

Abstract
Epstein-Barr virus (EBV) replication proteins are transported into the nucleus to synthesize viral genomes. We here report molecular mechanisms for nuclear transport of EBV DNA polymerase. The EBV DNA polymerase catalytic subunit BALF5 was found to accumulate in the cytoplasm when expressed alone, while the EBV DNA polymerase processivity factor BMRF1 moved into the nucleus by itself. Coexpression of both proteins, however, resulted in efficient nuclear transport of BALF5. Deletion of the nuclear localization signal of BMRF1 diminished the proteins' nuclear transport, although both proteins can still interact. These results suggest that BALF5 interacts with BMRF1 to effect transport into the nucleus. Interestingly, we found that Hsp90 inhibitors or knockdown of Hsp90β with short hairpin RNA prevented the BALF5 nuclear transport, even in the presence of BMRF1, both in transfection assays and in the context of lytic replication. Immunoprecipitation analyses suggested that the molecular chaperone Hsp90 interacts with BALF5. Treatment with Hsp90 inhibitors blocked viral DNA replication almost completely during lytic infection, and knockdown of Hsp90β reduced viral genome synthesis. Collectively, we speculate that Hsp90 interacts with BALF5 in the cytoplasm to assist complex formation with BMRF1, leading to nuclear transport. Hsp90 inhibitors may be useful for therapy for EBV-associated diseases in the future.
INTRODUCTION
The Epstein-Barr virus (EBV), a human lymphotropic herpesvirus featuring linear double-stranded DNA (dsDNA) 172 kb in length (1), infects resting B lymphocytes, inducing their continuous proliferation without production of virus particles, which is termed latent infection. Productive (lytic) infection, which occurs spontaneously or can be induced artificially, is characterized by the expression of lytic genes, leading to virus production. During productive infection, the EBV genome is amplified 100- to 1,000-fold by viral replication machinery composed of the BALF5 DNA polymerase catalytic subunit, the BMRF1 polymerase processivity factor, the BALF2 single-stranded DNA-binding protein, and the BBLF4-BSLF1-BBLF2/3 helicase-primase complex in discrete sites in nuclei, called replication compartments (2–4). With progression of lytic replication, replication compartments become enlarged and fuse to form large globular structures that eventually fill the nucleus in late stages (2).
The BALF5 protein, a DNA polymerase catalytic subunit with both DNA polymerase and 3′-to-5′ exonuclease activities, forms a complex with the BMRF1 polymerase processivity factor with 1-to-1 stoichiometry (5–7). The resultant holoenzyme is characterized by significantly elevated polymerase processivity (6, 7). BMRF1 is a major phosphoprotein demonstrating abundant expression in lytic replication-induced cells (8, 9) when the expression level of the BALF5 protein is low. The BMRF1 can form head-to-head homodimer or tetrameric ring in solution (10). Judging from the finding that almost all expressed BMRF1 proteins bind to viral genome DNA (2, 11), the factor could not only act as a polymerase processivity factor but also perform other unknown functions (2). We have recently reported subnuclear domains that are highly enriched in viral polymerase processivity factor BMRF1, designated BMRF1 cores, inside replication compartments (4). Viral genomes are synthesized mainly outside the core and then transported inward. Thus, each replication compartment can be partitioned into two subdomains, outside and inside the BMRF1 core.
Viral replication proteins are predominantly localized in nuclei of lytic replication-induced cells. BMRF1 possesses a nuclear localization signal (NLS) at the carboxy-terminal domain (amino acids [aa] 378 to 404) (12). BALF2 single-stranded DNA (ssDNA) binding protein is also transported into nucleus by itself (13). The concurrent presence of all three components of the helicase-primase complex is required for the nuclear localization of BBLF2/3 and BSLF1 proteins (14). On the other hand, the BBLF4 protein can be converted from a strictly cytoplasmic pattern to a strictly nuclear pattern simply by interacting with the BZLF1 oriLyt binding protein (14). However, the mechanisms of nuclear localization of the BALF5 DNA polymerase catalytic subunit remain unclear.
Regarding the nuclear transport mechanism of the herpesvirus DNA polymerase catalytic subunit, it was reported that UL30 of herpes simplex virus 1 (HSV-1), being complexed with its processivity factor UL42 as a holoenzyme, translocated to nuclei utilizing its own NLS (15). It was also demonstrated that nuclear translocation of HSV-1 UL30 was strongly inhibited by the inhibitors of heat shock protein 90 (Hsp90), resulting in decreased HSV-1 yields and viral DNA synthesis (16). The inhibition occurred even when UL30 was solely transduced in cells, indicating that Hsp90 directly participates in the nuclear translocation of UL30 (16).
Heat shock proteins (HSPs) induced by various stresses are known to be involved in quality control of cellular proteins and homeostasis in cells (17–19). One example, Hsp90, is one of the most abundant cellular proteins. An evolutionarily well-conserved molecular chaperone, Hsp90 has two cytosolic isoforms, Hsp90α (inducible form) and Hsp90β (constitutive form), both featuring ATP-binding domains in their N-terminal domains and demonstrating functional dependence on binding and hydrolyzing ATP. Through the ATPase cycle, Hsp90 facilitates conformational maturation, stabilization, protein interaction, and intracellular trafficking of many client proteins. Radicicol and 17-allylamino-17-demethoxygeldana-mycin (17-AAG), a geldanamycin derivative, are “Hsp90 inhibitors” that associate directly with the ATP-binding pocket of Hsp90, preventing ATP binding and interaction with client proteins (20).
In the present study, we demonstrate that the viral DNA polymerase BALF5 does not translocate to the nucleus by itself. Rather, BALF5 accumulates inside the nucleus only when viral polymerase processivity factor BMRF1 is coexpressed, making a clear contrast to the nuclear translocation mechanism of HSV-1 UL30 (16). Intriguingly, in spite of the apparently different translocation mechanisms employed by EBV and HSV-1 DNA polymerases, EBV replication was also strongly inhibited by Hsp90 inhibitors, in analogy to the case of HSV-1 replication. Our overall results indicate that EBV DNA polymerase interacts with the BMRF1 polymerase processivity factor via molecular chaperone Hsp90, resulting in its nuclear translocalization. The difference of nuclear transport mechanisms between EBV and HSV-1 is discussed.
MATERIALS AND METHODS
Cells.
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) and the B95-8 B lymphoblastoid cell line in RPMI 1640 medium, both supplemented with 10% fetal bovine serum. To induce lytic EBV replication of B95-8 cells, 12-O-tetradecanoylphorbol-13-acetate (TPA), A23187, and sodium butyrate were added to the culture medium at final concentrations of 20 ng/ml, 0.25 μM, and 5 mM, respectively. Tet-BZLF1/B95-8 cells (21) were maintained in RPMI 1640 medium supplemented with 1 μg puromycin and 250 μg of hygromycin B per milliliter and 10% tetracycline-free fetal bovine serum. To induce lytic EBV replication, the tetracycline derivative doxycycline (Dox) was added to the culture medium at a final concentration of 4 μg/ml. AGS-CR2/EGFP-EBV cells were maintained in RPMI 1640 medium containing 150 μg of hygromycin B and 400 μg G418 per milliliter. For their preparation, an EBV-negative cell line from gastric cancer, AGS, was stably transfected with CR2 (CD21, the receptor for EBV) expression vector (22) and infected with an enhanced green fluorescent protein-expressing EBV (EGFP-EBV) strain (23) followed by G418 selection.
Antibodies and reagents.
BMRF1 protein-specific rabbit polyclonal antibodies (Ab) were raised as described previously (11, 24, 25). Anti-BALF5 protein-specific rabbit polyclonal Ab (26) were affinity purified with BALF5 protein coupled-Sepharose 4B (27). An anti-EBV EA-D-p52/50 (BMRF1 gene product) protein-specific mouse monoclonal antibody (MAb), clone name R3 (Chemicon, Inc.), anti-Hsp90β mouse MAb (Enzo Life Science), antihemagglutinin (anti-HA) tag mouse MAb (Roche), anti-FLAG tag rabbit polyclonal Ab (Sigma), anti-Chk1 rabbit polyclonal Ab (Cell Signaling), anti-cdc2 mouse MAb (Oncogene), and anti-tubulin rabbit polyclonal Ab (Cell Signaling) were purchased and used as primary antibodies. Horseradish peroxidase-linked goat antibodies against mouse, rabbit, and rat IgG (Amersham Biosciences) and Alexa Fluor 488-, 594-, or 688-conjugated goat antibodies against rabbit, mouse, and rat IgG (Molecular Probes) were purchased for application as secondary antibodies.
Radicicol and 17-allylamino-17-demethoxygeldanamycin (17-AAG) were purchased from Wako Pure Chemicals (Osaka, Japan), with stock solutions being prepared at concentrations of 10 mM in dimethyl sulfoxide (DMSO). Stocks of these drugs were aliquoted and kept at −20°C until use. The final concentration of each drug used is indicated in the figure legends.
