

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV) stimulates proliferation, angiogenesis, and inflammation to promote Kaposi sarcoma (KS) tumor growth, which involves various growth factors and cytokines. Previously, we found that KSHV infection of human umbilical vein endothelial cells (HUVECs) induces a transcriptional induction of the proangiogenic and proinflammatory cytokine angiopoietin-2 (Ang-2). Here, we report that KSHV induces rapid release of Ang-2 that is presynthesized and stored in the Weibel-Palade bodies (WPB) of endothelial cells upon binding to its integrin receptors. Blocking viral binding to integrins inhibits Ang-2 release. KSHV binding activates the integrin tyrosine kinase receptor signaling pathways, leading to tyrosine phosphorylation of focal adhesion kinase (FAK), the tyrosine kinase Src, and the Calα2 subunit of the l-type calcium channel to trigger rapid calcium (Ca2+) influx. Pretreatment of endothelial cells with specific inhibitors of protein tyrosine kinases inhibits KSHV-induced Ca2+ influx and Ang-2 release. Inhibition of Ca2+ mobilization with calcium channel blockers also inhibits Ang-2 release. Thus, the interaction between KSHV and its integrin receptors plays a key role in regulating rapid Ang-2 release from endothelial cells. This finding highlights a novel mechanism of viral induction of angiogenesis and inflammation, which might play important roles in the early event of KS tumor development.
INTRODUCTION
Kaposi sarcoma (KS) is a neoplasia of endothelial cell origin that is etiologically associated with Kaposi sarcoma-associated herpesvirus (KSHV) infection (1). The development of KS depends on various growth factors and cytokines to support proliferation, angiogenesis, and inflammation (2, 3). Infiltrating inflammatory cells and KSHV-infected endothelial cells are major sources of growth factors and cytokines. Indeed, KSHV infection enhances expression of various growth factors and cytokines that include, but are not limited to, vascular endothelial growth factor (VEGF) (4, 5, 6), basic fibroblast growth factor (b-FGF) (7), growth-regulated oncogene alpha (GRO-α) (8), tumor necrosis factor alpha (TNF-α) (9, 10), interleukin-6 (IL-6) (11), interleukin-8 (IL-8) (12, 13), and COX-2 (14). KSHV itself also encodes several homologues of cellular cytokines and chemokines, such as viral interleukin-6 (vIL-6), viral G protein-coupled receptor (vGPCR), and viral macrophage inflammatory proteins 1 and 2 (vMIP-1 and vMIP-2) (15). Both cellular and virus-encoded cytokines and chemokines likely contribute to KS tumor development.
Previously, we found high levels of the proangiogenic and proinflammatory cytokine angiopoietin-2 (Ang-2) in KS tumors (16, 17). We also demonstrated that KSHV infection of endothelial cells enhances Ang-2 expression (16, 18). Although Ang-2 expression is minimal during quiescence under normal physiological conditions, a significant amount of Ang-2 is presynthesized and stored in the endothelial cell-specific organelles Weibel-Palade bodies (WPBs) and rapidly released via regulated exocytosis upon stimulation (19). Various factors, such as thrombin, histamine, and TNF-α, stimulate rapid Ang-2 release from the WPBs to promote angiogenesis and inflammation (19). However, no virus has been reported to act as such a stimulus to induce rapid Ang-2 release.
In this study, we report that KSHV binding to human umbilical vein endothelial cells (HUVECs) and primary lymphatic endothelial cells triggers rapid Ang-2 release. Our data suggest that the dynamic interaction between KSHV and its cellular receptor integrins plays a key role in inducing rapid Ang-2 release. This finding defines a novel mechanism of viral induction of angiogenesis and inflammation during acute KSHV infection, which might be a critical event in the onset of KS tumors.
MATERIALS AND METHODS
Cell culture.
HUVECs and primary human lymphatic endothelial cells were purchased from Lonza (Walkersville, MD) and cultured in EBM-2 medium supplemented with various growth factors, according to instructions by the manufacturer. Human primary effusion lymphoma (PEL) BCBL1-BAC36 cells and KSHV-negative B lymphoma BJAB cells were cultured in RPMI 1640 medium plus 15% fetal bovine serum (FBS). BCBL1-BAC36 cells carry a green fluorescent protein (GFP)-expressing recombinant KSHV, BAC36 (20).
Production and purification of KSHV and adenovirus virions.
