, the composition of the UDMs, and the molecular determinants for Gag association with these microdomains remain unknown. In this study, we found that Gag multimerization prior to budding but beyond dimerization is necessary for Gag localization to the uropods, indicating that uropod localization occurs early in the assembly process. We also found that prior to membrane curvature, Gag multimers associate with a specific subset of UDMs containing PSGL-1, CD43, and CD44 but not ICAM-1, ICAM-3, or CD59. Notably, upon association, Gag excludes ICAM-3 from this subset of UDMs, revealing an active and selective reorganization of these microdomains by Gag. This specific association between Gag and UDMs is dependent on the highly basic region (HBR) in the Gag matrix (MA) domain. The overall positive charge of the HBR was needed for the interaction with the specific UDM subset, while the exact HBR sequence was not, unlike that seen for MA binding to the plasma membrane phospholipid phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2]. Taken totogether, these findings revealed that HIV-1 Gag associates with specific microdomains present in polarized T cells in an MA-dependent manner, which results in modification of the microdomain constituents.
INTRODUCTION
T cells are among the natural targets of HIV-1. Accumulating evidence obtained using cultured T cells suggests that HIV-1 spreads efficiently via cell-to-cell transmission (1–5). T cells infected with HIV-1 or human T lymphotropic virus type I (HTLV-I) have been observed to form cell contact structures known as virological synapses at which massive transfer of newly assembled or assembling virus particles to uninfected cells takes place (3, 6, 7). A recent study observed formation of virological synapse by cells infected with murine leukemia virus (MLV) in vivo in lymphoid organs, where cell-to-cell transmission likely occurs frequently (8). In these locations, T cells, including HIV-1-infected ones (9), adopt a polarized morphology (9–14). Therefore, understanding the mechanisms of HIV-1 localization, assembly, and transmission in polarized T cells could provide better insights into how virological synapses form and how HIV-1 is spread in vivo.
Polarized T cells form two ends, the leading edge at the front and a protrusion at the rear called a uropod. Uropods are enriched in several adhesion molecules containing single transmembrane domains, including PSGL-1, CD43, CD44, ICAM-1, and ICAM-3 (15–19), as well as the lipid raft marker CD59 (a glycosylphosphatidylinositol [GPI]-anchored protein) (15). Functionally, uropods are important for T cell migration and are known to mediate cell-cell contacts (20–22). Previously, we showed that the HIV-1 structural protein Gag localizes to uropods in polarized T cells and that Gag-laden uropods then participate in virological synapses between HIV-1-infected and uninfected T cells (23).
HIV-1 Gag is synthesized as a polyprotein, the expression of which is sufficient for formation of virus-like particles (VLPs) (24, 25). Gag is composed of four structural domains, matrix (MA), capsid (CA), nucleocapsid (NC), and p6, as well as two spacer peptides, SP1 and SP2. These Gag domains promote well-defined steps during VLP assembly in cells. Briefly, MA mediates plasma membrane targeting and membrane binding of Gag (reviewed in reference 26). CA and NC promote Gag multimerization. The CA C-terminal domain (CTD) forms a dimerization interface (27–31), whereas NC binding to RNA facilitates higher-order multimerization of Gag by binding to RNA (29, 32–37). NC also specifically recognizes the unspliced viral RNA, a process that is essential for packaging of the viral genome (38). p6 facilitates pinching off virus particles from the plasma membrane by recruiting the cellular ESCRT complex (39).
MA mediates binding of Gag to the plasma membrane through a bipartite signal. The first part is an N-terminal myristate moiety that is normally sequestered within MA. Through a structural change, the myristate is exposed and inserted into the inner leaflet of the plasma membrane, which increases membrane binding of Gag (40–43). The second signal is a stretch of basic amino acids that form a highly basic region (HBR) on the surface of MA (44–49). The positive charge of the HBR is thought to increase Gag association with acidic lipids in the plasma membrane. In particular, residues found within the HBR mediate a direct interaction between Gag and a plasma-membrane-specific, negatively charged phospholipid, phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2]. This MA-PI(4,5)P2 interaction facilitates targeting and binding of Gag to the plasma membrane (50–55).
At the plasma membrane, HIV-1 Gag associates with submicroscopic and dynamic microdomains called lipid rafts (56–59), which are enriched in cholesterol and glycosphingolipids (60). In addition to lipid rafts, markers for other microdomains such as tetraspanins, which homo- or hetero-oligomerize to form tetraspanin-enriched microdomains (TEMs), have also been found to colocalize with Gag at the plasma membrane (61–63). Lipid rafts and TEMs are experimentally distinguishable by biochemical methods (e.g., by revealing differential sensitivities to cholesterol depletion [64]; for a review, see Charrin et al. [65]) and by microscopy (61, 66–69). Thus, it is notable that, at least in adherent cell lines, Gag is able to recruit lipid rafts and TEMs together at virus assembly sites in a manner dependent on membrane curvature induced by Gag multimerization or subsequent HIV assembly steps (66, 70). However, it remains unclear whether these and other microdomains on the plasma membrane accumulate to virus assembly sites without changes in their structures and components or whether assembling Gag actively reorganizes the structure or composition of microdomains accumulating to these sites.
In our previous studies, we determined that NC-mediated multimerization is essential for uropod localization of HIV-1 Gag and that Gag associates with uropod-directed microdomains (UDMs) that laterally comigrate with Gag to uropods as T cells polarize (23). Gag proteins of other retroviruses and related multimerizing proteins have also been shown to have similar properties (71, 72). These findings support a working model in which Gag multimerization and UDM association play key roles in Gag localization to uropods. However, several questions remain to be answered. First, at what stage of multimerization does Gag localize to uropods? Does Gag accumulate to uropods as a result of virus surfing of completed virions on the outer leaflet of the plasma membrane (73, 74)? Or conversely, is an early stage of Gag multimerization sufficient to drive uropod localization? Second, what is the composition of UDMs? Do UDMs exist in the absence of assembly events? Finally, how does Gag associate with UDMs? Is UDM association dependent on membrane curvature, as observed for recruitment of lipid rafts and TEMs to assembly sites, or dependent on the mode of Gag-membrane binding? Observations made in this study support a model in which uropod localization of Gag can occur early in assembly before completion of virus particles. Furthermore, we found that Gag associates with a specific subset of UDMs via a mechanism dependent on the positive charge of the MA HBR but not membrane curvature, revealing a new function of MA. Finally, we found that Gag excludes an otherwise UDM-associated protein from Gag-associated UDMs, demonstrating the ability of Gag to reorganize these microdomains upon their association with Gag multimers.
MATERIALS AND METHODS
Plasmids.
The HIV-1 molecular clone pNL4-3/Gag-YFP (pNL4-3/Gag-yellow fluorescent protein) was described previously (29, 50). The YFP-tagged budding-arrested Gag mutants containing CA mutation P99A (66) or EE75,76AA (30) (P99A/Gag-YFP or EE75,76AA/Gag-YFP) were described previously (30, 66, 75). Molecular clones encoding Fyn(10)/Gag-YFP and Fyn(10)/ΔMA/Gag-YFP were described previously (50). The LZ mutants 20LK/WMAA/LZ/Gag-YFP, 20LK/WMAA/LZ3/Gag-YFP, and 20LK/WMAA/LZ4/Gag-YFP (LZ2/Gag-YFP, LZ3/Gag-YFP, and LZ4/Gag-YFP, respectively) were constructed by first replacing NC-p1-p6 with the GCN4 LZ sequence derived from the Zwt construct (a kind gift from H. Gottlinger) described previously (76) in the context of 20LK/WMAA/Gag-YFP by standard molecular biology techniques, and subsequent amino acid substitutions were introduced into the LZ sequence by PCR mutagenesis. The amino acid sequences of LZ variants are as follows: for LZ2/Gag-YFP, RMKQLEDKVEELLSKNYHLENEVARLKKLVGER; for LZ3/Gag-YFP, RMKQLEDKVEELLSKNYHLENRVARLEKLVGER; and for LZ4/Gag-YFP, KVKQLVDKVEELLSKNYHLVNEVARLVKLVGER. In addition to the LZ mutants, 20LK/WMAA/14A1G Gag-YFP (CA/NC/Gag-YFP) was constructed by introducing 15 basic-to-neutral amino acid changes into NC in the 20LK/WMAA/Gag-YFP context using standard molecular biology techniques. The molecular clone pNL4-3/6A2T was described previously (48). The molecular clone encoding Fyn(10)/6A2T/Gag-YFP was constructed by introducing the 6A2T mutations into pNL4-3/Fyn(10)/Gag-YFP. Molecular clones encoding HBR/RKswitch/Gag-YFP and Fyn(10)/HBR/RKswitch/Gag-YFP were constructed by PCR mutagenesis of the 8 basic amino acids of the HBR. All Gag derivatives were encoded in the context of the HIV-1 molecular clone pNL4-3 (77). In addition, for the use in liposome binding assays, pGEMNLNR-based plasmids (50) encoding Gag-YFP, 6A2T/Gag-YFP, and HBR/RKswitch/Gag-YFP were constructed.
Copatching assay.
