

Abstract
The leader (L) protein of encephalomyocarditis virus (EMCV) shuts off host cell nucleocytoplasmic trafficking (NCT) by inducing hyperphosphorylation of nuclear pore proteins. This dramatic effect by a nonenzymatic protein of 6 kDa is not well understood but clearly involves L binding to cellular Ran GTPase, a critical factor of active NCT. Exogenous GDP and GTP are inhibitory to L-Ran binding, but the guanine-nucleotide exchange factor RCC1 can relieve this inhibition. In the presence of RCC1, L binds Ran with a KD (equilibrium dissociation constant) of ∼3 nM and reaches saturation within 20 min. The results of fluorescently tagged nucleotide experiments suggest that L-Ran interactions affect the nucleotide-binding pocket of Ran.
TEXT
Encephalomyocarditis virus (EMCV) infection shuts down active nucleocytoplasmic trafficking (NCT) by inducing the phosphorylation of PheGly-containing nuclear pore proteins (Nups), potentially blocking otherwise toxic antiviral responses (1–4). The leader (L) protein of EMCV alone is responsible for this activity. In the context of cytoplasm and nuclear pore targets, L induces irreversible NCT inhibition, new cellular mRNA transcripts are unable to be exported from the nucleus, and nuclear-targeted cytoplasmic proteins fail to be actively imported (3, 5, 6).
L is a small protein (67 amino acids) with a novel N-terminal CHCC zinc-finger motif and a highly acidic (pI 3.8) β-hairpin motif (7, 8). Despite being sufficient to shut down NCT, L itself has no predicted or observable enzymatic activity. It must act by binding or abrogating cellular and/or viral cofactors to bring about such significant cellular responses (9). Indeed, among the known binding partners of L is Ran GTPase, a central component of all active NCT (10).
Previous studies have shown that L not only interacts with Ran, it also inhibits critical events that are dependent on Ran GTP/GDP cycling (3, 10). To understand how L redirects Ran's role and triggers consequent Nup phosphorylation is of interest, not only to the field of virus-host interactions but also to the field of mitotic molecular biology, in which Ran serves other central functions.
GST-L interaction with His-Ran.
Glutathione S-transferase (GST)-tagged L and His-tagged Ran (10) were expressed and purified from Escherichia coli. GST-tagged RCC1 was generated from pGEX-RCC1 (a kind gift from C. Wiese). Previous data have shown that GST-L can pull down native Ran from HeLa cell lysates even in the presence of 300 mM NaCl, suggesting a tight interaction (10). But those experiments did not measure any kinetic parameters or explore equilibrium binding. Recombinant GST-L, prereacted with glutathione-Sepharose beads, was incubated 1:1 with His-Ran. After 1 h, Western blot analyses of the bound and unbound fractions showed that ∼32% of His-Ran was extracted by GST-L (Fig. 1), indicating that L and Ran do interact, but not particularly efficiently in this context (10). The addition of a 50× molar excess of GTP or GDP (2.5 μM) lowered the pulldown efficacy to 23% for GDP and to an undetectable level for GTP. However, when recombinant RCC1-GST (1 nM) was added, 100% of the His-Ran was recovered from the beads, even in the presence of GTP. Since GST-L and His-Ran are in 50× excess to RCC1-GST, the enhanced pulldown cannot be due solely to the RCC1 GST moiety. Indeed, by itself RCC1-GST did not pull down His-Ran (0%). Though Ran and RCC1 most certainly interact, the off-rate is very high (11) and those complexes should not survive the wash conditions. Rather, the data indicate that Ran structural shifts, catalyzed transiently by RCC1, made it more amenable to saturation binding by L, with simultaneous relief of GDP/GTP solution inhibition.
Fig 1.

GST-L–His-Ran binding. GST-L linked to glutathione-Sepharose beads (50 nM) was reacted (1 h, 25°C) with His-Ran (50 nM) in the presence or absence of GDP or GTP (2.5 μM) and/or RCC1-GST (1 nM). The clarified supernatant (Unbound) and bead-bound proteins (Bound) were precipitated (30% trichloroacetic acid), solubilized (alkaline SDS), fractionated by PAGE, and then transferred to polyvinylidene difluoride membranes. Western blot analyses used anti-Ran polyclonal antibody (product no. SC-1156; Santa Cruz Biotech) or anti-GST monoclonal antibody (product no. 71097; Novagen). Secondary antibodies were horseradish peroxidase-conjugated anti-goat antibody (product no. A5420; Sigma) or anti-mouse antibody (product no. A2554; Sigma). Relative pixel counts (% total) were determined by ImageQuant (GE Life Sciences) scanning of the membranes.
Nucleotide spectrofluorimetry of complexes.