Transfection.
All transfections were carried out using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. The total amounts of plasmid DNAs were standardized by addition of an empty vector.
Immunofluorescence analyses.
HeLa cells were grown on coverslips and transfected with pcDNA-FLAG-BALF5, pcDNA-BMRF1, pcDNA-BMRF1ΔC, and pcDNA-HA-Hsp90 (provided by Y. Matsuura, Osaka University). AGS-CR2/EGFP-EBV cells were grown on coverslips and transfected with pcDNA-BZLF1. At 24 h posttransfection, cells were washed with ice-cold phosphate-buffered saline (PBS) and fixed by 3.7% formalin for 15 min. Fixed cells were washed with PBS and incubated in a blocking buffer (2.5% bovine serum albumin [BSA], 0.2 M glycine, 0.1% Triton X-100, 0.02% sodium azide in PBS) for 30 min at room temperature. The samples were then incubated with the primary antibodies diluted in blocking buffer at 4°C overnight, followed by the incubation with diluted secondary goat anti-rabbit, anti-rat, and anti-mouse IgG antibodies conjugated with Alexa Fluor 488, 594, or 688 for 30 min at room temperature.
Lytic infection-induced Tet-BZLF1/B95-8 cells were treated with ice-cold modified cytoskeleton (mCSK) buffer [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.8), 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.5% Triton X-100] on ice for 10 min. Multiple protease inhibitor cocktails (Sigma; 25 μl/ml), 200 μM Na3VO4, and 20 mM NaF were also added to the buffer, followed by fixation with 70% ethanol at −20°C overnight. The cells then were washed with PBS containing 0.1% normal goat serum and 0.01% Tween 20, permeabilized with 0.5% Triton X-100 in PBS for 15 min, and blocked for 1 h in 10% normal goat serum in PBS. Samples were then incubated overnight with the primary antibodies diluted in PBS containing 0.1% normal goat serum and 0.01% Tween 20, followed by the incubation with diluted Alexa Fluor 488- or 594-conjugated secondary antibodies for 1 h.
All of the primary antibodies were employed at a 1:100 dilution, and the secondary antibodies were employed at a 1:200 dilution. All washes after antibody incubation were performed with PBS containing 0.1% normal goat serum and 0.01% Tween 20. Both samples were mounted in ProLong Gold antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Molecular Probes) and analyzed by laser-scanning confocal microscopy (Carl Zeiss MicroImaging, Inc.). Images were captured with a plan-Apochromat 100×/1.4-numerical aperture oil immersion objective and processed using an LSM510 Meta microscope.
IP analyses.
HEK293T cells in 6-cm dishes were transfected with pcDNA-FLAG-BALF5, pcDNA-BMRF1, and pcDNA-HA-Hsp90 and cultivated for 24 h. The cells were harvested, washed with cold PBS, and then lysed for 10 min on ice with 500 μl of ice-cold mCSK buffer containing 0.1% Triton X-100, 50 mM NaCl, and 25 μl/ml protease inhibitor cocktail (Sigma). When BMRF1 was expressed, the samples were treated with DNase I (1,000 U/ml) for 30 min at 25°C to solubilize BMRF1. After treatment, homogenates were centrifuged at 3,000 × g at 4°C for 5 min. The supernatant fraction was used as extracts for assays. One hundred microliters each of the Triton X-100-extractable fractions was diluted with mCSK buffer to 500 μl and then mixed with 1 μg each of anti-HA, anti-BALF5, or anti-BMRF1 antibodies or 10 μl anti-M2 beads (Sigma) for 3 h at 4°C. After the addition of 20 μl of protein G-Sepharose beads, the mixtures were further incubated for 1 h at 4°C. The beads were then washed with 0.1% mCSK buffer containing 50 mM NaCl, and the immunoprecipitates were boiled in SDS sample buffer and subjected to immunoblotting analysis. As the IgG control experiments, the same amounts of extracts (500 μl of diluted extracts) were subjected to immunoprecipitation (IP) using the same amounts (1 μg each) of normal IgG (Santa Cruz Biotechnology) and were precipitated with the same amounts of protein G-Sepharose beads.
Where indicated, 20 mM disodium molybdate dehydrate (Wako) was added in the mCSK buffer to stabilize and enhance the binding of Hsp90 to client proteins and included throughout the IP experiments (including the washing steps).
Immunoblotting analysis.
HeLa cells, 293T cells, short hairpin RNA (shRNA)-transduced AGS-CR2/EGFP-EBV cells, and luciferase shRNA (shLuc)-transduced HeLa cells were harvested at the indicated condition. Cells were washed with phosphate-buffered saline (PBS) and treated with lysis buffer (0.02% sodium dodecyl sulfate [SDS], 0.5% Triton X-100, 300 mM NaCl, 20 mM Tris-HCl [pH 7.6], 1 mM EDTA, 25 μl/ml protease inhibitor cocktail [Sigma]) for 20 min on ice. Samples were centrifuged at 12,000 × g for 10 min at 4°C, and the clarified cell extracts were measured for protein concentrations by using a Bio-Rad protein assay kit. Samples underwent in SDS–8% or –10% PAGE (acrylamide/bisacrylamide ratio, 29.2:0.8) and then were transferred to polyvinylidene difluoride membranes. After washing with a wash buffer (1× PBS containing 0.1% Tween 20) and blocking for 60 min in a wash buffer containing 10% low-fat powdered milk, the membranes were incubated with primary antibodies in wash buffer containing 5% low-fat powdered milk for 60 min at room temperature, followed by incubation with a horseradish peroxidase-conjugated secondary antibody at room temperature for 60 min. Target proteins were detected with an enhanced chemiluminescence detection system (Amersham).
Quantification of viral DNA synthesis during lytic replication.
Levels of viral DNA were determined by quantitative real-time PCR (28). Lytic replication induced B95-8 cells were harvested, suspended in 200 μl of a lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 0.001% Triton X-100, 0.001% SDS), sonicated, and then incubated at 50°C for 2 h with 50 μg/ml of proteinase K (Sigma). Primers and a probe for a sequence within the BALF2-coding region were designed using Primer Express (Applied Biosystems). The sequences were as follows: 5′-GCCCGTCCGGTTGTCA-3′ (forward primer), 5′-AATATCTGGTTGTTGCCGTTGA-3′ (reverse primer), and 5′-6-carboxyfluorescein (FAM)-CTGCCAGTGACCATCAACAAGTACACGG-6-carboxytetramethylrhodamine (TAMRA)-3′ (probe). PCR was performed in 10 μl of aqueous solution containing 1 μM each primer, 0.4 μM labeled probe, Fast Start Universal Probe Master (Roche), and 1 μl of DNA mixture using the 7300 real-time PCR system (Applied Biosystems). PCR included 2 min at 50°C, 10 min at 95°C, and 40 cycles at 95°C for 15 s followed by 1 min at 60°C.
Cell viability and cell growth.
Cell viability was determined by the trypan blue dye exclusion test. B95-8 cells were induced to enter lytic replication in either the presence or the absence of radicicol treatment. The induced cells were harvested at 24, 48, and 72 h postinduction and resuspended in trypan blue solution. Cell viability and cell growth were determined by counting the numbers of unstained (live) and stained (dead) cells by using a hemocytometer.
Knockdown of Hsp90β with an shRNA.
Knockdown of Hsp90β was carried out with a retrovirus shRNA system. The target sequence for the Hsp90β shRNA was 5′-AGCCTACGTTGCTCACTAT-3′. A retroviral vector expressing shRNA for luciferase (shLuc) was used as a control. AGS-CR2/EGFP-EBV cells and HeLa cells were infected with retroviral vectors expressing either luciferase shRNA or Hsp90β shRNA. Puromycin was added to the transduced cells at 24 h postinfection, and puromycin-resistant cell pools were obtained at 72 h postinfection.
RESULTS
Nuclear transfer of the BALF5 DNA polymerase catalytic subunit is dependent on the BMRF1 polymerase processivity factor.
All EBV DNA replication proteins localize to nuclei of lytic replication-induced cells. However, the nuclear localization of BBLF2/3 and BSLF1 proteins, constituting the viral helicase-primase complex, requires the concurrent presence of BBLF4 protein (14). Therefore, we first examined localization of the BALF5 DNA polymerase catalytic subunit in HeLa cells transfected with FLAG-tagged BALF5 and/or BMRF1 expression vectors, using immunofluorescence staining. As shown in Fig. 1A, when FLAG-BALF5 was expressed alone, it was detected predominantly in the cytoplasm. In contrast, BMRF1 was observed exclusively in the nucleus (Fig. 1B). When both proteins were coexpressed, BALF5 efficiently localized to the nucleus (Fig. 1C). It is known that the BMRF1 protein interacts with the BALF5 polymerase catalytic subunit to form a heterodimer complex, resulting in acquirement of enhanced polymerase processivity and exonuclease activities (7, 26). The coimmunoprecipitation assay confirmed that BALF5 interacted with BMRF1 (Fig. 1D). These results clearly indicate that BALF5 by itself cannot translocate to the nucleus, but it can do so in a BMRF1-dependent manner.