To produce KSHV, BCBL1-BAC36 cells (4 × 106/ml; 120 ml total) were stimulated with phorbol 12-tetradecanoate 13-acetate (TPA) (20 ng/ml), as described previously (16). The TPA-containing medium was replaced with 60 ml fresh RPMI 1640 plus 15% FBS 2 days after induction, and the cells were cultured for three more days before harvesting the virus. To determine the viral yield, 1 ml of culture supernatant was used to infect 5 × 105 HUVECs for 48 h, followed by determining the percentage of GFP-positive cells under a fluorescence microscope. To purify the virus, cell culture supernatant (60 ml) was collected, followed by low-speed centrifugation (5,000 × g; 15 min) to remove cells and cellular debris. The supernatant was then loaded onto a 30% sucrose cushion and subjected to high-speed (28,000 × g) centrifugation at 4°C for 2 h. The pellet was resuspended in 12 ml basic EBM2 medium without supplements, and the purified and concentrated viral stock solution was used to infect HUVECs or lymphatic endothelial cells. For mock infection, equivalent numbers of BJAB cells were subjected to the same viral induction and purification procedures, and the resulting pellet was resuspended in the same amount of basic EBM2 medium and used as a control solution for mock infection. UV light-inactivated KSHV stock solution was prepared by exposing the purified viral stock to UV irradiation for 5 min in a UV Cross-linker 2400 (Stratagene). Also, adenovirus (Stratagene) expressing DsRed fluorescent protein (RFP) was constructed and produced in 293 cells by following instructions provided by the manufacturer and used as a control. To determine the yield of the adenovirus, 1 ml of viral stock solution was used to infect healthy 293 cells, followed by counting RFP-positive cells under an AMG/EVOS-fi fluorescence microscope (AMG, Bothell, WA). The same procedure as for KSHV purification was used to prepare the adenoviral stock solution.
Real-time RT-PCR.
Total RNA was isolated by using an RNA purification kit (Promega, Madison, WI). Reverse transcription (RT) of total RNA was performed by using Superscript Transcriptase II (Invitrogen, Carlsbad, CA). The primers for Ang-2 transcript were 5′TGGAAGCTGGAGGAGGCGGGTGG3′ (forward) and 5′ATGTGGTGGAAGAGGACACAGTG3′ (reverse); the primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 5′CAATGACCCCTTCATTGACC3′ (forward) and 5′GATCTCGCTCCTGGAAGATG3′ (reverse). Each reaction was repeated three times. The relative levels of Ang-2 transcript were normalized to that of the GAPDH housekeeping gene.
Measurement of Ang-2 release by Western blotting and IFA staining.
Identical numbers (2 × 105/ml) of HUVECs and/or human primary lymphatic endothelial cells were loaded into each well of 6-well culture plates and cultured in supplement-enriched EBM2 medium for 24 h. To induce Ang-2 release, the culture medium was removed, followed by adding 0.5 ml viral or control stock solution. After incubation at 37°C for various times, the added solution was collected and centrifuged at 12,000 × g for 5 min to remove any viral and cellular debris. Cells from each well were separately collected and resuspended in protein lysis buffer containing 100 mM Tris, pH 7.5, 1 mM EDTA, 400 mM NaCl, and 2% sodium dodecyl sulfate (SDS). The supernatants and cell lysates were then subjected to standard procedures for SDS-polyacrylamide gel electrophoresis (PAGE) and Western blot analysis for detection of released Ang-2 and β-tubulin, respectively, using a rabbit anti-Ang-2 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and a mouse monoclonal anti-β-tubulin antibody (Sigma). The bound primary antibodies were revealed by incubation with horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgGs (Santa Cruz Biotechnology) and a chemiluminescence reaction. In a separate experiment, the purified KSHV glycoproteins gB and gP-K8.1A were incubated with HUVECs for 15 min, and detection of Ang-2 in the supernatants was analyzed by Western blotting. The characterization and purity of these two proteins have been described previously (21). To further confirm Ang-2 release, following exposure to viral or control stock solution, HUVECs were subjected to immunofluorescent antibody (IFA) staining, as described previously (22), with the rabbit anti-Ang-2 antibody to detect intracellular Ang-2 and the mouse anti-KSHV small capsid protein (ORF65) antibody to detect KSHV virions. The bound primary antibodies were revealed by AlexaFluo-430 (green)-labeled donkey anti-rabbit IgG and AlexaFluo-594 (red)-labeled donkey anti-mouse IgG (Molecular Probes) and imaged under a fluorescence microscope (Carl Zeiss, Inc., Thornwood, NY). To further confirm that KSHV induces the release of premade and stored Ang-2, HUVECs were pretreated with 25 μM cycloheximide (Sigma-Aldrich) for 1 h at 37°C, followed by incubation with mock or viral stock solutions for 15 min. Western blot measurement of Ang-2 levels in the supernatants was carried out as described previously.