P2 cells, a polarized T cell line derived from the A3.01 T cell line as described previously (23), were cultured in RPMI 1640 media containing 10% fetal bovine serum (FBS) (RPMI-10). Gag-YFP and its derivatives were expressed in P2 cells by spin infection with vesicular stomatitis virus G (VSV-G)-pseudotyped virus stocks encoding the Gag derivatives as described previously (23). Infected cells were maintained in RPMI-10 for 2 days. Cells were then resuspended in RPMI-10 containing primary antibody (diluted 1:200) against CD43, CD44, PSGL-1, ICAM-1, ICAM-3, or CD59 (BD Biosciences Pharmingen, San Diego, CA) and incubated for 30 min at 37°C. Cells were washed twice in RPMI-10 and resuspended in RPMI-10 containing Alexa Fluor 594-conjugated goat anti-mouse IgG secondary antibody (Invitrogen, Carlsbad, CA) (diluted 1:200) for 30 min at 37°C. Cells were then washed twice with RPMI-10, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (4% PFA), and processed for microscopy as previously described (23). For most Gag-YFP derivatives, we quantified copatching between Gag and a UDM marker in images of single focal planes set at the top surface of cells or three-dimensional (3D) maximum projections. The quantification was performed using the JACoP plugin for ImageJ with which we calculate Pearson's correlation coefficient after 10-pixel rolling ball background subtraction. Because Fyn(10)/ΔMA/Gag-YFP localizes to intracellular compartments as well as the plasma membrane, to ensure that only plasma-membrane-associated Gag was quantified for colocalization, images of cells expressing Fyn(10)/Gag-YFP and Fyn(10)/ΔMA/Gag-YFP were taken at the middle focal plane of the cell, and regions of interest corresponding to the cell periphery were used for quantification as described above.
Polarization calculation.
Cells expressing wild-type Gag-YFP, P99A/Gag-YFP, EE75,76AA/Gag-YFP, CA/NC/Gag-YFP, LZ2/Gag-YFP, LZ3/Gag-YFP, LZ4/Gag-YFP, or LZ/Gag-YFP were fixed in 4% PFA for 20 min, washed with PBS–2% FBS, and mounted on slides as described above. To quantify polarity of Gag localization, a 10-segmented grid was placed over each cell along the cell's longest axis as described previously (23). A value of 10 minus the number of segments that contained plasma-membrane-associated Gag was then used as the polarization index. Thus, a higher number corresponds to higher polarization, with 0 corresponding to Gag distributed over the entire plasma membrane.
Nucleofection.
For expression of Gag mutants containing the Fyn(10) sequence on their N terminus in P2 cells, nucleofection of the molecular clones was performed using Amaxa kit V because VSV-G-pseudotyped virus stocks that would transduce these mutants have low infectivity. Briefly, 1 × 106 P2 cells were resuspended in 100 μl Amaxa V solution with 2 μg DNA and transferred to a cuvette. Program C-016 was used to electroporate the samples. Nucleofected cells were added to 2 ml RPMI-10 and cultured for 3 days before immunostaining experiments were performed.
Liposome binding assays.
Liposome binding assays were performed as previously described (50).
Virus release assay.
P2 cells (3 × 105 cells) were infected with VSV-G-pseudotyped HIV-1 expressing Gag-YFP. Two days postinfection, metabolic labeling of infected cells with [35S]methionine-cysteine, preparation of cell and virus lysates, and immunoprecipitation of viral proteins using HIV immunoglobulin (HIV-Ig; AIDS Research and Reference Reagent Program) were performed as described previously (50). Virus release efficiency was calculated as the amount of virus-associated Gag as a fraction of the total (cell plus virus) Gag synthesized during a 3-h metabolic labeling period.
RESULTS
Virus surfing and budding are not required for uropod localization.
While previous studies showed that Gag multimerization is essential for its localization to the uropod (23, 71), the aspect of multimerization that drives uropod localization remained to be elucidated. Gag multimerization is required for membrane curvature and particle formation, which are prerequisites for virus surfing. Thus, it is possible that rearward surfing of formed virus particles on the plasma membrane, but not Gag multimerization per se, mediates uropod localization of HIV-1. To address this possibility, we examined two YFP-tagged Gag derivatives with CA amino acid substitutions, P99A and EE75,76AA. Both P99A/Gag-YFP and EE75,76AA/Gag-YFP show electron-dense patches underneath the plasma membrane in EM studies, suggesting that they multimerize on the plasma membrane. However, they do not efficiently curve the membrane or form virus particles (Fig. 1A) (30, 66, 75, 78). Mutations in the equivalent region of MLV CA were observed to show a similar budding-arrested phenotype (79). As expected, P99A and EE75,76AA mutants are deficient in virus release from T cells (Fig. 1B and C).
Fig 1.

Membrane curvature or particle formation is not required for uropod localization. (A) An illustration indicating the virus assembly step that is inhibited by the CA mutations P99A and EE75,76AA. (B) P2 cells expressing P99A/Gag-YFP and EE75,76AA/Gag-YFP were examined for virus release efficiency as described in Materials and Methods. (C) Quantification of virus release efficiency. Data from three different experiments are shown as means ± standard deviations. (D) P2 cells were infected with VSV-G-pseudotyped viruses encoding P99A/Gag-YFP (upper panel) or EE75,76AA/Gag-YFP (lower panel) and immunostained for PSGL-1 before fixation. Uropod localization of Gag-YFP was assessed using PSGL-1 as a uropod marker. (E) Gag polarity index values for wild-type Gag-YFP, P99A/Gag-YFP, and EE75,76AA/Gag-YFP in polarized P2 cells were determined as described in Materials and Methods. n, the number of cells used for quantification. Error bars represent standard errors of the means. ns, not significant.
P99A/Gag-YFP and EE75,76AA/Gag-YFP were expressed in P2 cells, a T cell line that adopts a polarized morphology frequently and spontaneously (23), and examined for localization to the uropod, which was identified by accumulation of the uropod marker PSGL-1. We observed that both of these budding-arrested mutants clearly localized to the uropod of polarized P2 cells (Fig. 1D) to the same extent as wild-type Gag-YFP (Fig. 1E). These observations indicate that complete particle formation, and hence virus surfing on the outer leaflet of the plasma membrane, is not required for uropod localization of Gag. Conversely, this also suggests that multimerization and association with the inner leaflet of the plasma membrane are sufficient for Gag to localize to the uropod.
A low level of multimerization is sufficient for Gag polarization toward the uropod.
The observation that budding-arrested mutants localize to the uropod suggests that uropod localization could occur before Gag multimers curve the membrane. To determine how early in assembly uropod localization occurs, we sought to construct and examine Gag mutants that form only low-order multimers. A Gag construct in which NC is replaced with a heterologous dimerizing leucine zipper (LZ) sequence (LZ/Gag) is known to multimerize and form virus-like particles (VLPs) (76, 80, 81). Previously, we showed that a YFP-tagged derivative of this construct, LZ/Gag-YFP, localizes to the uropod (23) (Fig. 2). To block higher-order multimerization of LZ/Gag-YFP, we introduced mutations that disrupt the CA dimerization interface, WM184,185AA. Previous studies showed that LZ/Gag constructs that contain WM184,185AA mutations are largely dimeric in solution (80) and form lower-order but not higher-order multimers in cells (82). To compensate for the membrane binding defect imposed by these CA mutations (29, 83), the MA mutation 20LK, which increases membrane binding via increased affinity to PI(4,5)P2 (84–86), was also introduced into this construct (20LK/WMAA/LZ/Gag-YFP). Specific mutations in the LZ sequence have been shown to change its oligomerization property from a dimer to a trimer (referred to as LZ3) (106) or tetramer (referred to as LZ4) (87). Thus, to express Gag derivatives that form lower-order multimers beyond dimers, these mutations were introduced into the 20LK/WMAA/LZ/Gag-YFP construct (20LK/WMAA/LZ3/Gag-YFP and 20LK/WMAA/LZ4/Gag-YFP, respectively; Fig. 2A). A Gag construct that contains the 20LK mutation and mutations in both the CA dimerization interface and the 15 basic residues of NC (20LK/WMAA/14A1G/Gag-YFP), which is similar to a previously described monomeric Gag mutant (88), was also examined. 20LK/WMAA/14A1G/Gag-YFP, 20LK/WMAA/LZ/Gag-YFP, 20LK/WMAA/LZ3/Gag-YFP, and 20LK/WMAA/LZ4/Gag-YFP are referred to here as CA/NC/Gag-YFP, LZ2/Gag-YFP, LZ3/Gag-YFP, and LZ4/Gag-YFP, respectively (Fig. 2A).
Fig 2.