Fig 1.
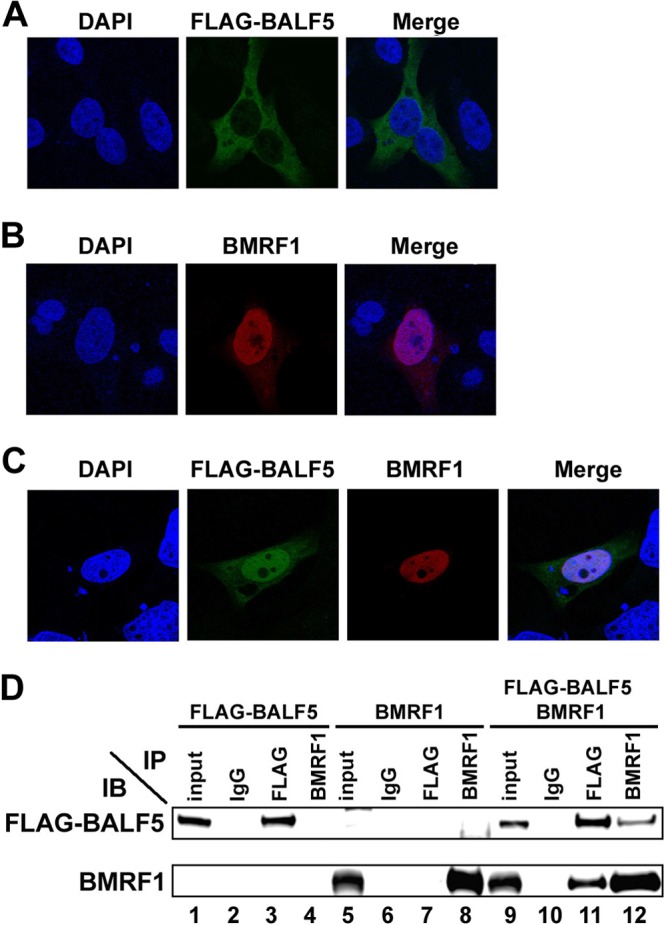
Nuclear transport of the BALF5 DNA polymerase catalytic subunit requires the BMRF1 polymerase accessory protein. HeLa cells were transfected with pcDNA-FLAG-BALF5 (A) or pcDNA-BMRF1 (B) or cotransfected with both expression vectors (C). Cells were fixed at 24 h posttransfection, immunostained with anti-FLAG (green) and anti-BMRF1 (red) antibodies, and examined by laser-scanning confocal microscopy. In each sample, nuclei were stained with DAPI (blue). The right panels are merged images. (D) Interaction between BALF5 and BMRF1. HEK293T cells were transfected with pcDNA-FLAG-BALF5, pcDNA-BMRF1, or both. Cells were harvested at 24 h posttransfection, and immunoprecipitation (IP) analysis was carried out, as described in Materials and Methods. The antibodies used for IP and subsequent immunoblot (IB) analysis are indicated.
It is known that BMRF1 protein possesses a nuclear localization signal (NLS) in its C-terminal domain (Fig. 2A) (29). A BMRF1 mutant lacking the C-terminal domain (BMRF1ΔC) still possesses double-stranded DNA binding activity and functions as a polymerase processivity factor in vitro, just like wild-type BMRF1 (10). Expectedly, unlike wild-type BMRF1, BMRF1ΔC was detected in the cytoplasm (Fig. 2B). This truncated mutant could also physically interact with BALF5 (Fig. 2C). When the BMRF1ΔC mutant and BALF5 were coexpressed, BALF5 remained in the cytoplasm. (Fig. 2D), suggesting that BALF5 utilizes the NLS function of BMRF1 for its transport into the nucleus.
Fig 2.
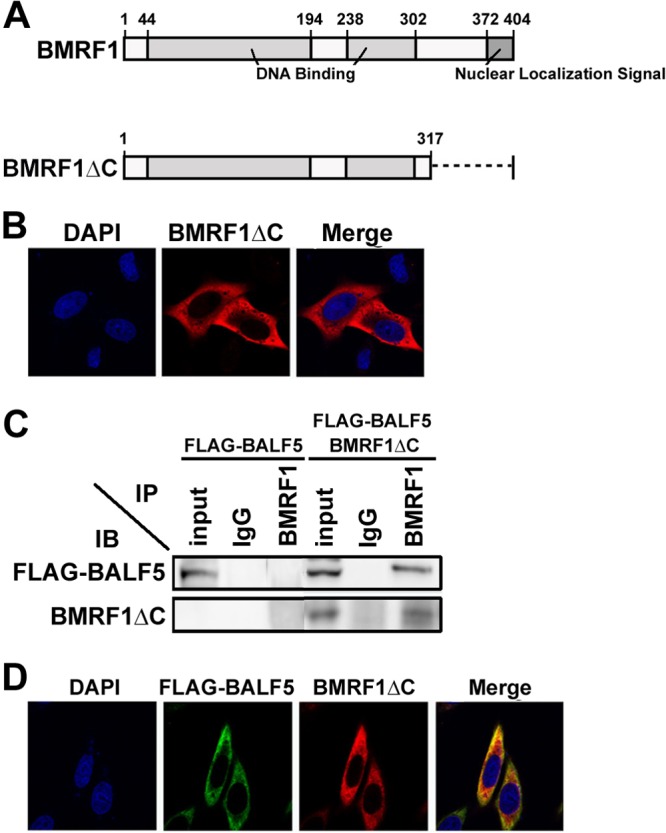
A BMRF1 mutant lacking the nuclear localization signal (NLS) does not support nuclear transport of the BALF5 DNA polymerase catalytic subunit. (A) Functional domains of BMRF1 protein. BMRF1 protein, consisting of 404 amino acids, has an NLS in its C terminus (amino acids 372 to 404). A BMRF1 mutant lacking the C-terminal 90 amino acids (aa 1 to 317; BMRF1ΔC) is indicated. (B) Subcellular localization of BMRF1ΔC. HeLa cells were transfected with pcDNA-BMRF1ΔC, fixed at 24 h posttransfection, immunostained with anti-BMRF1 (red), and examined by laser-scanning confocal microscopy. Nuclei were stained with DAPI (blue). (C) Physical interaction between BALF5 and BMRF1ΔC. HEK293T cells were transfected with pcDNA-FLAG-BALF5 alone or both pcDNA-FLAG-BALF5 and pcDNA-BMRF1ΔC. Cells were harvested at 24 h posttransfection, and immunoprecipitation (IP) analysis was carried out, as described in Materials and Methods. The antibodies used for IP and subsequent immunoblot (IB) analysis are indicated. (D) Subcellular localization of BALF5 in the presence of BMRF1ΔC. HeLa cells were transfected with pcDNA-FLAG-BALF5 and pcDNA-BMRF1ΔC, fixed at 24 h posttransfection, immunostained with anti-FLAG (green) and anti-BMRF1 (red), and examined by laser-scanning confocal microscopy. Nuclei were stained with DAPI (blue). The right panels are merged images.
Hsp90 inhibitors prevent BMRF1-dependent nuclear transport of the BALF5 protein.
Hsp90 is known to facilitate conformational maturation, stabilization, protein interaction, and intracellular trafficking of many client proteins. It was reported that Hsp90 was involved in nuclear translocation of UL30, a DNA polymerase catalytic subunit of HSV-1 (16). Therefore, we examined the possibility that it might be involved in the nuclear transport of BALF5 and BMRF1 proteins. In the presence or absence of an Hsp90 inhibitor, radicicol, BALF5 was detected in the cytoplasm when the protein was expressed alone (Fig. 3A), while BMRF1 was found in the nucleus (Fig. 3B), indicating at least that nuclear transport of the BMRF1 protein is independent of Hsp90. When both proteins were coexpressed, BMRF1-dependent nuclear transport of BALF5 was clearly inhibited by radicicol (Fig. 3C). Figure 3D shows a numerical analysis of the intracellular localization patterns of BALF5. In the absence of Hsp90 inhibitors, BALF5 was predominantly localized in the nucleus in almost all of the cells expressing both proteins (Fig. 3D). In the presence of radicicol or 17-AAG, cell populations localizing in cytoplasm increased vastly. Thus, BALF5 nuclear localization is absolutely dependent on BMRF1 and blocked by Hsp90 inhibitors. It should be noted that the expressed Hsp90 was localized predominantly in the cytoplasm in all cases.
Fig 3.