To examine the effects of viral binding on Ang-2 release, HUVECs were exposed to stock solutions of KSHV, UV-treated KSHV, and KSHV preincubated (30 min at 37°C) with soluble heparin sulfate, and the preincubated viral stock was then used to infect HUVECs in the presence of soluble heparin sulfate (10 μg/ml; Sigma-Aldrich, St. Louis, MO), 10 μM RGD and RAD peptides (AnaSpec Inc., Fremont, CA), or 10 μM cyclo-RGD and cyclo-RAD peptides (Peptides International, Louisville, KY), respectively, for 15 min. Alternatively, HUVECs were pretreated with 5 μg/ml of mouse monoclonal antibodies to human integrins CD29/β1 (Beckman Coulter, Brea, CA), CD61/β3 (Abcam), CD49c/α3 (Millipore, Billerica, MA), and CD51-CD61/αVβ3 (Millipore), or mouse IgG (Sigma-Aldrich) at 37°C for 30 min and then exposed to mock or viral stock solution for 15 min in the presence of 1 μg/ml of the corresponding blocking antibodies. To examine the effects of protein tyrosine phosphorylation on Ang-2 release, HUVECs were pretreated with the protein tyrosine kinase inhibitors PP2 (R&D Systems Inc., Minneapolis, MN) and genistein (Sigma-Aldrich) at final concentrations of 10 nM and 2 μM, respectively, for 2 h (23), followed by exposure to viral stock solution for 15 min. To study protein tyrosine phosphorylation, cell lysates from mock- or KSHV-infected HUVECs were analyzed by Western blot detection, using mouse monoclonal antibodies against tyrosine kinases Src, FAK, and phospho-FAK (Y397) and a rabbit polyclonal antibody against phospho-Src (Y419) from R&D Systems. To test if protein tyrosine phosphorylation is also involved in rapid Ang-2 release by other known factors, such as thrombin (19), we pretreated HUVECs with 2 μM genistein for 2 h, followed by stimulation with 2 U/ml thrombin purified from human plasma (Sigma-Aldrich) for 15 min. To examine the effects of Ca2+ mobilization on Ang-2 release, HUVECs were pretreated with 100 μM Ca2+ chelator BAPTA, BAPTA-AM (Sigma-Aldrich), or EGTA (Sigma-Aldrich); 20 μM Ca2+ entry blocker SKF 96365 (abcamBiochemicals, Cambrige, MA); 5 μM Ca2+ channel blocker nifedipine (Sigma-Aldrich) or amlodipine (Pfizer, New York, NY); and 5 μM thapsigargin (Sigma-Aldrich) for 30 min, followed by exposure to viral stock solution for 15 min. The levels of released Ang-2 in the supernatants of these differently treated cells were analyzed by SDS-PAGE and Western blot detection, as described above.
Cytotoxicity test.
The Vybrant cytotoxicity assay kit (Molecular Probes), which monitors the release of the cytosolic enzyme glucose 6-phosphate dehydrogenase (G6PD) from damaged cells into the surrounding medium, was used to assess the toxicities of the tyrosine kinase inhibitors PP2 and genistein. The assay detects G6PD through a two-step enzymatic process that leads to the reduction of resazurin into red-fluorescent resorufin. The resulting fluorescence intensity is proportional to the amount of released G6PD and the cytotoxicity of the inhibitors. Thus, identical numbers (2 × 104/well) of HUVECs were seeded in 96-well plates and grown in EBM2 medium with all supplements for 24 h, followed by treatment with different concentrations of PP2 and genistein for 2 h at 37°C. Measurement of the fluorescence intensity of each well and assessment of the toxicity of the inhibitors were performed by following the instructions provided by the manufacturer.
Coimmunoprecipitation.