In cells, RCC1 localizes to nuclei, accelerating by about 200,000-fold the exchange of GDP for GTP on nuclear Ran (11). Nucleotide spectrofluorimetry was used to evaluate the interchange potential of recombinant His-Ran in solution. A small N-methyl-anthraniloyl tag on the 2′ or 3′ OH of GDP (mant-GDP) is quenched in solution but fluoresces strongly when trapped in a hydrophobic environment (11). In the absence of RCC1-GST, His-Ran showed a low level of mant-GDP fluorescence relative to the background, indicating the expected slow exchange of the tagged nucleotide for coisolated native nucleotides or limited tag entry into proteins having initially empty nucleotide pockets (Fig. 2). RCC1-GST increased the fluorescence by ∼2.5-fold relative to His-Ran alone. Therefore, the recombinant enzyme was catalytically active, could help His-Ran exchange nucleotides, and could pick up more tagged nucleotide.
Fig 2.
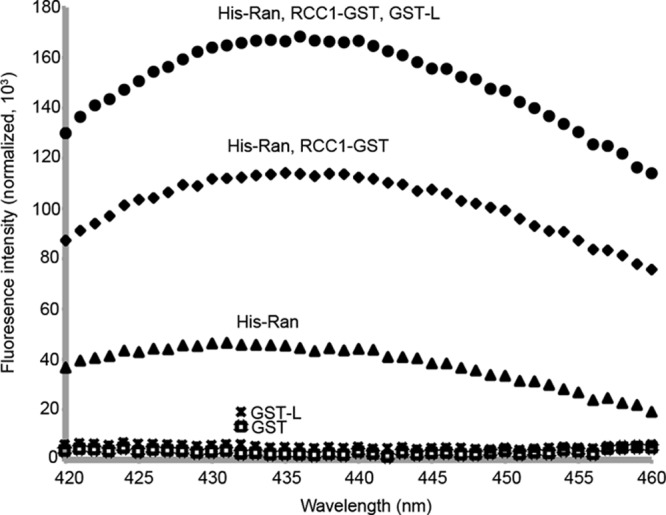
RCC1-GST is catalytically active. The fluorescence of nucleotide analog mant-GDP (N-methyl-anthraniloyl-tagged GDP; λex = 355 nm, λem = 420 to 460 nm) was measured by spectrofluorimetry after 15 min. Reactions were performed at 25°C with His-Ran, GST, or GST-L (1.5 μm each). The analogue was at 50× molar excess (75 μm). The reactions were repeated in the presence of RCC1-GST (30 nM) or a combination of RCC1-GST (30 nM) and GST-L (1.5 μm).
Without RCC1, L-Ran binding is inhibited by free nucleotides (Fig. 1). If this inhibition were strictly competitive, GST-L should cause L-dependent exclusion of mant-GDP and subsequent decreased fluorescence. Instead, GST-L, when added with RCC1-GST, more than doubled the fluorescence. Since virtually all of the His-Ran becomes tied up with GST-L (Fig. 1), increased fluorescence can happen only if mant-GDP and GST-L cobind the same His-Ran protein. Mutual binding sites very near each other might allow GST-L to provide enhanced hydrophobic shielding. Alternatively, if L binding were at a more distant noncompetitive site, it must necessarily induce a transformative conformational change proximal to the Ran nucleotide-binding pocket, with consequent increased shielding or a faster mant-GDP on-rate. Either scenario requires an exceptionally tight, long-lived L-Ran interaction, facilitated by RCC1, that is not displaced by nucleotides once formed.
Equilibrium constants determined by SPR.
Although GST-L and His-Ran do not by themselves reach saturation in a typical 1-h reaction (e.g., 32% in the results shown in Fig. 1), RCC1-GST accelerated the binding to where >90% of the His-Ran could be pulled down in less than 20 min (not shown). The increased rate was sufficient for real-time kinetic experiments via surface plasmon resonance (SPR). GST antibodies coupled to the chip surface(s) were reacted with GST-L. His-Ran did not bind this surface unless L sequences were present (Fig. 3A). RCC1-GST added alone only slightly increased the chip mass, presumably as residual antibodies slowly saturated or exchanged with new GST. However, when His-Ran and RCC1-GST were flowed together over the surface, the rate and degree of resonance units increased significantly, to the point where the normalized sensorgrams approached equilibrium and saturation (Fig. 3B). This ensured that the collective binding data (triplicate experiments) were accurate and suitable for regression analysis of relative interaction constants. Calculations using the BIAevaluation software determined the KD (equilibrium dissociation constant) of GST-L–His-Ran in the presence of GST-RCC1 to be ∼3 nM (Table 1).
Fig 3.