Prevention of BMRF1-dependent nuclear transport of BALF5 DNA polymerase catalytic subunit by the Hsp90 inhibitor radicicol. HeLa cells were transfected with the indicated combinations of pcDNA-FLAG-BALF5, pcDNA-BMRF1, and pcDNA-HA-Hsp90 in the absence or presence of 1 μM radicicol. After fixation 24 h posttransfection, the cells were immunostained with anti-FLAG (green) and anti-HA (yellow) antibodies (A), with anti-BMRF1 (red) and anti-HA (yellow) antibodies (B), and with anti-FLAG (green), anti-BMRF1 (red), and anti-HA (yellow) antibodies(C). Nuclei were stained with DAPI (blue). (D) Numerical analysis of the localization pattern of BALF5 in transfected cells. The proportions of cells showing each localization pattern are expressed as percentages out of the examined cells (more than 100). The images in the upper panels represent the typical BALF5 localization patterns. (E) Effect of Hsp90 inhibitor. HeLa cells were treated with 0.5 μM radicicol for 24 h and harvested. Clarified cell lysates were prepared, separated by SDS–10% PAGE, and applied for Western blot analyses with anti-Chk1 protein polyclonal antibody.
Hsp90 inhibitor, in general, decreases the expression levels of client proteins. To verify the efficacy of the Hsp90 inhibitor in this experimental system, we examined whether the Hsp90 inhibitor affected the expression level of Chk1, a host Hsp90 client protein. The result revealed that the level of Chk1 was greatly decreased by radicicol treatment (Fig. 3E), confirming that the inhibitor was working efficiently in this system. From these results, we speculate that Hsp90 is involved in the association between the BALF5 and BMRF1 proteins in the cytoplasm.
Next, to confirm whether Hsp90 inhibition prevents nuclear transport of the BALF5 DNA polymerase catalytic subunit in lytic replication-induced cells, subcellular localizations of BALF5 and BMRF1 were examined in lytic infection-induced Tet-BZLF1/B95-8 cells with or without Hsp90 inhibitors. Tet-BZLF1/B95-8 cells treated with doxycycline were harvested at 24 h postinduction, extracted with 0.5% Triton X-100 in mCSK buffer, and fixed with 70% ethanol (EtOH), followed by immunostaining with anti-BALF5 polyclonal or anti-BMRF1 monoclonal antibodies. In the absence of Hsp90 inhibitors, both BALF5 and BMRF1 proteins were observed within replication compartments in the nucleus [Fig. 4, Dox(+)]. In their presence, BALF5 exhibited cytoplasmic localization (Fig. 4A). Accordingly, replication compartments diminished, and BMRF1 was distributed throughout nuclei (Fig. 4B). Thus, Hsp90 inhibitors prevented nuclear localization of the BALF5 protein not only in an overexpression system but also in the context of cells featuring lytically replicating viruses.
Fig 4.
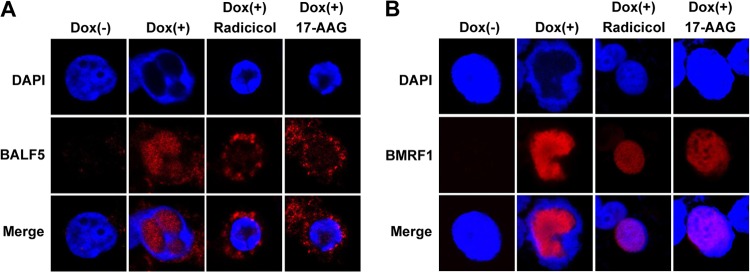
Effects of Hsp90 inhibitors on the subcellular localization of BALF5 and BMRF1 proteins in lytic replication-induced cells. Tet-BZLF1/B95-8 cells were treated with doxycycline (4 μg/ml) to induce lytic replication in the absence or the presence of 1 μM radicicol or 1 μM 17-AAG, and the cells were processed for immunostaining at 24 h postinduction with anti-BALF5 (A; red) or anti-BMRF1 (B; red) antibodies. DAPI-stained images (upper panels) and merged images (lower panels) are also shown.
BALF5 is a possible client protein of Hsp90.
To validate our findings, 293T cells were transfected with combinations of HA-tagged Hsp90, FLAG-tagged BALF5, and BMRF1 expression vectors in the absence or presence of the Hsp90 inhibitor radicicol, and protein-protein interactions were examined by immunoprecipitation analysis (Fig. 5). Cells were extracted with 0.1% Triton X-100–mCSK buffer containing 50 mM NaCl (and treated with DNase I only when BMRF1 was expressed), and soluble fractions were reacted with anti-HA or anti-FLAG antibody-conjugated beads, anti-BMRF1 IgG, or nonimmune IgG as a control. With the combination of HA-Hsp90 and FLAG-BALF5, anti-HA antibodies could coimmunoprecipitate FLAG-BALF5 protein (Fig. 5A, lane 11). Conversely, anti-FLAG antibodies could coimmunoprecipitate the HA-Hsp90 protein (Fig. 5A, lane 12). Although the interaction between FLAG-BALF5 and HA-Hsp90 was fairly weak (Fig. 5A), it is known that interactions of Hsp90 and client proteins are generally not so strong as to be detected easily by immunoprecipitation analysis. The interaction was diminished by radicicol treatment (Fig. 5B, compare lanes 3 and 4 with lanes 7 and 8), supporting the idea that Hsp90 and BALF5 interact with each other. The amount of BALF5 protein did not change by Hsp90 inhibitor (radicicol) treatment, while the amount of cdc2 protein, a host Hsp90 client protein, was decreased (Fig. 5C). This result indicates that the diminished interaction between Hsp90 and BALF5 is not due to the reduction of BALF5 protein. We further examined immunoprecipitation analysis in the presence of molybdate to confirm the interaction between BALF5 and Hsp90. It is known that molybdate stabilizes the interaction of Hsp90 and its client proteins (30, 31). The Hsp90-BALF5 interaction was significantly stabilized in the presence of molybdate (Fig. 5D, compare lane 3 with lane 6). In contrast, when HA-Hsp90 and BMRF1 were coexpressed, BMRF1 did not coimmunoprecipitate with Hsp90 (Fig. 5E, lane 3) and vice versa (Fig. 5E, lane 4). Any association between BMRF1 and Hsp90 was not detected even in the presence of molybdate (Fig. 5E, lanes 7 and 8). Taken together, the results indicate that BALF5, but not BMRF1, is a possible client protein of Hsp90.
Fig 5.