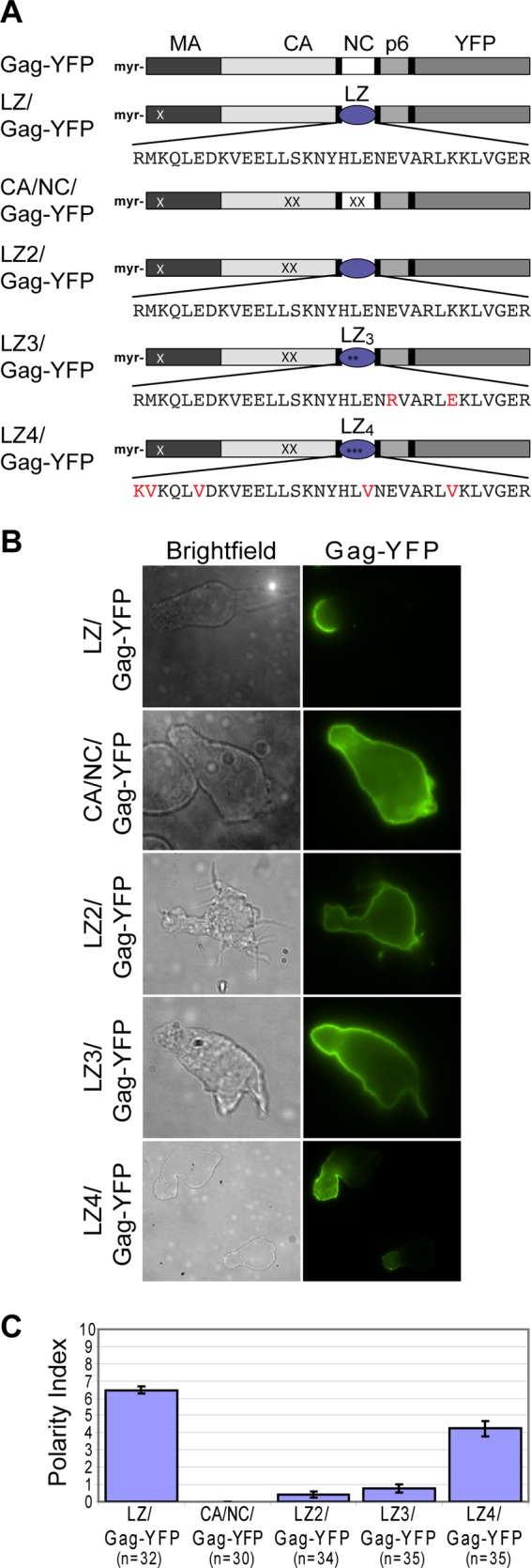
LZ4/Gag-YFP polarizes more extensively than CA/NC/Gag-YFP, LZ2/Gag-YFP, or LZ3/Gag-YFP. (A) Illustration of Gag derivatives containing leucine zipper (LZ) sequences in place of NC. LZ/Gag-YFP is a prototype LZ-containing Gag that multimerizes and forms particles efficiently. Red letters in LZ sequences and asterisks denote mutations that are expected to convert dimeric LZ into trimeric (LZ3) or tetrameric (LZ4) ones. The CA mutation, WM184,185AA (WMAA; denoted by XX in CA), which prevents CA-mediated dimerization, was introduced into the LZ2/Gag-YFP, LZ3/Gag-YFP, and LZ4/Gag-YFP constructs so that LZ sequences provide the only major Gag-Gag interaction activity in these constructs. In CA/NC/Gag-YFP, the 14A1G change was introduced into NC (all NC basic residues substituted with Ala or Gly; denoted by XX in NC) in addition to the WMAA change to eliminate major Gag-Gag interaction domains. All constructs contain the MA mutation 20LK (denoted by a white X in MA), which increases membrane binding. (B) CA/NC/Gag-YFP, LZ2/Gag-YFP, LZ3/Gag-YFP, LZ4/Gag-YFP, and LZ/Gag-YFP were expressed in P2 cells via infection with VSV-G-pseudotyped viruses encoding these Gag constructs. (C) The Gag polarity index was determined for the mutants in polarized P2 cells as described in Materials and Methods. n, the number of cells used for quantification. Error bars represent standard errors of the means.
When expressed in polarized P2 cells, LZ2/Gag-YFP localized over the entire cell surface, consistent with our previous observation that a CA-CTD-deleted LZ/Gag-YFP construct with a heterologous triple acylation signal [Fyn(10)] (added to augment membrane binding) does not localize to the uropod (23). These results suggest that the dimerizing LZ sequence is insufficient for uropod localization of Gag in the absence of a functional CA dimerization interface. Similarly, the CA/NC/Gag-YFP and LZ3/Gag-YFP constructs were observed to localize around the entire plasma membrane, although LZ3/Gag-YFP often showed a gradual increase in Gag accumulation toward one end of the cell. In contrast, LZ4/Gag-YFP was significantly more polarized than CA/NC/Gag-YFP, LZ2/Gag-YFP, or LZ3/Gag-YFP, although slightly less so than LZ/Gag-YFP containing the 20LK change (Fig. 2B and C). These findings further show that uropod localization of Gag depends on the level of its multimerization. Although the extent of oligomerization of these Gag constructs remains to be directly verified, these results, together with the observation obtained with the budding-arrested Gag mutants, suggest that this process occurs early in assembly.
Gag associates with a specific subset of uropod-directed microdomains.
Multimerization of membrane-associated proteins could stabilize association of these proteins with membrane microdomains (reviewed in references 56, 89, and 90). Previously, we showed a high level of copatching between multimerized Gag and uropod markers PSGL-1 and CD43 (23). In the copatching assay, antibodies directed to cell surface markers are added prior to fixation so as to detect otherwise submicroscopic microdomain association of the markers via cross-linking-dependent patching (discussed in more detail in reference 66). Therefore, the observed copatching between Gag and the uropod markers suggests that Gag associates with UDMs. However, whether the composition of these Gag-associated UDMs simply reflects the composition of uropods or whether Gag associates with UDMs that contain specific subsets of uropod-associated proteins remained to be elucidated. Thus, we analyzed the copatching between Gag and other uropod-associated proteins, CD44, CD59, ICAM-1, and ICAM-3 (15–18), in P2 cells (Fig. 3A to D). In order to observe the prepolarization status of Gag association with UDMs, we examined copatching between Gag and uropod-associated proteins in unpolarized P2 cells (Fig. 3A to D, bottom rows). The dispersed distribution of patches of Gag and uropod-associated proteins in these cells also allowed us to assess colocalization of Gag and uropod protein clusters unambiguously, in contrast to assessing colocalization in polarized cells in which Gag and uropod proteins could concentrate to the small uropod area and overlap each other. We found that, in unpolarized T cells, Gag-YFP efficiently copatched with CD44, as was observed previously (and confirmed here) for PSGL-1 and CD43 (Fig. 3A, bottom rows, and 3E). However, Gag-YFP did not efficiently copatch with CD59, ICAM-1, or ICAM-3 in these unpolarized cells (Fig. 3B to D, bottom rows, and 3E) even though these proteins are also directed to uropods in polarized T cells (Fig. 3B to D, top rows). Thus, these observations indicate that Gag associates with UDMs containing a specific subset of uropod-associated proteins.
Fig 3.

Gag associates with a specific subset of uropod-directed microdomains in unpolarized P2 cells. (A to D) P2 cells were infected with a VSV-G-pseudotyped virus encoding Gag-YFP. Cells were immunostained before fixation for CD44 (A), CD59 (B), ICAM-1 (C), and ICAM-3 (D). Polarized cells were observed to confirm that the uropod proteins were enriched at the uropod (top rows). To determine copatching of Gag and uropod markers, which reflects copartitioning of the markers with Gag in the same microdomains, unpolarized cells were examined (bottom rows). For unpolarized cells, the top surface of cells is shown. (E) Copatching between Gag and uropod markers in unpolarized cells was quantified using the square of Pearson's correlation coefficient (R2). n, the number of cells used for quantification. Error bars represent standard errors of the means. P values were calculated based on comparisons between Gag copatching with PSGL-1 and other uropod markers. ***, P < 0.001; ns, not significant.
Gag actively reorganizes UDMs.
As described above, Gag associates with a specific subset of UDMs containing CD43, PSGL-1, and CD44. We wondered whether these UDM proteins associate with each other in the absence of Gag or whether Gag reorganizes UDMs. To distinguish between these possibilities, an analysis of copatching between CD44 and the other UDM proteins was performed in uninfected P2 cells. As expected, CD44 patches induced by two different specific antibodies (IgG1 and IgG2b) displayed almost complete colocalization (Fig. 4A and G). In addition, we found that CD44 copatches with PSGL-1 and CD43 at high levels (Pearson's correlation coefficient [R2] > 0.5) (Fig. 4B, C, and G), similar to the Gag copatching seen with these uropod proteins. In contrast, CD44 showed markedly less copatching with CD59 or ICAM-1 (Fig. 4D, E, and G). This pattern of separation into two groups mirrors the copatching pattern of Gag with these uropod-directed proteins (Fig. 3). However, interestingly, we also found that CD44 copatches with ICAM-3 (Fig. 4F and G) even though ICAM-3 does not copatch well with Gag (Fig. 3). Thus, these observations suggest that, while PSGL-1, CD43, CD44, and ICAM-3 associate with the same microdomain that exists in the absence of Gag, Gag does not simply associate with this UDM passively. Rather, Gag appears to modify this UDM by excluding ICAM-3. Supporting the latter interpretation, three-color imaging revealed that in cells expressing Gag-CFP, Gag copatches well with CD44 but these CD44 patches no longer colocalize with patches of ICAM-3 (Fig. 5). These data suggest that, in addition to the coalescence of disparate microdomains observed previously (66, 70), Gag is also able to mediate modification of existing microdomains.
Fig 4.
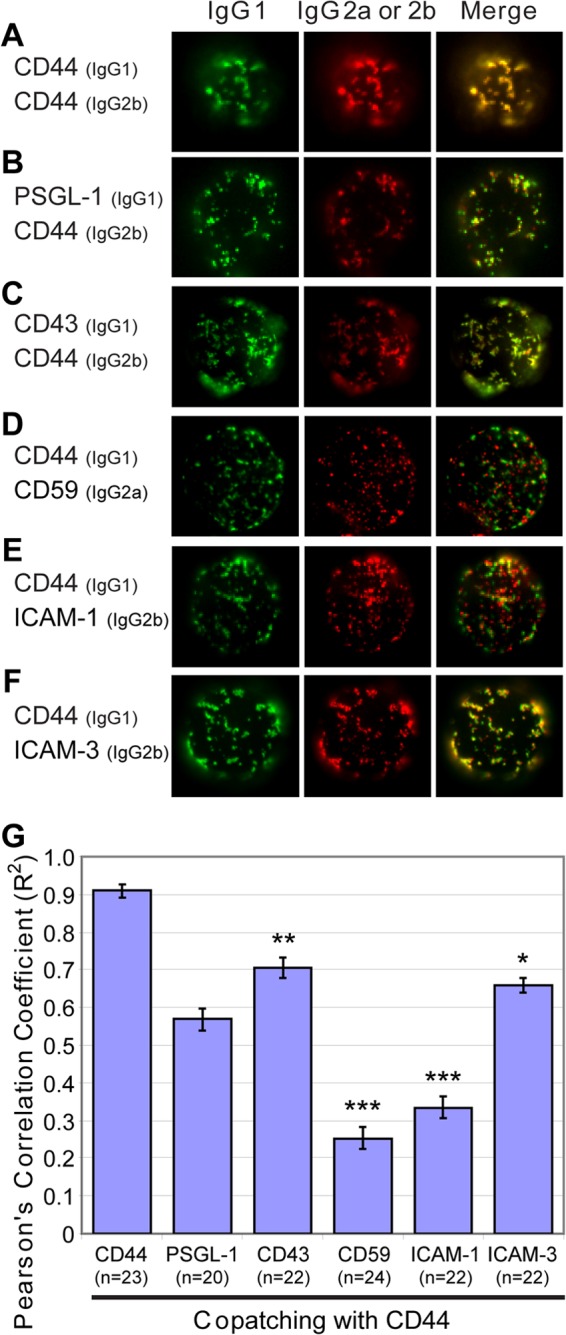
CD44 and other uropod-associated proteins copatch in the absence of Gag. (A to F) Uninfected P2 cells were immunostained prior to fixation using two antibodies differing in their isotypes, one antibody recognizing CD44 and the other recognizing CD44 (A), PSGL-1 (B), CD43 (C), CD59 (D), ICAM-1 (E), or ICAM-3 (F). The top surface of cells is shown. (G) The level of copatching between CD44 and uropod markers was quantified with the square of Pearson's correlation coefficient (R2). n, the number of cells used for quantification. Error bars represent standard errors of the means. P values were calculated based on comparisons between CD44 copatching with PSGL-1 and other uropod markers. ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, not significant.