Identical numbers (8 × 105/ml) of HUVECs were pretreated with dimethyl sulfoxide (DMSO) (placebo) or the protein tyrosine kinase inhibitors PP2 and genistein for 2 h, followed by exposure to control (mock) or viral stock solution for 15 min. The cells from each treatment were then resuspended in 2 ml of lysis buffer containing 0.2 mM NaCl, 0.01 mM sodium phosphate, pH 7.2, 1 mM EDTA, 1% NP-40, 1% aprotinin (Trasylol), and 0.05% sodium fluoride. After centrifugation at 12,000 × g for 5 min, the supernatants were collected, and 1 ml of each supernatant was incubated with a rabbit polyclonal antibody (2 μg/ml) against the human calcium channel subunit Calα2 (Biocompare, South San Francisco, CA) at 4°C with shaking overnight. The antigen-antibody complexes were pulled down with protein A/G-agarose beads (Fisher Scientific) after 1 h of incubation at ambient temperature and three rounds of washing with the protein lysis buffer. The immunoprecipitated antigen-antibody complexes were eluted in 200 μl of lysis buffer (100 mM Tris, pH 7.5, 1 mM EDTA, 400 mM NaCl, and 2% SDS) and subjected to SDS-PAGE and Western blot analysis to detect the Ca2+ channel subunit Calα2 with the rabbit anti-Calα2 antibody, tyrosine-phosphorylated Calα2 with mouse monoclonal anti-phosphotyrosine antibody (4G10; Millipore), and the tyrosine kinase Src and phospho-Src with the specific antibodies described previously.
Measurement of intracellular Ca2+.
Changes in intracellular Ca2+ concentrations were measured with the calcium-sensitive Fluo-4NW dye assay kit (Molecular probes). Briefly, HUVECs were seeded in a 96-well plate at a density of 10,000 cells per well and cultured in supplement-enriched EBM2 medium for 24 h. The cell monolayers were washed with Hanks' balanced salt solution (HBSS), loaded with 100 μl Fluo-NW dye mixture, and incubated at 37°C for 30 min and at ambient temperature for another 30 min. After removing the dye and filling the wells with 50 μl of basic EBM2 medium in each well, the cells were subjected to stimulation with 150 μl of mock or viral stock solution. The plate was read at multiple time intervals in a Synergy microplate reader (BioTek, Winooski, VT) with an excitation wavelength of 494 nm and an emission wavelength of 516 nm. Alternatively, HUVECs pretreated with the Fluo-NW dye were also imaged under the AMG/EVOS-fi fluorescence microscope before and 1 min after addition of mock or viral stock solution to monitor intracellular Ca2+ changes.
RESULTS
KSHV induces rapid Ang-2 release from endothelial cells.
A significant amount of Ang-2 is presynthesized and stored in the endothelial cell-specific organelles WPBs under normal conditions and is rapidly released upon stimulation with various factors, such as thrombin and histamine. To examine if KSHV induces rapid Ang-2 release, we stimulated identical numbers of HUVECs with identical amounts (0.5 ml) of purified KSHV or mock stock solution and measured the levels of released Ang-2 by Western blotting. KSHV strongly induces Ang-2 release from HUVECs, which starts as early as 1 min postinfection, peaks at 15 min postinfection, and decreases dramatically at 120 min postinfection (Fig. 1A). KSHV also induces rapid release of IL-8, another cytokine that is costored in WPBs in endothelial cells, along with Ang-2, P-selectin, eotaxin-3, endothelin, and Von Willebrand factor (VWF) (24, 25, 26). In contrast, mock infection causes little Ang-2 release. To confirm KSHV induction of Ang-2 release, we performed dual-color IFA staining on HUVECs following mock or KSHV infection for 15 min, with a polyclonal rabbit anti-Ang-2 antibody to detect intracellular Ang-2 and a mouse monoclonal anti-KSHV small capsid protein (ORF65) to detect the infecting virions. In mock-infected HUVECs, Ang-2 is detected in the cytoplasm in what was previously described as the WPBs (Fig. 1B). However, the level of intracellular Ang-2 decreases dramatically upon KSHV stimulation, which is consistent with the reduced Ang-2 levels in the cell lysates of KSHV-infected HUVECs shown in Fig. 1A. To examine if the high levels of Ang-2 in the supernatants of KSHV-infected HUVECs result from transcriptional induction, as reported previously (16), we conducted real-time RT-PCR analysis of the Ang-2 transcript from mock- and KSHV-infected HUVECs. As shown in Fig. 1C, the Ang-2 transcript level does not change at 1 h postinfection but increases by 6.7-fold at 54 h postinfection, suggesting that the high levels of Ang-2 in the supernatants of KSHV-induced HUVECs (Fig. 1A) come from rapid release of presynthesized Ang-2. Further supporting this result, the Ang-2 level in the cell lysate of KSHV-infected HUVECs is substantially lower than that in mock-infected HUVECs at 1 h postinfection (Fig. 1D). In contrast, the intracellular level of Ang-2 increases at 54 h postinfection, which is consistent with previous results (16). In addition, pretreatment of HUVECs with the protein synthesis inhibitor cycloheximide has little effect on KSHV-induced Ang-2 release (Fig. 1E), thus further confirming that the source of Ang-2 is storage rather than de novo protein synthesis.