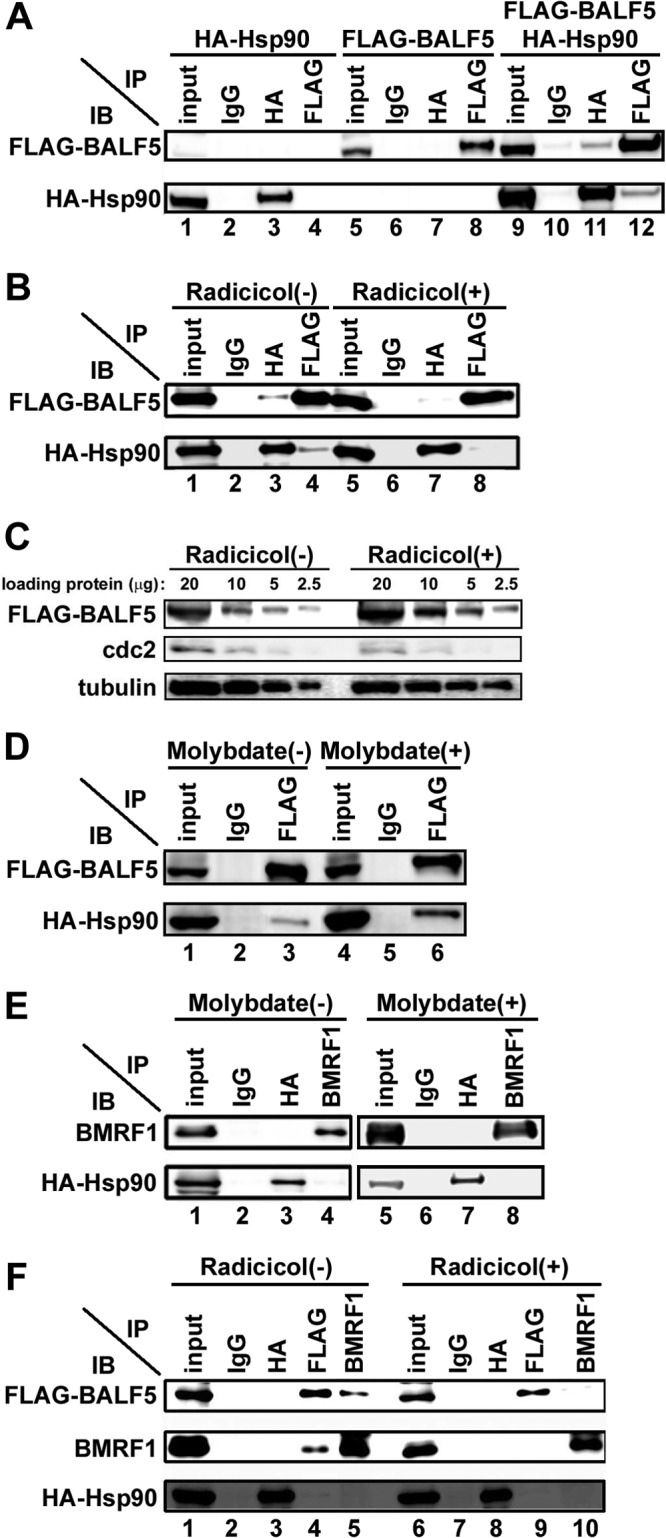
Interaction of Hsp90 with the BALF5 DNA polymerase catalytic subunit. (A) Coimmunoprecipitation of Hsp90 with BALF5. HEK293T cells were transfected with pcDNA-FLAG-BALF5 alone, pcDNA-HA-Hsp90 alone, or both pcDNA-FLAG-BALF5 and pcDNA-HA-Hsp90. Cells were harvested at 24 h posttransfection, and immunoprecipitation (IP) analysis was carried out as described in Materials and Methods. The antibodies used for IP and subsequent immunoblot (IB) analysis are indicated. (B) Disruption of the interaction between BALF5 and Hsp90 by radicicol. HEK293T cells were transfected with pcDNA-FLAG-BALF5 and pcDNA-HA-Hsp90 in the absence or the presence of 0.5 μM radicicol. Cells were harvested at 24 h posttransfection, and IP was carried out, as described in Materials and Methods. Antibodies used for IP and subsequent IB are indicated. (C) The indicated amounts of input samples of panel B were electrophoresed and analyzed by immunoblotting with anti-FLAG, anti-cdc2, and antitubulin antibodies. (D) Stabilization of the interaction between BALF5 and Hsp90 by molybdate. HEK293T cells transfected with pcDNA-FLAG-BALF5 and pcDNA-HA-Hsp90 were lysed in 0.5% mCSK buffer in the presence or absence of 20 mM molybdate. Lysates were subjected to IP and subsequent IB analyses. The antibodies used for IP and IB are indicated. (E) Hsp90 does not associate with BMRF1. HEK293T cells were transfected with pcDNA-BMRF1 and pcDNA-HA-Hsp90 in the absence or presence of 20 mM molybdate. Cells were harvested at 24 h posttransfection, and IP was carried out as described in Materials and Methods. The antibodies used for IP and subsequent IB analyses are indicated. (F) Hsp90 contributes to BALF5-BMRF1 complex formation. HEK293T cells were transfected with pcDNA-FLAG-BALF5, pcDNA-HA-Hsp90, and pcDNA-BMRF1. Cells were harvested at 24 h posttransfection, and IP was carried out as described in Materials and Methods. The antibodies used for IP and subsequent IB analyses are indicated. On each panel, the input lane contains 10% of the amount of the soluble fractions that were subjected to immunoprecipitation.
Hsp90 assists complex formation between the BALF5 and BMRF1 proteins.
Next, we examined whether Hsp90 is included in the BALF5-BMRF1 complex. When three factors were overexpressed, protein-to-protein interactions of BALF5 and BMRF1 proteins could be clearly demonstrated by immunoprecipitation using each antibody (Fig. 5F, lanes 4 and 5). However, association of Hsp90 with the BALF5-BMRF1 complex could barely be detected (Fig. 5F, lane 3), suggesting that Hsp90 is absent in the BALF5-BMRF1 complex. Radicicol treatment attenuated the BALF5-BMRF1 association (Fig. 5F, lanes 9 and 10). Considering these observations, we speculate that Hsp90 interacts at least with BALF5 protein to facilitate BALF5-BMRF1 complex formation.
Hsp90 inhibition suppresses EBV DNA replication.
Because Hsp90 inhibitors changed localization of EBV DNA polymerase catalytic subunit, we speculated that such treatment might suppress EBV DNA replication. With quantitative real-time PCR, levels of viral DNA increased with time (Fig. 6A, closed squares) in lytic replication-induced B95-8 cells. Treatment of cells with 1 μM radicicol almost completely blocked viral genome synthesis at each time point (Fig. 6A, closed triangles). Similar effects were observed when cells were treated with another Hsp90 inhibitor, 17-AAG (data not shown). Importantly, Hsp90 inhibitor did not affect the expression level of BZLF1 (Fig. 6B). Furthermore, the Hsp90 inhibitor did not affect cell viability (Fig. 6C), although the induction of lytic replication resulted in complete growth arrest (Fig. 6D). These observations indicate that the inhibition of EBV DNA replication by the Hsp90 inhibitor was not due to cell death. Thus, Hsp90 inhibitors can completely inhibit EBV DNA replication.
Fig 6.
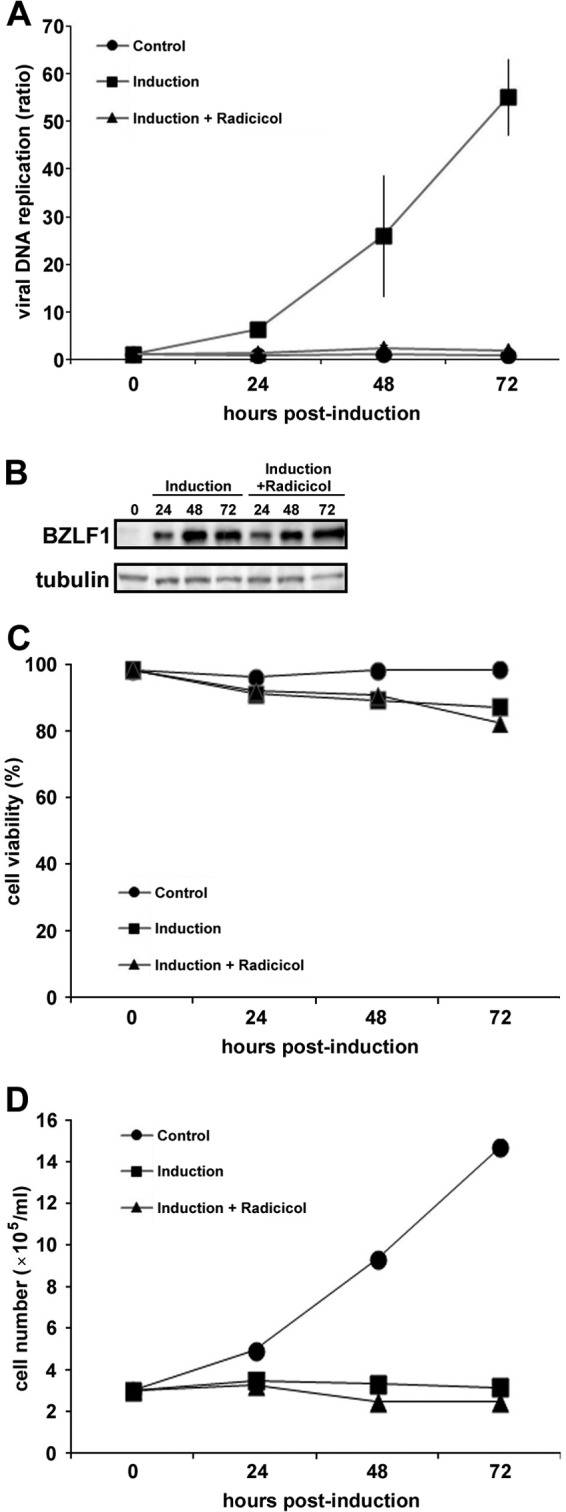
Effects of radicicol on viral DNA synthesis in lytic replication-induced cells. (A) Lytic replication was induced in B95-8 cells with the addition of doxycycline in the presence or the absence of 1 μM radicicol, and the cells were harvested at the indicated hours postinduction. The levels of viral DNA synthesis were determined by quantitative real time-PCR assay and plotted as ratios to the value at 0 h ± the standard error of the mean from three independent experiments. (B) The expression levels of BZLF1 were determined by Western blotting analyses using anti-BZLF1 protein polyclonal antibody. (C) Cell viabilities were determined by trypan blue dye exclusion test. The percentages of viable cells out of total cells (more than 100 cells counted for each sample) are indicated. (D) The growth curves of the cells in panel C are shown.
Knockdown of Hsp90β prevents EBV DNA replication.