Fig 5.
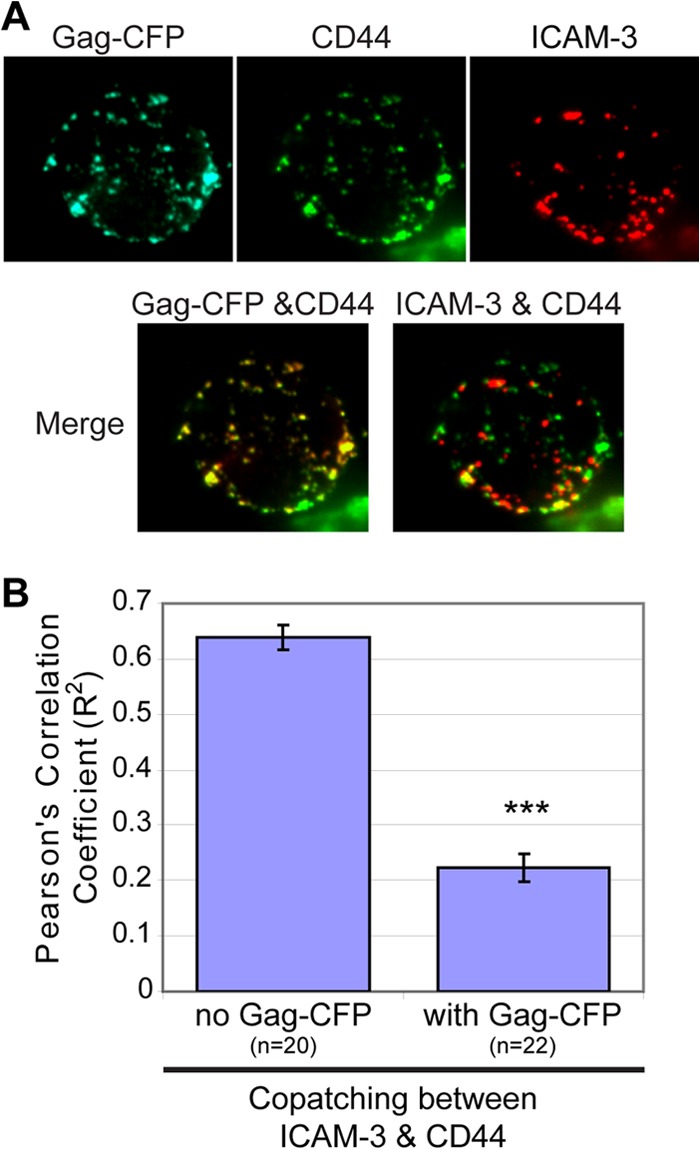
Gag reorganizes UDMs by excluding ICAM-3. (A) P2 cells were infected with VSV-G-pseudotyped virus particles encoding Gag-CFP. Two days postinfection, cells were immunostained prior to fixation for CD44 and ICAM-3. (B) The level of copatching between CD44 and ICAM-3 in the absence or presence of Gag was quantified with the square of Pearson's correlation coefficient (R2). ***, P < 0.001.
Completion of particle formation is not required for recruitment of specific UDMs.
Membrane curvature has been implicated in membrane domain organization (91). Furthermore, we recently observed that Gag-dependent coalescence of lipid rafts and TEMs that do not normally colocalize with each other correlates with the ability of Gag to induce membrane budding (66). Therefore, it is conceivable that Gag associates with specific UDMs that are attracted to membrane curvature.
To investigate whether specific UDM association of Gag is determined by the ability of Gag to bud, copatching between the budding-arrested mutants P99A/Gag-YFP and EE75,76AA/Gag-YFP and the uropod markers PSGL-1, CD43, and CD59 was examined in unpolarized P2 cells. Almost identically to wild-type Gag-YFP, both mutants were observed to copatch with PSGL-1 and CD43 but not with CD59 (Fig. 6). These observations indicate that membrane curvature is not required for Gag-YFP association with specific UDMs. Thus, these results also suggest that the mechanism of UDM recruitment to assembly sites is different from the curvature-dependent recruitment of rafts and TEMs observed in HeLa cells (66).
Fig 6.
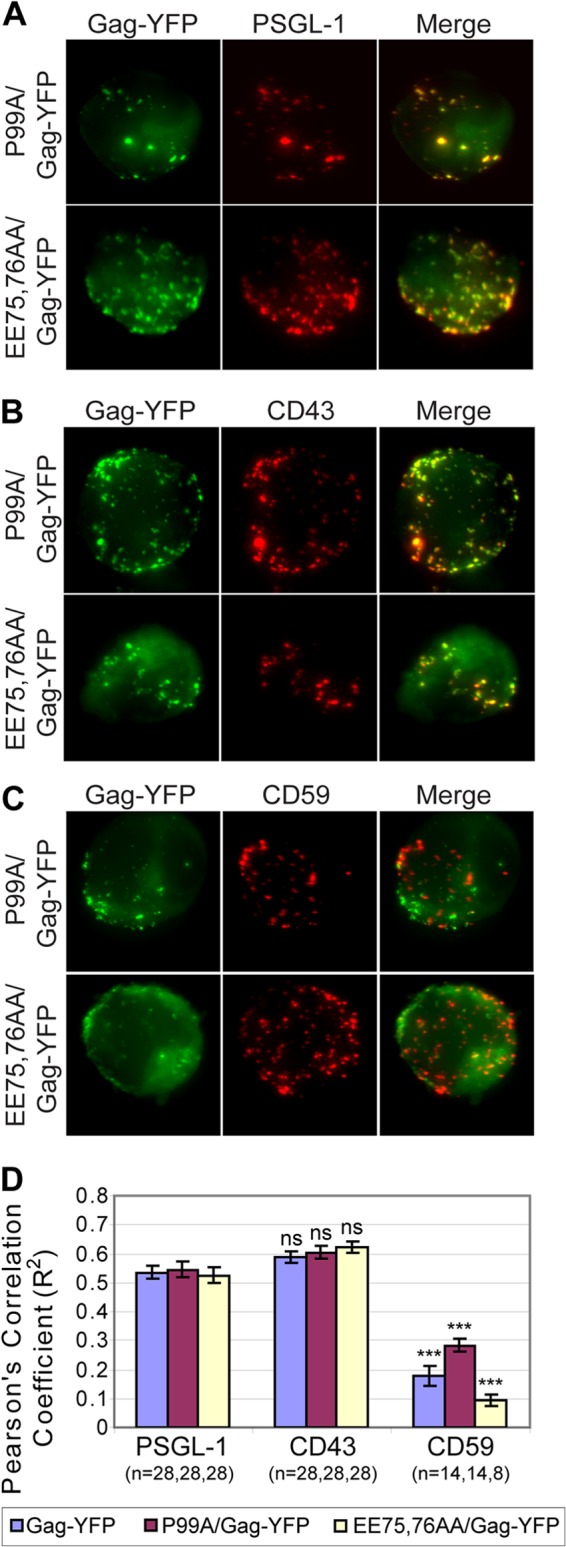
Membrane curvature is not required for specific UDM association. (A to C) P2 cells were infected with VSV-G-pseudotyped virus particles encoding the budding-deficient mutants P99A/Gag-YFP (top rows) and EE75,76AA/Gag-YFP (bottom rows). These cells were immunostained for PSGL-1 (A), CD43 (B), or CD59 (C) prior to fixation. Maximum projections reconstructed from z-stack images of cells are shown. (D) Copatching between Gag and uropod markers was quantified with the square of Pearson's correlation coefficient (R2). n, the number of cells used for quantification. Error bars represent standard errors of the means. P values were calculated based on comparisons between Gag copatching with PSGL-1 and other uropod markers. ***, P < 0.001; ns, not significant.
MA determines the specificity of UDM association.