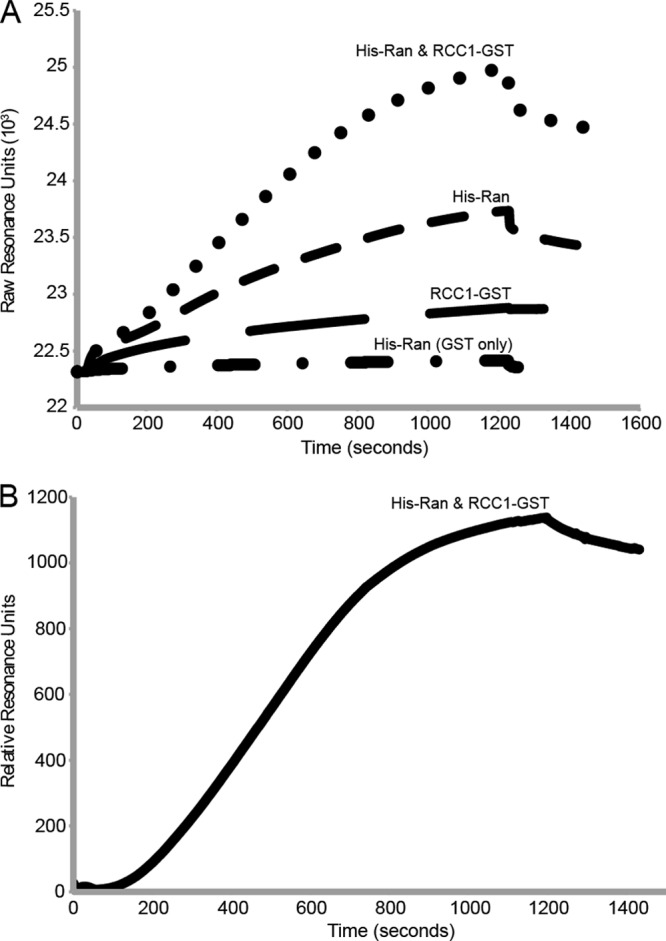
Measurement of GST-L–His-Ran affinity by SPR. (A) Representative SPR sensorgram curves for His-Ran (5 μg/ml, flowed over both GST and GST-L surfaces), RCC1-GST (0.5 μg/ml), and combined binding to GST-L (initially, 10 μl/min, 75 μg total, 25°C), using amine-coupled monoclonal antibody-GST surfaces on CM5 chips (GE Healthcare) and a BIAcore 2000 instrument. Only His-Ran in the presence of RCC1-GST over a GST-L chip reached equilibrium binding when measured over 20 min (1,200 s). At this point, the buffer was switched to one without protein (for 300 s), and monitoring was continued. (B) His-Ran binding to the GST-L surface in the presence of RCC1-GST is shown as normalized values, after taking into account all nonspecific binding controls (i.e., RCC1-GST and antibody only). Triplicate SPR determinations like this, with and without added GDP/GTP (10 μM), gave similar processed curves and were used for the KD calculations whose results are shown in Table 1.
Table 1.
Equilibrium constants for GST-L–His-Ran bindinga
| Inhibitor | Mean value (±SD) |
||||
|---|---|---|---|---|---|
| kon (M−1 s−1) | koff (s−1) | KD (nM) | Ki (μM) | IC50 (mM) | |
| None | 1.01 (±0.01) × 105 | 3.39 (±0.54) × 10−4 | 3.25 (±0.63) | NA | NA |
| GDP, 10 μM | 2.33 (±0.06) × 104 | 4.98 (±0.36) × 10−4 | 22.0 (±1.4) | 1.73 (±0.11) | 1.0 (±0.2) |
| GTP, 10 μM | 1.79 (±0.01) × 104 | 8.81 (±0.07) × 10−4 | 48.8 (±0.2) | 0.714 (±0.003) | 2.5 (±0.1) |
On and off rates were determined through Langmuir fitting of normalized sensorgrams. KD was determined as the ratio of koff/kon when adjusted for the concentration of His-Ran in solution. Uncertainty was determined as the standard deviation of triplicate measurements. Significant figures were limited by the precision of protein concentrations. Ki was calculated using the equation Ki = [GXP]/[(KDGXP/KD) −1], where GXP represents either GDP or GTP. IC50 was calculated using the equation IC50 = Ki(1 + [Rancell]/KD). NA, not applicable.
GDP and GTP were then added to equivalent chip eluents in the presence of His-Ran and RCC1-GST (sensorgrams not shown). As summarized in Table 1, the inclusion of GDP or GTP increased the SPR-determined KD of GST-L–His-Ran by 7-fold (to ∼22 nM) or 15-fold (to ∼49 nM), respectively, relative to the KD of the nucleotide-free samples. Further extrapolation from the slopes of the normalized curves (as shown in Fig. 3B) showed that, for this concentration of proteins, the presence of either nucleotide decreased the on-rate constants by ∼10-fold relative to the constants in the absence of GDP/GTP. The off-rates also increased correspondingly, with GDP at about 1.5-fold and GTP at about 2.5-fold. The changes translate into inhibitory constants of around 1 μM for either nucleotide, values far below the measured ∼1 mM (12) cellular concentrations. The 50% inhibitory concentrations (IC50), as calculated with the Cheng-Prusoff equation (13), were estimated at 1 mM and 2.5 mM for GDP and GTP, respectively (Table 1), much closer to the actual intracellular concentrations.