To elucidate whether the results obtained by Hsp90 inhibitors are actually due to Hsp90 functional block, we examined the effect of Hsp90 knockdown. First, we confirmed EBV DNA replication by transducing Hsp90 shRNA into virus-producing cells. Since it is known that Hsp90α is not induced during EBV lytic infection, we focused on the effect of Hsp90β knockdown. The Hsp90β expression level was significantly reduced in shHsp90β-transduced AGS-CR2/EGFP-EBV cells harboring recombinant EBV genomes, compared to the level in shLuc-transduced cells (Fig. 7A). When lytic replication was induced in shLuc-transduced cells by BZLF1 transfection as a positive control, the level of EBV DNA markedly increased (more than 40-fold) (Fig. 7B, shLuc+BZLF1). In contrast, when lytic replication was induced in shHsp90β-transduced cells, the increase in viral DNA synthesis was far less than that of BZLF1-transfected shLuc-transduced cells (Fig. 7B). Thus, knockdown of Hsp90β prevented EBV DNA replication.
Fig 7.
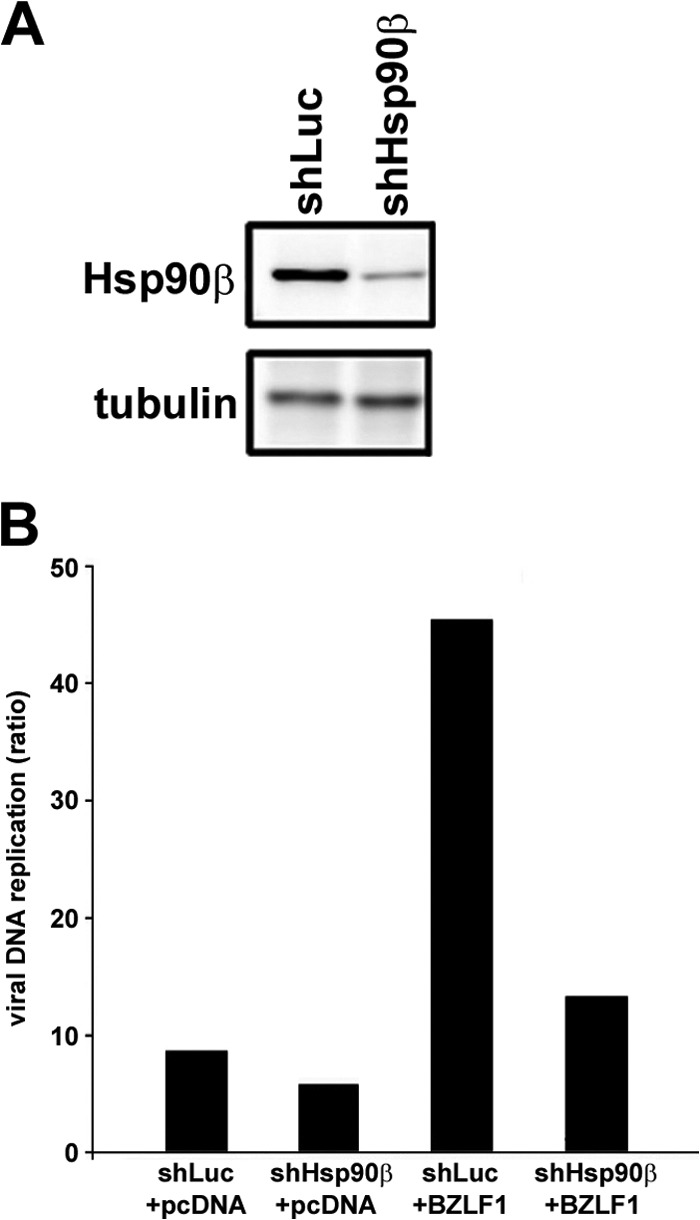
Hsp90β knockdown prevents viral DNA synthesis in lytic replication-induced AGS-CR2/EGFP-EBV cells. (A) AGS-CR2/EGFP-EBV cells expressing either luciferase shRNA (shLuc) or Hsp90β shRNA (shHsp90β) were subjected to immunoblotting analysis with anti-Hsp90β and antitubulin antibodies. (B) Cells transduced with shLuc or shHsp90β were transfected with either pcDNA or pcDNA-BZLF1 to induce lytic replication and were harvested at 72 h posttransfection. Levels of viral DNA synthesis were determined by quantitative real time-PCR assay. Bars indicate ratios compared to the value of the pcDNA-transfected shLuc cells at 0 h.
Knockdown of Hsp90β prevents BMRF1-dependent nuclear translocalization of BALF5.
Next, the effects of Hsp90β knockdown on localization of BALF5 and BMRF1 proteins were examined. When lytic replication was induced in shLuc-transduced AGS-CR2/EGFP-EBV cells by BZLF1 transfection, both BALF5 and BMRF1 proteins were accumulated in the nucleus (Fig. 8A-a). On the other hand, in the shHsp90β-transduced AGS-CR2/EGFP-EBV cells, BALF5 protein remained in the cytoplasm (Fig. 8A-b). We also observed the localization of both proteins on the overexpressed system in Hsp90β-knockdown HeLa cells. The Hsp90β expression level was significantly reduced in shHsp90β-transduced HeLa cells compared to that in shLuc-transduced cells (Fig. 8B-c). In the shLuc-transduced cells expressing both proteins, BALF5 efficiently localized to the nucleus as well as BMRF1 (Fig. 8B-a). Hsp90β mainly localized in the cytoplasm. In contrast, even in the presence of BMRF1, BALF5 localized to the cytoplasm in shHsp90β-transduced cells (Fig. 8B-b). These results correlated well with results of experiments with Hsp90 inhibitors. Thus, constitutively expressed Hsp90β is involved in the BMRF1-dependent nuclear transport of the BALF5 DNA polymerase catalytic subunit.
Fig 8.
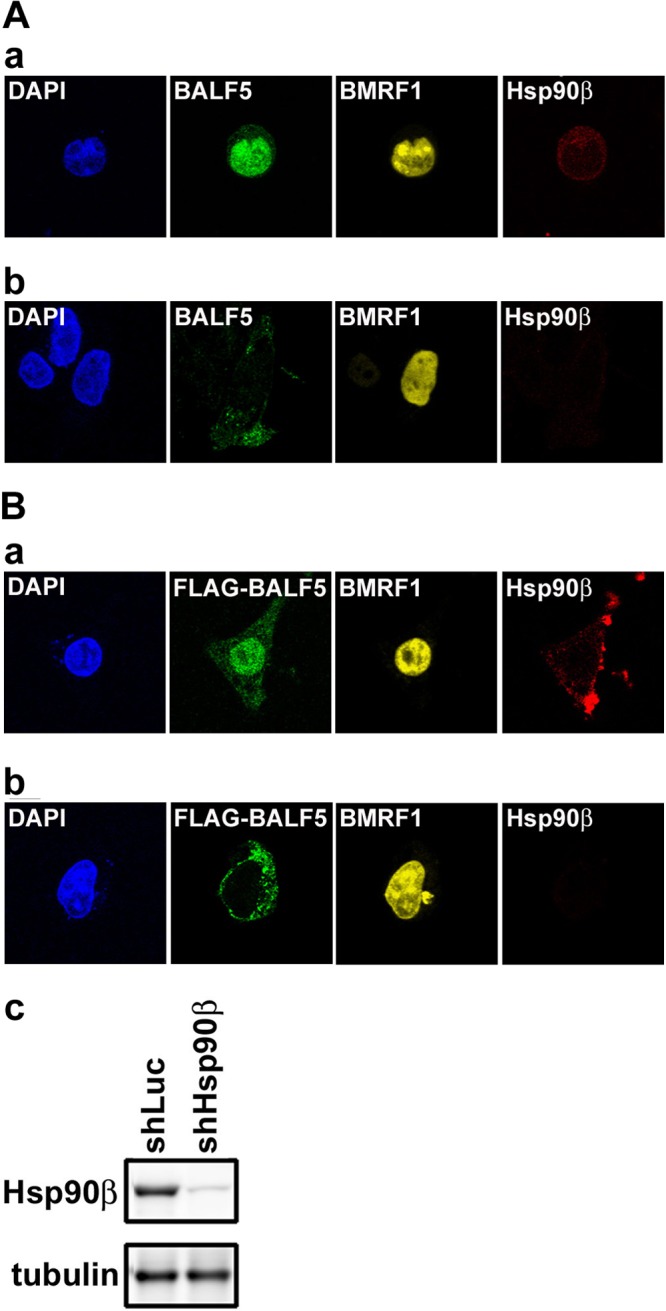
Hsp90β knockdown blocks nuclear transport of BALF5 DNA polymerase catalytic subunit. (A) AGS-CR2/EGFP-EBV cells expressing either luciferase shRNA (shLuc) (a) or Hsp90β shRNA (shHsp90β) (b) were subjected to immunofluorescence analysis. Each cell was transfected with pcDNA-BZLF1 to induce lytic replication and harvested 24 h posttransfection. Harvested cells were fixed and immunostained with anti-BALF5 (green), anti-Hsp90β (red), and anti-BMRF1 (yellow) antibodies. Nuclei were stained with DAPI (blue). (B) HeLa cells expressing either luciferase shRNA (shLuc) (a) or Hsp90β shRNA (shHsp90β) (b) were transfected with pcDNA-FLAG-BALF5 and pcDNA-BMRF1. At 24 h posttransfection, cells were fixed and immunostained with anti-FLAG (green), anti-Hsp90β (red), and anti-BMRF1 (yellow) antibodies. Nuclei were stained with DAPI (blue). Hsp90β expression levels of each cell were confirmed by immunoblot analysis (c).