Fig 1.

KSHV induces rapid Ang-2 release from endothelial cells. (A) Western blot detection of Ang-2 and IL-8 in the supernatants (Sup) and total cell lysates of mock- or KSHV-infected HUVECs at various time points. (B) IFA staining of intracellular Ang-2 (green) and KSHV (red) virions in mock- or KSHV-infected HUVECs at 15 min postinfection, using a rabbit polyclonal antibody against Ang-2 and a monoclonal antibody against KSHV small capsid protein (ORF65). DAPI was used to stain cellular nuclei (dark blue). (C) Real-time RT-PCR measurement of Ang-2 transcript from mock- and KSHV-infected HUVECs at 1 h postinfection and 54 h postinfection. The error bars indicate standard deviations. (D) Western blot detection of intracellular Ang-2 from mock- and KSHV-infected HUVECs at 1 h postinfection and 54 h postinfection. (E) Western blot detection of Ang-2 in the supernatants of HUVECs that were pretreated with 25 μM cycloheximide or placebo (ethanol) for 1 h, followed by incubation with mock and KSHV viral stock for 15 min. (F) Western blot detection of Ang-2 in the supernatants of HUVECs or human primary lymphatic endothelial cells (Lymph-EC) that were collected 15 min postinfection with mock, KSHV, and adenovirus stock solutions, respectively. Equivalent amounts of mock and KSHV stock solutions were also loaded as controls to confirm the absence of Ang-2 in the stock solutions. For all experiments, identical numbers of cells were infected with identical amounts of mock or viral stock solutions. Also, β-tubulin from the cell lysates was used as a reference to calibrate loading for all Western blot analyses.
To examine if KSHV induction of rapid Ang-2 release is virus and host specific, we also mock, KSHV, and adenovirus infected HUVECs and primary lymphatic endothelial cells. As shown in Fig. 1F, KSHV induces rapid Ang-2 release from both HUVECs and lymphatic endothelial cells. In contrast, adenovirus does not induce Ang-2 release at all. Collectively, these data strongly suggest that KSHV specifically induces rapid Ang-2 release from both vascular and lymphatic endothelial cells.
KSHV binding to endothelial cells is responsible for induction of Ang-2 release.
Because KSHV-induced Ang-2 release occurs so rapidly, we reasoned that the event must take place at the viral entry level. To test this hypothesis, we prepared a “replication-deficient” KSHV viral stock solution by UV light irradiation of the purified viral stock solution. The UV-treated KSHV has been shown to still bind to cells but is unable to cause infection because of a damaged genome (27). We also utilized soluble heparin sulfate (HS) to block KSHV entry (28). As visualized in Fig. 2A and summarized in Fig. 2B, infection with KSHV stock solution results in 59.5% GFP-positive cells, while infection with UV-treated KSHV or KSHV preincubated with HS gives rise to only 1.2% and 1.4% GFP-positive cells. As shown in Fig. 2C, both KSHV and UV-treated KSHV induce Ang-2 release from HUVECs. The fact that the UV-treated KSHV gives rise to a reduced level of Ang-2 release suggests that UV treatment may have caused partial damage to the viral structure. In contrast, preincubation with and infection in the presence of HS abolish KSHV induction of Ang-2 release. These results suggest that KSHV binding to endothelial cells is responsible for induction of Ang-2 release.
Fig 2.

KSHV binding to endothelial cells induces rapid Ang-2 release. (A) Representative images of HUVECs infected with recombinant KSHV, BAC36, UV-treated KSHV, and KSHV pretreated with and in the presence of soluble heparin sulfate (10 μg/ml). KSHV-infected cells were GFP positive at 48 h postinfection. (B) Viral infection rates represented as percentages of GFP-positive cells at 48 h postinfection. The error bars indicate standard deviations. (C and D) Western blot detection of Ang-2 released from HUVECs exposed to mock infection, KSHV, UV-treated KSHV, KSHV in the presence of HS, and different doses of KSHV glycoproteins gB and gP-K8.1A for 15 min. For all experiments, identical numbers of cells were infected with identical amounts of mock or KSHV stock solution. Also, β-tubulin from the cell lysates was used as a reference to calibrate loading for all Western blot analyses.