It is conceivable that, while multimerization is required for uropod localization and UDM association, an additional function of Gag may be necessary for determining the specificity of UDM association. The Gag region most likely to determine which UDMs Gag associates with is MA, because this domain interacts directly with the plasma membrane. To examine this possibility, a Gag mutant in which MA was deleted [Fyn(10)/ΔMA/Gag-YFP] was expressed in P2 cells and examined for UDM association by copatching analysis. In this construct, MA was replaced with the triple acylation signal Fyn(10), which compensates for the loss of MA-dependent membrane binding. To control for the addition of the Fyn(10) sequence, Fyn(10)/ΔMA/Gag-YFP was compared with Fyn(10)/Gag-YFP, which contains the full MA sequence. Gag puncta on the plasma membrane detected in the middle focal plane of cells were used to quantify colocalization (see Materials and Methods). Strikingly, while Fyn(10)/Gag-YFP associated with the same PSGL-1/CD43-enriched subset of UDMs as Gag-YFP (Fig. 7A), the loss of MA caused Gag to segregate from PSGL-1 and CD43 (Fig. 7B). As observed for wild-type Gag-YFP, both Fyn(10)/Gag-YFP and Fyn(10)/ΔMA/Gag-YFP did not copatch with ICAM-3 efficiently (data not shown). Interestingly, copatching between Fyn(10)/ΔMA/Gag-YFP and CD59 was enhanced compared with Fyn(10)/Gag-YFP results (Fig. 7). Together, these data indicate that despite Fyn(10)/Gag-YFP and Fyn(10)/ΔMA/Gag-YFP having the same Fyn acylation signals on their N terminus, these two mutants associate with different UDMs. Thus, the specificity of UDM association is likely determined by the MA sequence itself.
Fig 7.
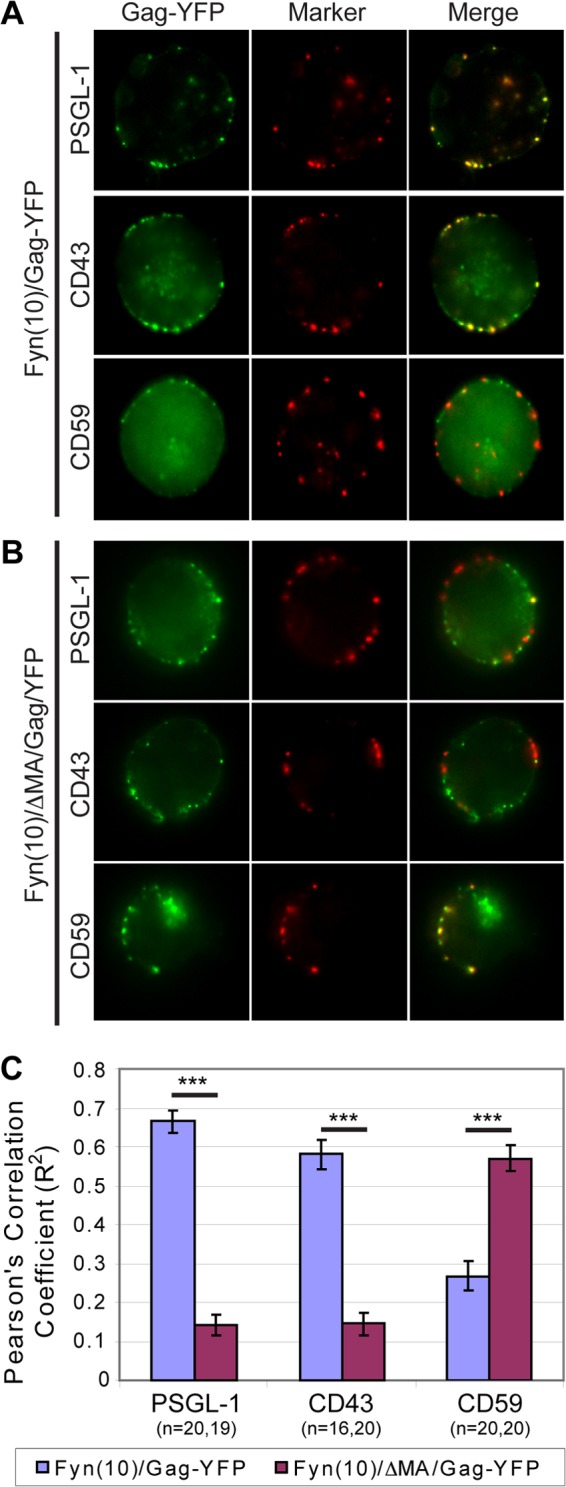
The MA sequence promotes Gag association with specific UDMs. (A and B) P2 cells were nucleofected with molecular clones encoding Fyn(10)/Gag-YFP (A) or Fyn(10)/ΔMA/Gag-YFP (B). After 3 days, cells were immunostained for PSGL-1, CD43, or CD59 before fixation. The middle focal plane of cells is shown. Prominent cytoplasmic signals are observed in some cells. (C) The level of copatching between Gag and uropod markers was quantified with the square of Pearson's correlation coefficient (R2). To ensure that only plasma-membrane-associated Gag was quantified for colocalization, regions of interest corresponding to the cell periphery at the middle focal plane of the cell were used for quantification as described in Materials and Methods. n, the number of cells used for quantification. Error bars represent standard errors of the means. ***, P value < 0.001.
A positive charge of the MA HBR is required for specific UDM association.
One of the prominent features in the MA sequence is the HBR. It is conceivable that the positive charge of the HBR mediates association of Gag with specific UDMs by binding to PI(4,5)P2 or other negatively charged phospholipids. The HBR could also mediate direct interaction with negatively charged residues in UDM-associated proteins. Thus, to determine whether the HBR plays a role in specific UDM association, the 8 basic residues within the HBR of Fyn(10)/Gag-YFP were mutated to the neutral amino acid alanine or threonine [Fyn(10)/6A2T/Gag-YFP]. To examine the role of charge in any HBR-mediated phenotype, copatching of Fyn(10)/6A2T/Gag-YFP with PSGL-1, CD43, and CD59 was compared with that of another HBR mutant in which the six Lys (K) were mutated to Arg (R) and the two Arg were mutated to Lys [Fyn(10)/HBR/RKswitch/Gag-YFP] (Fig. 8A). In this construct, the same basic residues as in Fyn(10)/6A2T/Gag-YFP were substituted but, unlike in Fyn(10)/6A2T/Gag-YFP, the overall positive charge of the HBR was maintained. Both Fyn(10)/6A2T/Gag-YFP and Fyn(10)/HBR/RKswitch/Gag-YFP showed cell surface punctate signals, suggesting that they form higher-order Gag multimers at the plasma membrane. When Fyn(10)/6A2T/Gag-YFP was expressed in P2 cells, we observed that Gag lost its association with PSGL-1 and CD43 but gained an association with CD59, as was observed for Fyn(10)/ΔMA/Gag-YFP (Fig. 8B and D), indicating that the HBR is required for specific UDM association. In contrast, we found that Fyn(10)/HBR/RKswitch/Gag-YFP copatched with PSGL-1 and CD43 but not with CD59 in a manner similar to that seen with wild-type Gag-YFP (Fig. 8C and D). Together, these observations indicate that the positive charge of the HBR plays a major role in specific UDM association of Gag.
Fig 8.
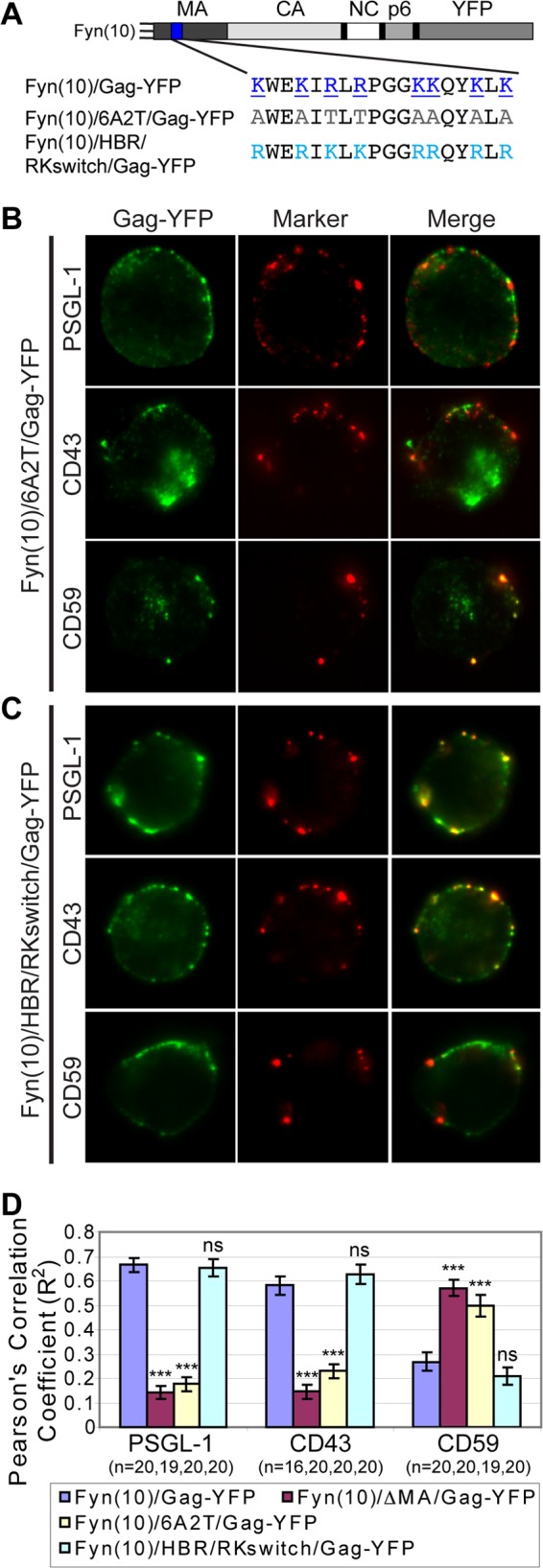
A positive charge of the MA HBR is required for specific UDM association. (A) Illustration depicting the MA HBR amino acid sequences and changes introduced in the MA mutants 6A2T and HBR/RKswitch. The basic amino acids of the wild-type HBR (blue, underlined) were changed to neutral amino acids (gray) in 6A2T or switched from R to K and from K to R (cyan) in HBR/RKswitch. (B and C) P2 cells were nucleofected with molecular clones encoding Fyn(10)/6A2T/Gag-YFP (B) or Fyn(10)/HBR/RKswitch/Gag-YFP (C). After 3 days, cells were immunostained before fixation for PSGL-1, CD43, or CD59. The middle focal plane of cells is shown. (D) Copatching between Gag and uropod markers was quantified with the square of Pearson's correlation coefficient (R2) as described for Fig. 7. n, the number of cells used for quantification. Error bars represent standard errors of the means. Data for Fyn(10)/Gag-YFP and Fyn(10)ΔMA/Gag-YFP from Fig. 7 are included for comparison. P values were calculated based on comparisons between Fyn(10)/Gag-YFP and its derivatives. ***, P < 0.001; ns, not significant.