DISCUSSION
The present investigation of mechanisms of nuclear transport of the EBV DNA polymerase catalytic subunit BALF5 demonstrated a dependence on BMRF1 polymerase processivity factor and molecular chaperone Hsp90.
It is well known that DNA polymerase processivity factors of many human herpesviruses such as UL42 of herpes simplex virus 1 (HSV-1) (32), BMRF1 of EBV (33), UL44 of human cytomegalovirus (HCMV) (34), PF-8 of Kaposi's sarcoma-associated herpesvirus (KSHV) (35), p41 of human herpesvirus 6 (HHV-6) (36), and U27 of human herpesvirus 7 (HHV-7) (37) localize to the nucleus when expressed alone. Processivity factors other than p41 of HHV-6 have been shown to contain a functional NLS (12, 35, 37–39). Indeed, we observed that BMRF1 protein, when it was expressed alone, localized to the nucleus (Fig. 1B), while C-terminally truncated BMRF1 lacking NLS did not (Fig. 2B). On the other hand, nuclear transport mechanisms of DNA polymerase catalytic subunits differ among herpesviruses. In the case of HSV-1 DNA polymerase (UL30), it was previously reported that UL30 possesses an NLS and can by itself be transferred to the nucleus, even in the absence of UL42 polymerase processivity factor (15, 16, 40). Also, HCMV DNA polymerase catalytic subunit (UL54) possesses an NLS in the C-terminal domain and can itself move to the nucleus (40). Unlike HSV-1 UL30 and HCMV UL54, the Pol-8 of the KSHV DNA polymerase catalytic subunit alone remains localized in cytoplasm and translocates to the nucleus with the help of PF-8 polymerase processivity factor, although involvement of Hsp90 in their association has not been clarified (35). We here have demonstrated that coexpression of BMRF1 possessing an NLS is required for nuclear translocation of BALF5 with help from Hsp90.
Hsp90 inhibitors are small molecules that specifically inhibit the Hsp90 chaperone machinery by binding to the ATP-binding domain (20). It is assumed that Hsp90 contributes to the stabilization, maturation, and trafficking of client proteins. Our results revealed that, in analogy to HSV-1 DNA replication, EBV DNA replication is inhibited by Hsp90 inhibitors. Their inhibitory effects are through the inhibition of DNA polymerases in both cases. However, it has become apparent that there are several critical differences regarding the nuclear translocation mechanisms of the DNA polymerase of EBV (BALF5) and that of HSV-1 (UL30). First, UL30 has its own NLS and by itself translocates to nuclei, while BALF5 does not have one. Second, UL30 appeared to interact with Hsp90 and get stabilized. This is because the addition of a proteasome inhibitor, MG132, antagonized the destabilization of UL30 caused by Hsp90 inhibitors (16). In contrast, MG132 treatment did not affect the level of BALF5 in the presence of Hsp90 inhibitors (data not shown). Thus, it appears that Hsp90 is involved in both trafficking and stabilization of HSV-1 UL30, while Hsp90 is not involved in stabilization of EBV BALF5. Rather, the Hsp90 inhibitor radicicol prevented the complex formation between BALF5 and BMRF1 (Fig. 5F). Thus, Hsp90 promotes the association of BALF5 with BMRF1, and consequently BALF5 translocates to nuclei.
Immunoprecipitation analyses demonstrated that BALF5 interacted with Hsp90 and that the BALF5-BMRF1 complex, however, did not contain Hsp90 (Fig. 5D). It is reported that aryl hydrocarbon receptor (AhR) interacts with Hsp90 for its stabilization and heterodimerizes with the AhR nuclear translocator (Arnt), resulting in dissociation of Hsp90 (41). Molecular chaperones such as Hsp90 were originally thought to mediate the posttranslational assembly of protein complexes, without becoming actual components of complexes (42). From this point of view, since Hsp90 was not detected at all in the BALF5-BMRF1 complex, it is possible that Hsp90 dissociates from BALF5 when BALF5 forms a complex with BMRF1.
Overall, Hsp90 is an essential host factor for the nuclear transfer of EBV DNA polymerase. As a result, Hsp90 inhibitors might efficiently prevent EBV genome replication. Sun et al. previously reported that Hsp90 inhibitors decreased expression of EBNA1 in EBV-infected cells, retarding the growth of EBV latently infected malignant cells via an EBNA1-dependent mechanism (43). Jeon et al. described geldanamycin induction of apoptotic cell death in Epstein-Barr virus-positive NK/T-cell lymphoma cells by Akt downregulation (44). It is possible that inhibition of EBV DNA replication by the Hsp90 inhibitor may be due to effects not only on EBV DNA polymerase but also on other essential factors. For instance, cyclin-dependent kinases (CDKs) are known to interact with Hsp90, and their activities were essential for the expression of viral immediate early and early lytic proteins (45). Retardation of BALF5 nuclear transport might contribute, at least partly, to the inhibition of EBV DNA replication. Hsp90 inhibitors are effective with both latent and lytic EBV infections. Geldanamycin derivatives are in fact currently being examined in clinical trials for the treatment of various cancers (46, 47). Rapid progress in the field of Hsp90 biology has brought about more potent and less toxic inhibitors. These may become useful as antiviral drugs against EBV-associated disorders.
ACKNOWLEDGMENTS
We are grateful to Y. Matsuura (Osaka University) for providing pcDNA3.1-HA-Hsp90. We also thank S. Tsuzuki (Aichi Cancer Center Research Institute) for assistance with shRNA technology.
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology of Japan (no. 23114512, 3390118, and 24659213 to T.T.), the Ministry of Health, Labor and Welfare (to T.T.), and partly by the Takeda Foundation (to T.T.). A.S. is a Research Fellow of the Japanese Society for the Promotion of Science for Young Scientists.