Therefore, within EMCV-infected cells, native L-Ran interactions should occur readily and very tightly and be practically nondissociable, even if there are high local concentrations of GNPs. This binding will happen preferentially, however, only when the initial contacts are localized to sites where RCC1 is prevalent and able to facilitate complex formation, that is, in the vicinity of the nuclear pores. Once the interaction has occurred, the low KD of L-Ran binding is actually tighter than those of many antibody-antigen complexes, explaining previous findings that GST-L–Ran (native) can withstand very high salt concentrations without dissociation (10) and becomes impervious to added nucleotides.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI017331 to A.C.P. and NIH Training Grant GM07215 to R.V.P.
We thank Darrell McCaslin for his advice and expertise. Instrument data were obtained at the University of Wisconsin—Madison Biophysics Instrumentation Facility, which was established with support from the University of Wisconsin—Madison and grants BIR-9512577 (NSF) and S10 RR13790 (NIH). We also thank Chris Wiese for her generous donation of plasmid pGEX-RCC1.
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Hato SV, Ricour C, Schulte BM, Lanke KH, de Bruijni M, Zoll J, Melchers WJ, Michiels T, van Kuppeveld FJ. 2007. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 9:2921–2930 [DOI] [PubMed] [Google Scholar]
- 2. Hato SV, Sorgeloos F, Ricour C, Zoll J, Melchers WJ, Michiels T, van Kuppeveld FJ. 2010. Differential IFN-alpha/beta production suppressing capacities of the leader proteins of mengovirus and food-and-mouth disease virus. Cell. Microbiol. 12:310–317 [DOI] [PubMed] [Google Scholar]
- 3. Porter FW, Palmenberg AC. 2009. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition of cardioviruses. J. Virol. 83:1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zoll J, Melchers WJ, Galama JM, van Kuppeveld FJ. 2002. The mengovirus leader protein suppresses alpha/beta interferon production by inhibition of the iron/ferritin-mediated activation of NF-kappa B. J. Virol. 76:9664–9672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardina MV, Lidsky P, Sheval EV, Fominykh KV, van Kuppeveld FJ, Polyakov VY, Agol V. 2009. Mengovirus-induced rearrangements of the nuclear pore complex: hijacking cellular phosphorylation machinery. J. Virol. 83:3150–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lidsky PL, Hato S, Bardina MV, Aminev AG, Palmenberg AC, Sheval EV, Polyakov VY, van Kuppeveld FJ, Agol V. 2006. Nucleo-cytoplasmic traffic disorder induced by cardioviruses. J. Virol. 80:2705–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cornilescu CC, Porter FW, Zhao Q, Palmenberg AC, Markley JL. 2008. NMR structure of the mengovirus leader protein zinc-finger domain. FEBS Lett. 582:896–900 [DOI] [PubMed] [Google Scholar]
- 8. Dvorak CMT, Hall DJ, Hill M, Riddle M, Pranter A, Dillman J, Deibel M, Palmenberg AC. 2001. Leader protein of encephalomyocarditis virus binds zinc, is phosphorylated during viral infection and affects the efficiency of genome translation. Virology 290:261–271 [DOI] [PubMed] [Google Scholar]
- 9. Porter FW, Brown B, Palmenberg A. 2010. Nucleoporin phosphorylation triggered by the encephalomyocarditis virus leader protein is mediated by mitogen-activated protein kinases. J. Virol. 84:12538–12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Porter FW, Bochkov YA, Albee AJ, Wiese C, Palmenberg AC. 2006. A picornavirus protein interacts with Ran-GTPase and disrupts nucleocytoplasmic transport. Proc. Natl. Acad. Sci. U. S. A. 103:12417–12422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klebe C, Prinz H, Wittinghofer A, Goody RS. 1995. The kinetic mechanism of Ran-nucleotide exchange catalyzed by RCC1. Biochemistry 34:12543–12552 [DOI] [PubMed] [Google Scholar]
- 12. Gorlich D, Seewald MJ, Ribbeck K. 2003. Characterization of Ran-driven transport and the RanGTPase system by kinetic measurements and computer simulation. EMBO J. 22:1088–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng Y, Prusoff WH. 1973. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22:3099–3108 [DOI] [PubMed] [Google Scholar]