Gag-PI(4,5)P2 binding is not required for HBR-charge-mediated association of Gag with specific UDMs.
A recent report suggests that interactions with a protein can induce enrichment of PI(4,5)P2 in some microdomains, which in turn facilitates accumulation of the protein (92). It is conceivable that Gag, through its HBR, induces formation of such PI(4,5)P2-enriched microdomains to which some uropod proteins with a short basic stretch in juxtamembrane cytoplasmic regions, such as CD43, PSGL-1, and CD44, are also attracted. Alternatively, the HBR could play a similar role via recruitment of other negatively charged phospholipids in a more nonspecific and charge-dependent manner. Thus, using a liposome binding assay, we investigated whether HBR/RKswitch/Gag-YFP and 6A2T/Gag-YFP interact with PI(4,5)P2 or other negatively charged phospholipids. For this set of experiments, Gag constructs with the native N-terminal sequence but not the Fyn(10) sequence were used, because the strong membrane binding mediated by the Fyn(10) sequence (i.e., triple acylation) obscures the difference in Gag-liposome binding. We found that, unlike wild-type Gag-YFP, both 6A2T/Gag-YFP and HBR/RKswitch/Gag-YFP do not bind PI(4,5)P2-containing liposomes (Fig. 9A and B). This indicates that PI(4,5)P2 binding requires not only the overall basic charge of the HBR but also the specific amino acid sequence in the HBR.
Fig 9.

Gag binding to PI(4,5)P2-containing liposomes requires the specific HBR amino acid sequence, whereas the overall positive charge of the HBR is sufficient for RNA binding and acidic lipid binding in the absence of RNA. (A) Liposome binding assays were performed to examine binding of wild-type Gag-YFP, HBR/RKswitch/Gag-YFP, and 6A2T/Gag-YFP to control liposomes that contain PC and PS in a 2:1 ratio [PC;PS (2:1)] or liposomes that contain 7.25 mol% PI(4,5)P2 in addition to PC:PS (2:1). M, membrane-bound Gag; NM, non-membrane-bound Gag. (B) The amount of labeled Gag in each fraction was quantified using a phosphorimager, and the percentage of labeled Gag in the membrane-bound fraction versus the total amount of labeled Gag was calculated. The relative liposome-binding efficiency for each condition was calculated in comparison to the binding efficiency of wild-type Gag-YFP with PI(4,5)P2-containing liposomes. Data from three different experiments are shown as means ± standard deviations. Liposome binding efficiency was quantified, with all conditions normalized to Gag-YFP binding to PI(4,5)P2-containing liposomes. (C) Liposome binding assays were performed to examine binding of wild-type Gag-YFP, HBR/RKswitch/Gag-YFP, and 6A2T/Gag-YFP to PC:PS (2:1) liposomes with or without RNase treatment. M, membrane-bound Gag; NM, non-membrane-bound Gag. (D) Liposome binding efficiency was quantified as described for panel B, and the relative liposome-binding efficiency for each condition was calculated in comparison to the binding efficiency of RNase-treated Gag-YFP with PC:PS (2:1) liposomes. Data from three different experiments are shown as means ± standard deviations. Note that results shown in panels A and C were obtained in the same experiment, and therefore the same gel images are shown for the negative-control conditions (left side).
It was previously observed that, upon treatment with RNase, wild-type Gag is able to bind liposomes containing phosphatidylcholine (PC) and the negatively charged phospholipid phosphatidylserine (PS), presumably via electrostatic interactions, even when PI(4,5)P2 is absent (48). Similarly, we observed that wild-type Gag-YFP binds the PC-plus-PS liposomes upon RNase treatment (Fig. 7C and D). In contrast, 6A2T/Gag-YFP failed to exhibit PC-plus-PS binding regardless of RNase treatment, underlining the importance of electrostatic interactions in this mode of Gag-membrane binding (Fig. 9C and D). Interestingly, we found that, upon treatment with RNase, HBR/RKswitch/Gag-YFP binds PC-plus-PS liposomes nearly as efficiently as wild-type Gag-YFP (Fig. 9C and D). These results indicate that switching the Lys and Arg in the HBR had little impact on the ability of MA to bind RNA or negatively charged, non-PI(4,5)P2 phospholipids. Since the Lys-Arg switching has no impact on Gag-UDM copatching (Fig. 8), these data suggest that Gag association with specific UDMs is dependent on the positive charge of the HBR but not on HBR binding to PI(4,5)P2. Taken together, these results reveal a novel property of MA in determining the association of Gag with different microdomains.
DISCUSSION
Microdomains, such as lipid rafts and TEMs, have been implicated in virus assembly/release and cell-to-cell transmission (56, 58, 59, 63, 93–99). However, despite the potential importance of microdomain association in HIV-1 replication and spread, molecular determinants of HIV-1 Gag association with different microdomains, particularly in T cells, remain poorly understood. Here, we found that HIV-1 Gag multimers associate with microdomains that contain a specific subset of uropod-directed proteins. Previous studies using adherent cell lines (66, 70) have provided evidence that Gag multimerization and subsequent membrane budding are able to stabilize and coalesce lipid rafts and TEMs in a MA-sequence-independent manner. In contrast, in this study we found that the MA HBR (Fig. 7 and 8), but not membrane curvature (Fig. 6), plays a key role in association of Gag multimers with a specific subset of uropod-localizing proteins in T cells. As these proteins copatch with each other in the absence of virus expression (Fig. 4), they are likely to share affinity to common microdomains directed to the uropod in T cells. Notably, we also found that Gag is able to reorganize these UDMs by actively excluding proteins originally associated with these microdomains (e.g., ICAM-3) (Fig. 5). These findings demonstrate that Gag drives reorganization of the plasma membrane in a variety of ways.
Analysis of a panel of uropod proteins revealed that Gag associates with a specific UDM enriched in PSGL-1, CD43, and CD44 but not ICAM-1, ICAM-3, or CD59 in unpolarized P2 cells. As discussed below, it is unlikely that a specific subset of UDMs plays an irreplaceable role in movement of Gag multimers to the uropod. Nonetheless, it is conceivable that a specific UDM protein modulates HIV-1 release or cell-to-cell transmission. For example, a recent study showed that increased expression of the UDM protein CD43 correlates with enhanced cell-to-cell transmission of HTLV-1, another retrovirus that replicates in T cells (100). Thus, although it is not known whether association of CD43 and HTLV-1 in microdomains plays a role in this increase of cell-to-cell transmission, it is conceivable that specific UDMs or UDM-associated proteins contribute to cell-to-cell transmission of HIV-1. It is also of interest to determine why HIV-1 evolved to exclude ICAM-3 from the UDMs with which the virus associates. A previous study showed that HIV-1 replication in Jurkat T cells is inefficient when ICAM-3 is expressed and that ICAM-3 appears to exert the negative effect during the late phase of the virus replication cycle (101). Thus, it is tempting to speculate that reducing the ICAM-3 association with assembling viruses at the plasma membrane is potentially advantageous for HIV-1 assembly and/or the subsequent transmission. Further analyses of Gag-negative and Gag-positive UDM components are likely to provide us with insights into the advantages for HIV-1 to associate with, and modify the components of, the specific subset of UDMs.
Interestingly, while Fyn(10)/ΔMA/Gag-YFP did not associate with CD43/PSGL-1/CD44-enriched UDMs as wild-type Gag-YFP does, we found that it did associate with an alternate UDM enriched in the lipid raft marker CD59. It is likely that the association of Fyn(10)/ΔMA/Gag-YFP with CD59 is driven by the Fyn(10) sequence, which encodes a myristoylation and two palmitoylation signals. These acylation signals have been previously observed to facilitate association with cholesterol-dependent lipid raft microdomains (102). Thus, it is conceivable that, in the context of Fyn(10)/Gag-YFP, MA-mediated interactions with specific UDMs are dominant over Fyn(10)-mediated association with CD59-enriched lipid rafts. Conversely, in the absence of MA, Fyn(10) is able to facilitate Gag association with CD59-enriched lipid rafts. Thus, if UDM association is one of the prerequisites for uropod localization, it is conceivable that Gag that contains wild-type MA utilizes CD43/PSGL-1/CD44-enriched UDMs for this process, while this process is replaced by UDMs containing CD59-enriched lipid rafts in the context of Fyn(10)/ΔMA/Gag-YFP, which also localizes to the uropod (23).