KSHV interacts with multiple integrins, such as α3β1 and αVβ3, to attach to and enter the target cells (29, 30, 31). Several viral envelope glycoproteins, such as gB (ORF8), gH (ORF22), gL (ORF47), gM (ORF39), and gN (ORF53), play critical roles in mediating viral attachment and entry (32). However, among the five envelope glycoproteins, only gB carries an RGD motif that is critical for direct interaction with integrins (30). KSHV also encodes several unique lytic-cycle-associated glycoproteins, such as ORF4, gpK8.1A and gpK8.1B, K1, K14, and K15. ORF4 and gpK8.1A are also associated with the viral envelope but do not carry an RGD motif. To further confirm that viral binding to endothelial cells is responsible for Ang-2 release and to examine if these viral glycoproteins have similar effects on Ang-2 release, we treated identical numbers of HUVECs with different doses of purified KSHV glycoproteins gB and gP-K8.1A. Interestingly, only gB induced Ang-2 release, while gp-K8.1A had very little effect on Ang-2 release (Fig. 2D). Collectively, these results indicate that KSHV binding to endothelial cells causes rapid Ang-2 release and that the interaction between the KSHV glycoprotein gB and the integrin receptors on the surfaces of endothelial cells is critical for this event.
Blocking viral binding to integrin receptors inhibits KSHV-induced Ang-2 release.
To further confirm that KSHV binding to its integrin receptors triggers rapid Ang-2 release, we pretreated identical numbers of HUVECs with integrin-specific antibodies that had been previously shown to inhibit KSHV infection by blocking viral entry (29, 30) and then stimulated the cells with mock or viral stock solution in the presence of the blocking antibodies. Compared to the control IgG, all of the anti-integrin antibodies inhibit KSHV induction of Ang-2 release (Fig. 3A). In a separate experiment, we pretreated identical numbers of HUVECs with RGD and cyclo-RGD peptides, as well as RAD and cyclo-RAD control peptides, and then stimulated the cells with mock or viral stock solution in the presence of the respective peptides. Similar to the anti-integrin antibodies, both RGD and cyclo-RGD peptides, but not RAD and cyclo-RAD peptides, inhibit KSHV induction of Ang-2 release (Fig. 3B). To demonstrate that the antibodies against integrins and the RGD peptides are indeed functional in blocking viral binding, we pretreated HUVECs with the different antibodies and RGD and RAD peptides and then infected the cells with KSHV in the presence of these reagents. As reported previously, all of the anti-integrin antibodies and the RGD and cyclo-RGD peptides, but not IgG and the RAD and cyclo-RAD peptides, inhibit KSHV infection (Fig. 3C and D). Therefore, KSHV binding to integrin receptors indeed plays a key role in inducing Ang-2 release.
Fig 3.

Anti-integrin antibodies and RGD peptides inhibit KSHV-induced Ang-2 release. (A and B) Western blot detection of Ang-2 released from HUVECs pretreated with blocking antibodies specific for integrins β1, β3, α3, and αVβ3, control IgG, RGD, and cyclo-RGD (cRGD) peptides and RAD and cyclo-RAD (cRAD) control peptides for 30 min and then exposed to mock or viral stock solution in the presence of the corresponding blocking and control reagents for 15 min. For all experiments, identical numbers of HUVECs were infected with identical amounts of KSHV stock solution. Also, β-tubulin from the cell lysates was used as a reference to calibrate loading for all Western blot analyses. (C and D) Infection of HUVECs pretreated with and in the presence of anti-integrin antibodies, control IgG, RGD and cRGD peptides, and RAD and cRAD control peptides with KSHV. The viral infection rate is represented as percentages of GFP-positive cells at 48 h postinfection. The error bars indicate standard deviations.
KSHV induces protein tyrosine phosphorylation to promote Ang-2 release.