Footnotes
Published ahead of print 3 April 2013
REFERENCES
- 1. Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310:207–211 [DOI] [PubMed] [Google Scholar]
- 2. Daikoku T, Kudoh A, Fujita M, Sugaya Y, Isomura H, Shirata N, Tsurumi T. 2005. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J. Virol. 79:3409–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fixman ED, Hayward GS, Hayward SD. 1995. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 69:2998–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sugimoto A, Kanda T, Yamashita Y, Murata T, Saito S, Kawashima D, Isomura H, Nishiyama Y, Tsurumi T. 2011. Spatiotemporally different DNA repair systems participate in Epstein-Barr virus genome maturation. J. Virol. 85:6127–6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsurumi T. 2001. EBV replication enzymes. Curr. Top. Microbiol. Immunol. 258:65–87 [DOI] [PubMed] [Google Scholar]
- 6. Tsurumi T, Daikoku T, Kurachi R, Nishiyama Y. 1993. Functional interaction between Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit in vitro. J. Virol. 67:7648–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsurumi T, Daikoku T, Nishiyama Y. 1994. Further characterization of the interaction between the Epstein-Barr virus DNA polymerase catalytic subunit and its accessory subunit with regard to the 3′-to-5′ exonucleolytic activity and stability of initiation complex at primer terminus. J. Virol. 68:3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cho MS, Milman G, Hayward SD. 1985. A second Epstein-Barr virus early antigen gene in BamHI fragment M encodes a 48- to 50-kilodalton nuclear protein. J. Virol. 56:860–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li JS, Zhou BS, Dutschman GE, Grill SP, Tan RS, Cheng YC. 1987. Association of Epstein-Barr virus early antigen diffuse component and virus-specified DNA polymerase activity. J. Virol. 61:2947–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murayama K, Nakayama S, Kato-Murayama M, Akasaka R, Ohbayashi N, Kamewari-Hayami Y, Terada T, Shirouzu M, Tsurumi T, Yokoyama S. 2009. Crystal structure of Epstein-Barr virus DNA polymerase processivity factor BMRF1. J. Biol. Chem. 284:35896–35905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Daikoku T, Kudoh A, Sugaya Y, Iwahori S, Shirata N, Isomura H, Tsurumi T. 2006. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J. Biol. Chem. 281:11422–11430 [DOI] [PubMed] [Google Scholar]
- 12. Zhang Q, Holley-Guthrie E, Dorsky D, Kenney S. 1999. Identification of transactivator and nuclear localization domains in the Epstein-Barr virus DNA polymerase accessory protein, BMRF1. J. Gen. Virol. 80:69–74 [DOI] [PubMed] [Google Scholar]
- 13. Tsurumi T, Kobayashi A, Tamai K, Yamada H, Daikoku T, Yamashita Y, Nishiyama Y. 1996. Epstein-Barr virus single-stranded DNA-binding protein: purification, characterization, and action on DNA synthesis by the viral DNA polymerase. Virology 222:352–364 [DOI] [PubMed] [Google Scholar]
- 14. Gao Z, Krithivas A, Finan JE, Semmes OJ, Zhou S, Wang Y, Hayward SD. 1998. The Epstein-Barr virus lytic transactivator Zta interacts with the helicase-primase replication proteins. J. Virol. 72:8559–8567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alvisi G, Musiani D, Jans DA, Ripalti A. 2007. An importin alpha/beta-recognized bipartite nuclear localization signal mediates targeting of the human herpes simplex virus type 1 DNA polymerase catalytic subunit pUL30 to the nucleus. Biochemistry 46:9155–9163 [DOI] [PubMed] [Google Scholar]
- 16. Burch AD, Weller SK. 2005. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 79:10740–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bukau B, Weissman J, Horwich A. 2006. Molecular chaperones and protein quality control. Cell 125:443–451 [DOI] [PubMed] [Google Scholar]
- 18. Morimoto RI. 2008. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 22:1427–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Young JC, Agashe VR, Siegers K, Hartl FU. 2004. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 5:781–791 [DOI] [PubMed] [Google Scholar]
- 20. Whitesell L, Lindquist SL. 2005. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5:761–772 [DOI] [PubMed] [Google Scholar]
- 21. Kudoh A, Fujita M, Kiyono T, Kuzushima K, Sugaya Y, Izuta S, Nishiyama Y, Tsurumi T. 2003. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J. Virol. 77:851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katsumura KR, Maruo S, Wu Y, Kanda T, Takada K. 2009. Quantitative evaluation of the role of Epstein-Barr virus immediate-early protein BZLF1 in B-cell transformation. J. Gen. Virol. 90:2331–2341 [DOI] [PubMed] [Google Scholar]
- 23. Maruo S, Yang L, Takada K. 2001. Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J. Gen. Virol. 82:2373–2383 [DOI] [PubMed] [Google Scholar]
- 24. Tsurumi T. 1993. Purification and characterization of the DNA-binding activity of the Epstein-Barr virus DNA polymerase accessory protein BMRF1 gene products, as expressed in insect cells by using the baculovirus system. J. Virol. 67:1681–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsurumi T, Kishore J, Yokoyama N, Fujita M, Daikoku T, Yamada H, Yamashita Y, Nishiyama Y. 1998. Overexpression, purification and helix-destabilizing properties of Epstein-Barr virus ssDNA-binding protein. J. Gen. Virol. 79:1257–1264 [DOI] [PubMed] [Google Scholar]
- 26. Tsurumi T, Kobayashi A, Tamai K, Daikoku T, Kurachi R, Nishiyama Y. 1993. Functional expression and characterization of the Epstein-Barr virus DNA polymerase catalytic subunit. J. Virol. 67:4651–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fujita N, Kubo A, Francisco PB, Jr, Nakakita M, Harada K, Minaka N, Nakamura Y. 1999. Purification, characterization, and cDNA structure of isoamylase from developing endosperm of rice. Planta 208:283–293 [DOI] [PubMed] [Google Scholar]
- 28. Isomura H, Stinski MF, Kudoh A, Nakayama S, Murata T, Sato Y, Iwahori S, Tsurumi T. 2008. A cis element between the TATA box and the transcription start site of the major immediate-early promoter of human cytomegalovirus determines efficiency of viral replication. J. Virol. 82:849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Czar MJ, Galigniana MD, Silverstein AM, Pratt WB. 1997. Geldanamycin, a heat shock protein 90-binding benzoquinone ansamycin, inhibits steroid-dependent translocation of the glucocorticoid receptor from the cytoplasm to the nucleus. Biochemistry 36:7776–7785 [DOI] [PubMed] [Google Scholar]
- 30. Csermely P, Kajtar J, Hollosi M, Jalsovszky G, Holly S, Kahn CR, Gergely P, Jr, Soti C, Mihaly K, Somogyi J. 1993. ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J. Biol. Chem. 268:1901–1907 [PubMed] [Google Scholar]
- 31. Sullivan W, Stensgard B, Caucutt G, Bartha B, McMahon N, Alnemri ES, Litwack G, Toft D. 1997. Nucleotides and two functional states of hsp90. J. Biol. Chem. 272:8007–8012 [DOI] [PubMed] [Google Scholar]
- 32. Goodrich LD, Rixon FJ, Parris DS. 1989. Kinetics of expression of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type 1. J. Virol. 63:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiehl A, Dorsky DI. 1991. Cooperation of EBV DNA polymerase and EA-D(BMRF1) in vitro and colocalization in nuclei of infected cells. Virology 184:330–340 [DOI] [PubMed] [Google Scholar]
- 34. Plachter B, Nordin M, Wirgart BZ, Mach M, Stein H, Grillner L, Jahn G. 1992. The DNA-binding protein P52 of human cytomegalovirus reacts with monoclonal antibody CCH2 and associates with the nuclear membrane at late times after infection. Virus Res. 24:265–276 [DOI] [PubMed] [Google Scholar]
- 35. Chen Y, Ciustea M, Ricciardi RP. 2005. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 340:183–191 [DOI] [PubMed] [Google Scholar]
- 36. Agulnick AD, Thompson JR, Iyengar S, Pearson G, Ablashi D, Ricciardi RP. 1993. Identification of a DNA-binding protein of human herpesvirus 6, a putative DNA polymerase stimulatory factor. J. Gen. Virol. 74:1003–1009 [DOI] [PubMed] [Google Scholar]
- 37. Takeda K, Haque M, Nagoshi E, Takemoto M, Shimamoto T, Yoneda Y, Yamanishi K. 2000. Characterization of human herpesvirus 7 U27 gene product and identification of its nuclear localization signal. Virology 272:394–401 [DOI] [PubMed] [Google Scholar]
- 38. Alvisi G, Avanzi S, Musiani D, Camozzi D, Leoni V, Ly-Huynh JD, Ripalti A. 2008. Nuclear import of HSV-1 DNA polymerase processivity factor UL42 is mediated by a C-terminally located bipartite nuclear localization signal. Biochemistry 47:13764–13777 [DOI] [PubMed] [Google Scholar]
- 39. Alvisi G, Jans DA, Guo J, Pinna LA, Ripalti A. 2005. A protein kinase CK2 site flanking the nuclear targeting signal enhances nuclear transport of human cytomegalovirus ppUL44. Traffic 6:1002–1013 [DOI] [PubMed] [Google Scholar]
- 40. Loregian A, Piaia E, Cancellotti E, Papini E, Marsden HS, Palu G. 2000. The catalytic subunit of herpes simplex virus type 1 DNA polymerase contains a nuclear localization signal in the UL42-binding region. Virology 273:139–148 [DOI] [PubMed] [Google Scholar]
- 41. Hur E, Kim HH, Choi SM, Kim JH, Yim S, Kwon HJ, Choi Y, Kim DK, Lee MO, Park H. 2002. Reduction of hypoxia-induced transcription through the repression of hypoxia-inducible factor-1alpha/aryl hydrocarbon receptor nuclear translocator DNA binding by the 90-kDa heat-shock protein inhibitor radicicol. Mol. Pharmacol. 62:975–982 [DOI] [PubMed] [Google Scholar]
- 42. Ellis RJ, Hemmingsen SM. 1989. Molecular chaperones: proteins essential for the biogenesis of some macromolecular structures. Trends Biochem. Sci. 14:339–342 [DOI] [PubMed] [Google Scholar]
- 43. Sun X, Barlow EA, Ma S, Hagemeier SR, Duellman SJ, Burgess RR, Tellam J, Khanna R, Kenney SC. 2010. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 107:3146–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeon YK, Park CH, Kim KY, Li YC, Kim J, Kim YA, Paik JH, Park BK, Kim CW, Kim YN. 2007. The heat-shock protein 90 inhibitor, geldanamycin, induces apoptotic cell death in Epstein-Barr virus-positive NK/T-cell lymphoma by Akt down-regulation. J. Pathol. 213:170–179 [DOI] [PubMed] [Google Scholar]
- 45. Kudoh A, Daikoku T, Sugaya Y, Isomura H, Fujita M, Kiyono T, Nishiyama Y, Tsurumi T. 2004. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J. Virol. 78:104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neckers L. 2006. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb. Exp. Pharmacol. 172:259–277 [DOI] [PubMed] [Google Scholar]
- 47. Pearl LH, Prodromou C, Workman P. 2008. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 410:439–453 [DOI] [PubMed] [Google Scholar]