In this study, we identified the MA HBR as a Gag determinant that mediates Gag association with some UDM proteins and not others. The HBR is known to promote membrane binding through its interaction with acidic phospholipids, including PI(4,5)P2 (44, 45, 48, 50, 52, 53, 55). Some plasma membrane proteins with basic regions can mediate clustering of PI(4,5)P2, which in turn can mediate clustering of these proteins into microdomains (92, 103). Notably, analyses of the HBR amino acid substitution mutants showed that basic-to-neutral substitutions (6A2T) block association of Fyn(10)/Gag-YFP with the specific UDMs containing CD43 and PSGL-1, whereas switching of Lys and Arg (HBR/RKswitch) has no impact on this association. As shown in liposome binding experiments (Fig. 9), the HBR/RKswitch mutant Gag is likely to maintain electrostatic interactions with negatively charged phospholipids such as PS. Therefore, it is plausible that Gag multimers, via the HBR, induce clustering of certain negatively charged phospholipids to which some, but not all, uropod proteins (e.g., PSGL-1, CD43, and CD44) are also attracted via electrostatic interactions. It should be noted, however, that our data do not rule out a direct interaction between Gag and a UDM-associated protein via an interface heavily dependent on the charge of the HBR. While it is unlikely that the HBR alone serves as a direct protein-protein interaction interface that would depend on the exact amino acid sequence, further studies are necessary to determine how the MA HBR mediates specific UDM association.
The role played by the MA HBR in HIV-1 Gag interaction with PI(4,5)P2 is well recognized (26). However, whether this interaction is solely dependent on electrostatic interactions between HBR basic residues and the high-density negative charge of the PI(4,5)P2 head group or whether the interface structure promotes specific recognition of the head group had not been determined (44, 50, 104). In this regard, the phenotype we observed in this study of HBR/RKswitch/Gag-YFP in liposome binding assays is noteworthy. Although the switching of Arg and Lys does not alter the ability of the HBR to support electrostatic interactions, in contrast to wild-type Gag-YFP, HBR/RKswitch/Gag-YFP failed to bind to PI(4,5)P2-containing liposomes (Fig. 9). Therefore, it is unlikely that electrostatic interactions solely promote the MA-PI(4,5)P2 binding: rather, a structural element of the HBR plays a major role in recognition of PI(4,5)P2. Ongoing studies are aimed at determining whether such a structural element is directly involved in PI(4,5)P2 interaction or whether it indirectly modulates PI(4,5)P2 interaction via RNA binding.
While completed HIV-1 virus particles could accumulate to the uropod by surfing on the outer leaflet of the plasma membrane, virus surfing (73, 74) is not required for uropod localization of Gag in polarized T cells. We observed that P99A/Gag-YFP and EE75,76AA/Gag-YFP, which can multimerize but are defective in budding and hence in particle formation, localize to the uropod efficiently. Moreover, LZ4/Gag-YFP, despite the disruption of the CA dimerization interface, also polarizes on the plasma membrane. Therefore, it is likely that uropod localization begins at an early stage of the assembly process before spherical particle formation and surfing would occur. Notably, in contrast to LZ4/Gag-YFP, LZ2/Gag-YFP failed to localize to the uropod, suggesting an interesting possibility that the size of Gag oligomers determines the polarized localization. Oligomerization and the resulting microdomain association of membrane-associated proteins are thought to facilitate confinement of these proteins in compartments lined by the cortical actin cytoskeleton due to the increased size of the protein entity (105). Thus, it is conceivable that LZ4/Gag-YFP is more susceptible than LZ2/Gag-YFP to such oligomerization-enhanced confinement, which in turn promotes uropod localization in polarized T cells where rearward actin flow exists. Lower-order Gag multimers were not analyzed using the copatching assay in the current study, since they do not form distinct clusters distinguishable in conventional microscopy. However, superresolution analysis of lower-order Gag multimers with regard to their relationships with different UDMs and the rearward actin flow, which is under way in our laboratory, is likely to elucidate the mechanism by which multimerization mediates Gag accumulation to uropods and virological synapses.
ACKNOWLEDGMENTS
We thank members of our laboratory for helpful discussions and critical reviews of the manuscript and H. Gottlinger for providing the Zwt construct.
This work was supported by NIH grant R01 AI071727 to A.O. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: HIV-Ig from NABI and NHLBI.
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582–12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 67:2182–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hübner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FYS, Li XD, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martin N, Welsch S, Jolly C, Briggs JAG, Vaux D, Sattentau QJ. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 84:3516–3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 6:e1000788 doi:10.1371/journal.ppat.1000788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716 [DOI] [PubMed] [Google Scholar]
- 8. Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat. Commun. 3:1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. 2012. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490:283–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bajénoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. 2006. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 25:989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hugues S, Fetler L, Bonifaz L, Helft J, Amblard F, Amigorena S. 2004. Distinct T cell dynamics in lymph nodes during the induction of tolerance and immunity. Nat. Immunol. 5:1235–1242 [DOI] [PubMed] [Google Scholar]
- 12. Miller MJ, Wei SH, Cahalan MD, Parker I. 2003. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc. Natl. Acad. Sci. U. S. A. 100:2604–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miller MJ, Wei SH, Parker I, Cahalan MD. 2002. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science 296:1869–1873 [DOI] [PubMed] [Google Scholar]
- 14. Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG, Weninger W. 2006. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J. Exp. Med. 203:2749–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alonso-Lebrero JL, Serrador JM, Dominguez-Jimenez C, Barreiro O, Luque A, del Pozo MA, Snapp K, Kansas G, Schwartz-Albiez R, Furthmayr H, Lozano F, Sanchez-Madrid F. 2000. Polarization and interaction of adhesion molecules P-selectin glycoprotein ligand 1 and intercellular adhesion molecule 3 with moesin and ezrin in myeloid cells. Blood 95:2413–2419 [PubMed] [Google Scholar]
- 16. Sánchez-Madrid F, del Pozo MA. 1999. Leukocyte polarization in cell migration and immune interactions. EMBO J. 18:501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosenman SJ, Ganji AA, Tedder TF, Gallatin WM. 1993. Syn-capping of human T lymphocyte adhesion/activation molecules and their redistribution during interaction with endothelial cells. J. Leukoc. Biol. 53:1–10 [DOI] [PubMed] [Google Scholar]
- 18. del Pozo MA, Sánchez-Mateos P, Nieto M, Sánchez-Madrid F. 1995. Chemokines regulate cellular polarization and adhesion receptor redistribution during lymphocyte interaction with endothelium and extracellular matrix. Involvement of cAMP signaling pathway. J. Cell Biol. 131:495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serrador JM, Urzainqui A, Alonso-Lebrero JL, Cabrero JR, Montoya MC, Vicente-Manzanares M, Yanez-Mo M, Sanchez-Madrid F. 2002. A juxta-membrane amino acid sequence of P-selectin glycoprotein ligand-1 is involved in moesin binding and ezrin/radixin/moesin-directed targeting at the trailing edge of migrating lymphocytes. Eur. J. Immunol. 32:1560–1566 [DOI] [PubMed] [Google Scholar]
- 20. McFarland W, Heilman DH. 1965. Lymphocyte foot appendage: its role in lymphocyte function and in immunological reactions. Nature 205:887–888 [DOI] [PubMed] [Google Scholar]
- 21. del Pozo MA, Cabanas C, Montoya MC, Ager A, Sanchez-Mateos P, Sanchez-Madrid F. 1997. ICAMs redistributed by chemokines to cellular uropods as a mechanism for recruitment of T lymphocytes. J. Cell Biol. 137:493–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sánchez-Madrid F, Serrador JM. 2009. Bringing up the rear: defining the roles of the uropod. Nat. Rev. Mol. Cell Biol. 10:353–359 [DOI] [PubMed] [Google Scholar]
- 23. Llewellyn GN, Hogue IB, Grover JR, Ono A. 2010. Nucleocapsid promotes localization of HIV-1 gag to uropods that participate in virological synapses between T cells. PLoS Pathog. 6:e1001167 doi:10.1371/journal.ppat.1001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balasubramaniam M, Freed EO. 2011. New insights into HIV assembly and trafficking. Physiology (Bethesda) 26:236–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bieniasz PD. 2009. The cell biology of HIV-1 virion genesis. Cell Host Microbe 5:550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chukkapalli V, Ono A. 2011. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 410:512–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Datta SA, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, Kvaratskhelia M, Lebowitz J, Rein A. 2007. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J. Mol. Biol. 365:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gamble TR, Yoo SH, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853 [DOI] [PubMed] [Google Scholar]
- 29. Hogue IB, Hoppe A, Ono A. 2009. Quantitative fluorescence resonance energy transfer microscopy analysis of the human immunodeficiency virus type 1 Gag-Gag interaction: relative contributions of the CA and NC domains and membrane binding. J. Virol. 83:7322–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang WH, Hockley DJ, Nermut MV, Morikawa Y, Jones IM. 1996. Gag-Gag interactions in the C-terminal domain of human immunodeficiency virus type 1 p24 capsid antigen are essential for Gag particle assembly. J. Gen. Virol. 77:743–751 [DOI] [PubMed] [Google Scholar]
- 32. Burniston MT, Cimarelli A, Colgan J, Curtis SP, Luban J. 1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol. 73:8527–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Campbell S, Vogt VM. 1995. Self-assembly in vitro of purified Ca-Nc proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487–6497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cimarelli A, Sandin S, Hoglund S, Luban J. 2000. Basic residues in human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol. 74:3046–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawson L, Yu XF. 1998. The role of nucleocapsid of HIV-1 in virus assembly. Virology 251:141–157 [DOI] [PubMed] [Google Scholar]
- 36. Campbell S, Rein A. 1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol. 73:2270–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muriaux D, Mirro J, Harvin D, Rein A. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. U. S. A. 98:5246–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D'Souza V, Summers MF. 2005. How retroviruses select their genomes. Nat. Rev. Microbiol. 3:643–655 [DOI] [PubMed] [Google Scholar]