Integrins are protein tyrosine kinase receptors. Upon ligand binding, the activated integrins phosphorylate multiple downstream target proteins, such as the tyrosine kinases Src and focal adhesion kinase (FAK), to transduce multiple cellular signaling pathways. As shown in Fig. 4A, KSHV binding to HUVECs enhances tyrosine phosphorylation of both Src and FAK, which could be inhibited by the tyrosine kinase-specific inhibitors genistein (2 μM) and PP2 (10 nM). Interestingly, pretreatment of HUVECs with these inhibitors also abolishes KSHV-induced Ang-2 release. To rule out the possibility that the abolishment of Ang-2 release by genistein and PP2 was due to cell death caused by the inhibitors, we performed a cell toxicity assay by treating identical numbers of HUVECs with different doses of the inhibitors. As shown in Fig. 4B, no significant cell toxicities were seen in the ranges of 0 to 20 nM PP2 and 0 to 2.0 μM genistein.
Fig 4.

KSHV induction of rapid Ang-2 release is abolished by protein tyrosine kinase inhibitors. (A) Western blot detection of Ang-2 in the supernatants and β-tubulin, phosphorylated protein tyrosine kinases p-Src and p-FAC, and total Src and FAC in the cell lysates of HUVECs that were pretreated with the protein tyrosine phosphorylation inhibitors genistein (2 μM) and PP2 (10 nM) or DMSO (placebo) for 2 h, followed by mock or KSHV infection for 15 min. (B) Cytotoxicities of genistein and PP2 to HUVECs at different concentrations, which was measured with the Vybrant cytotoxicity assay kit (Molecular Probes) and represented as the average fluorescence intensity values (in RFU) of the differently treated cells from three repeated experiments. (C) Western blot detection of Ang-2 in the supernatants and β-tubulin, phosphorylated protein tyrosine kinase p-Src, and total Src in the lysates of HUVECs that were pretreated with genistein (2 μM) or DMSO (placebo) for 2 h, followed by incubation with basic EBM2 medium with 2 U thrombin or medium only as a control for 15 min.
Certain physiological factors, such as thrombin, have been previously shown to induce rapid Ang-2 release (19). Interestingly, interaction between thrombin and its receptors also modulates the integrin signaling pathways (33). To examine if protein tyrosine phosphorylation through integrin signaling is involved in thrombin induction of Ang-2 release, we pretreated HUVECs with genistein, followed by incubation with basic EBM2 medium with or without 2 U/ml thrombin for 15 min. As shown in Fig. 4C, thrombin causes both tyrosine phosphorylation of Src and rapid Ang-2 release. However, inhibition of protein tyrosine phosphorylation by genistein strongly reduces thrombin induction of rapid Ang-2 release. Taken together, these results suggest that KSHV binding to endothelial cells results in enhanced protein tyrosine phosphorylation through integrin signaling, which plays a key role in regulating rapid Ang-2 release by KSHV and other factors, such as thrombin.
KSHV infection induces Ca2+ influx to mediate Ang-2 release.
Previous studies have shown that Ca2+ is a key mediator of cytokine release from WPBs through regulated exocytosis (34). To test if Ca2+ also mediates KSHV induction of Ang-2 release, we pretreated HUVECs with different Ca2+ chelators and entry blockers. As shown in Fig. 5, the Ca2+ chelators BAPTA and EGTA strongly inhibit KSHV-induced Ang-2 release. The membrane-permeable intracellular Ca2+ chelator BAPTA-AM has lower activity in inhibiting Ang-2 release than BAPTA and EGTA, suggesting that Ca2+ influx plays a more important role than intracellular Ca2+ release. In full support of this conjecture, the Ca2+ channel blockers nifedipine, amlodipine, and SKF96365 all strongly inhibit Ang-2 release. In contrast, thapsigargin, an agent that raises cytosolic Ca2+ by blocking the ability of the cell to pump Ca2+ into the endoplasmic reticulum, only slightly inhibits Ang-2 release. These results support the idea that Ca2+ mediates KSHV-induced Ang-2 release.
Fig 5.
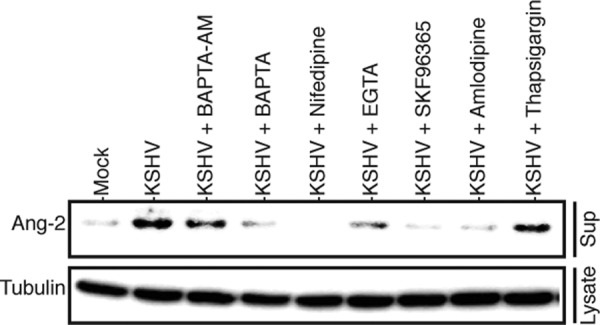
Ca2+ chelators and Ca2+ channel blockers inhibit KSHV-induced Ang-2 release. Identical numbers of HUVECs were pretreated with the Ca2+ chelators BAPTA (100 μM), BAPTA-AM (100 μM), and EGTA (100 μM); the Ca2+ channel blockers nifedipine (5 μM), amlodipine (5 μM), and SKF96365 (20 μM); and thapsigargin (5 μM) for 30 min, followed by mock or KSHV infection for 15 min in the presence of these reagents. The levels of Ang-2 in the supernatants and β-tubulin in the cell lysates of the differently treated cells were measured by Western blotting, as described previously.