- 39. Weiss ER, Gottlinger H. 2011. The role of cellular factors in promoting HIV budding. J. Mol. Biol. 410:525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. U. S. A. 87:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Göttlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 86:5781–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. 2004. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U. S. A. 101:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou W, Resh MD. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 70:8540–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dalton AK, Ako-Adjei D, Murray PS, Murray D, Vogt VM. 2007. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J. Virol. 81:6434–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou WJ, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yuan X, Yu XF, Lee TH, Essex M. 1993. Mutations in the N-terminal region of human immunodeficiency virus type 1 matrix protein block intracellular transport of the Gag precursor. J. Virol. 67:6387–6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ono A, Orenstein JM, Freed EO. 2000. Role of the gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J. Virol. 74:2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chukkapalli V, Oh SJ, Ono A. 2010. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. U. S. A. 107:1600–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. 1996. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U. S. A. 93:3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chukkapalli V, Hogue IB, Boyko V, Hu WS, Ono A. 2008. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol. 82:2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 101:14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 103:11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alfadhli A, Still A, Barklis E. 2009. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J. Virol. 83:12196–12203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. 2008. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J. Virol. 82:11228–11238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. 2006. Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45:4077–4083 [DOI] [PubMed] [Google Scholar]
- 56. Hogue IB, Llewellyn GN, Ono A. 2012. Dynamic association between HIV-1 Gag and membrane domains. Mol. Biol. Int. 2012:979765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lindwasser OW, Resh MD. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913–7924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nguyen DH, Hildreth JE. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J. Virol. 74:3264–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ono A, Freed EO. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. U. S. A. 98:13925–13930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lingwood D, Simons K. 2010. Lipid rafts as a membrane-organizing principle. Science 327:46–50 [DOI] [PubMed] [Google Scholar]
- 61. Nydegger S, Khurana S, Krementsov DN, Foti M, Thali M. 2006. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 173:795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. 2006. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 172:923–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thali M. 2009. The roles of tetraspanins in HIV-1 replication. Curr. Top. Microbiol. Immunol. 339:85–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Claas C, Stipp CS, Hemler ME. 2001. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J. Biol. Chem. 276:7974–7984 [DOI] [PubMed] [Google Scholar]
- 65. Charrin S, Le Naour F, Silvie O, Milhiet PE, Boucheix C, Rubinstein E. 2009. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem. J. 420:133–154 [DOI] [PubMed] [Google Scholar]
- 66. Hogue IB, Grover JR, Soheilian F, Nagashima K, Ono A. 2011. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 85:9749–9766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ley K, Zhang H. 2008. Dances with leukocytes: how tetraspanin-enriched microdomains assemble to form endothelial adhesive platforms. J. Cell Biol. 183:375–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barreiro O, Zamai M, Yanez-Mo M, Tejera E, Lopez-Romero P, Monk PN, Gratton E, Caiolfa VR, Sanchez-Madrid F. 2008. Endothelial adhesion receptors are recruited to adherent leukocytes by inclusion in preformed tetraspanin nanoplatforms. J. Cell Biol. 183:527–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Espenel C, Margeat E, Dosset P, Arduise C, Le Grimellec C, Royer CA, Boucheix C, Rubinstein E, Milhiet PE. 2008. Single-molecule analysis of CD9 dynamics and partitioning reveals multiple modes of interaction in the tetraspanin web. J. Cell Biol. 182:765–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Krementsov DN, Rassam P, Margeat E, Roy NH, Schneider-Schaulies J, Milhiet PE, Thali M. 2010. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 11:1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shen B, Fang Y, Wu N, Gould SJ. 2011. Biogenesis of the posterior pole is mediated by the exosome/microvesicle protein-sorting pathway. J. Biol. Chem. 286:44162–44176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fang Y, Wu N, Gan X, Yan W, Morrell JC, Gould SJ. 2007. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol. 5:e158 doi:10.1371/journal.pbio.0050158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 170:317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sherer NM, Jin J, Mothes W. 2010. Directional spread of surface-associated retroviruses regulated by differential virus-cell interactions. J. Virol. 84:3248–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Grover JR, Llewellyn GN, Soheilian F, Nagashima K, Veatch SL, Ono A. 2013. Roles played by capsid-dependent induction of membrane curvature and Gag-ESCRT interactions in tetherin recruitment to HIV-1 assembly sites. J. Virol. 87:4650–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Accola MA, Strack B, Gottlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kong LB, An D, Ackerson B, Canon J, Rey O, Chen IS, Krogstad P, Stewart PL. 1998. Cryoelectron microscopic examination of human immunodeficiency virus type 1 virions with mutations in the cyclophilin A binding loop. J. Virol. 72:4403–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Auerbach MR, Brown KR, Kaplan A, de Las Nueces D, Singh IR. 2006. A small loop in the capsid protein of Moloney murine leukemia virus controls assembly of spherical cores. J. Virol. 80:2884–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Crist RM, Datta SA, Stephen AG, Soheilian F, Mirro J, Fisher RJ, Nagashima K, Rein A. 2009. Assembly properties of human immunodeficiency virus type 1 Gag-leucine zipper chimeras: implications for retrovirus assembly. J. Virol. 83:2216–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang Y, Qian H, Love Z, Barklis E. 1998. Analysis of the assembly function of the human immunodeficiency virus type 1 gag protein nucleocapsid domain. J. Virol. 72:1782–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Klein KC, Reed JC, Tanaka M, Nguyen VT, Giri S, Lingappa JR. 2011. HIV Gag-leucine zipper chimeras form ABCE1-containing intermediates and RNase-resistant immature capsids similar to those formed by wild-type HIV-1 Gag. J. Virol. 85:7419–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ono A, Waheed AA, Joshi A, Freed EO. 2005. Association of human immunodeficiency virus type 1 gag with membrane does not require highly basic sequences in the nucleocapsid: use of a novel gag multimerization assay. J. Virol. 79:14131–14140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guo XF, Roldan A, Hu J, Wainberg MA, Liang C. 2005. Mutation of the SP1 sequence impairs both multimerization and membrane-binding activities of human immunodeficiency virus type 1 Gag. J. Virol. 79:1803–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed EO, Summers MF. 2007. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 366:574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Liu J, Deng Y, Zheng Q, Cheng CS, Kallenbach NR, Lu M. 2006. A parallel coiled-coil tetramer with offset helices. Biochemistry 45:15224–15231 [DOI] [PubMed] [Google Scholar]
- 88. Dou J, Wang JJ, Chen X, Li H, Ding L, Spearman P. 2009. Characterization of a myristoylated, monomeric HIV Gag protein. Virology 387:341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Langhorst MF, Reuter A, Stuermer CA. 2005. Scaffolding microdomains and beyond: the function of reggie/flotillin proteins. Cell. Mol. Life Sci. 62:2228–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Parton RG, Simons K. 2007. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8:185–194 [DOI] [PubMed] [Google Scholar]
- 91. Ursell TS, Klug WS, Phillips R. 2009. Morphology and interaction between lipid domains. Proc. Natl. Acad. Sci. U. S. A. 106:13301–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, Dier M, Hell SW, Grubmuller H, Diederichsen U, Jahn R. 2011. Membrane protein sequestering by ionic protein-lipid interactions. Nature 479:552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Grigorov B, Attuil-Audenis V, Perugi F, Nedelec M, Watson S, Pique C, Darlix JL, Conjeaud H, Muriaux D. 2009. A role for CD81 on the late steps of HIV-1 replication in a chronically infected T cell line. Retrovirology 6:28 doi:10.1186/1742-4690-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jolly C, Sattentau QJ. 2005. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 79:12088–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jolly C, Sattentau QJ. 2007. Human immunodeficiency virus type 1 assembly, budding, and cell-cell spread in T cells take place in tetraspanin-enriched plasma membrane domains. J. Virol. 81:7873–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Krementsov DN, Weng J, Lambele M, Roy NH, Thali M. 2009. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology 6:64 doi:10.1186/1742-4690-6-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prevost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J. Virol. 83:6234–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lindwasser OW, Resh MD. 2002. Myristoylation as a target for inhibiting HIV assembly: unsaturated fatty acids block viral budding. Proc. Natl. Acad. Sci. U. S. A. 99:13037–13042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ono A. 2010. Relationships between plasma membrane microdomains and HIV-1 assembly. Biol. Cell 102:335–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mazurov D, Ilinskaya A, Heidecker G, Filatov A. 2012. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J. Virol. 86:2447–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Biggins JE, Biesinger T, Yu Kimata MT, Arora R, Kimata JT. 2007. ICAM-3 influences human immunodeficiency virus type 1 replication in CD4(+) T cells independent of DC-SIGN-mediated transmission. Virology 364:383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zacharias DA, Violin JD, Newton AC, Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296:913–916 [DOI] [PubMed] [Google Scholar]
- 103. McLaughlin S, Murray D. 2005. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 438:605–611 [DOI] [PubMed] [Google Scholar]
- 104. Chan J, Dick RA, Vogt VM. 2011. Rous sarcoma virus gag has no specific requirement for phosphatidylinositol-(4,5)-bisphosphate for plasma membrane association in vivo or for liposome interaction in vitro. J. Virol. 85:10851–10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kusumi A, Suzuki KG, Kasai RS, Ritchie K, Fujiwara TK. 2011. Hierarchical mesoscale domain organization of the plasma membrane. Trends Biochem. Sci. 36:604–615 [DOI] [PubMed] [Google Scholar]
- 106. Ciani B, Bjelic S, Honnappa S, Jawhari H, Jaussi R, Payapilly A, Jowitt T, Steinmetz MO, Kammerer RA. 2010. Molecular basis of coiled-coil oligomerization-state specificity. Proc. Natl. Acad. Sci. U. S. A. 107:19850–19855 [DOI] [PMC free article] [PubMed] [Google Scholar]