KSHV induces Ca2+ influx through integrin signaling.
Integrin signaling through protein tyrosine phosphorylation is known to induce Ca2+ influx in endothelial cells (35). To examine if KSHV binding to endothelial cells induces Ca2+ influx in a protein tyrosine phosphorylation-dependent manner, we pretreated HUVECs with the tyrosine kinase inhibitor genistein, PP2, or DMSO (placebo) and then measured the relative intracellular Ca2+ concentrations before and after exposure to mock or viral stock solution. As shown in Fig. 6A and B, exposure of HUVECs to KSHV instantly increased the intracellular Ca2+ level. However, both genistein and PP2 significantly inhibited the rapid increase of the intracellular Ca2+ concentration following KSHV infection. Therefore, KSHV-induced protein tyrosine phosphorylation is indeed involved in controlling Ca2+ influx.
Fig 6.

KSHV binding to endothelial cells induces Ca2+ influx that can be inhibited by protein tyrosine kinase inhibitors. (A) Representative images showing changes in intracellular Ca2+ concentrations in HUVECs before and 60 s after exposure to mock or KSHV stock solution. The images were taken under an AMG/EVOS-fi fluorescence microscope. (B) Measurement of intracellular Ca2+ concentrations in HUVECs before and after mock and KSHV infection, using the calcium-sensitive dye Fluo-4NW assay kit (Molecular Probes). Identical numbers of HUVECs in 96-well plates were pretreated with the protein tyrosine phosphorylation inhibitors genistein (2 μM) and PP2 (10 nM) or DMSO (placebo) for 2 h, followed by exposure to mock or KSHV solution. The intracellular Ca2+ concentrations of the differently treated cells were read as fluorescence intensities at multiple time intervals in a Synergy microplate reader (BioTek, Winooski, VT), with an excitation wavelength of 494 nm and an emission wavelength of 516 nm. (C) Western blot detection of proteins resulting from coimmunoprecipitation with an antibody against the Calα2 subunit of the l-type Ca2+ channel or control IgG. Identical numbers of HUVECs were pretreated with PP2, genistein, or DMSO (placebo) for 2 h, followed by mock or KSHV infection for 15 min, respectively. Total cell lysates from each treatment were immunoprecipitated with a rabbit anti-Calα2 antibody or rabbit IgG (negative control), and the resulting products were analyzed by SDS-PAGE and Western blot detection with antibodies to Calα2, phosphotyrosine (for p-Calα2), Src, and p-Src, respectively.
Previous studies have suggested that protein tyrosine phosphorylation regulates Ca2+ channel opening and that the Calα2 subunit responsible for the opening of the l-type Ca2+ channel is physically associated with and phosphorylated by the tyrosine kinase Src (36, 37). We conducted a coimmunoprecipitation assay to pull down Calα2 and its associated proteins from identical numbers of HUVECs that were untreated or pretreated with the inhibitors genistein and PP2 and then mock infected or infected with KSHV for 15 min. As shown in Fig. 6C, Calα2 was pulled down by the anti-Calα2 antibody, but not by the control IgG. Phosphorylation of Calα2 in HUVECs was enhanced upon exposure to KSHV, which was strongly inhibited by pretreatment with genistein and PP2. The anti-Calα2 antibody also pulled down Src, thus confirming previous reports that Src is associated with Calα2 and plays a key role in regulating l-type Ca2+ channel opening.
DISCUSSION
KS is a highly angiogenic and inflammatory tumor (38). The growth of early-stage KS heavily depends on various growth factors, cytokines, and chemokines. The fact that the proangiogenic and proinflammatory cytokine Ang-2 is highly expressed in KS tumors suggests that the cytokine plays an important role in KS tumor development (16, 39). The high expression level of Ang-2 in KS tumors is most likely attributable to KSHV infection. Indeed, KSHV infection of primary endothelial cells upregulates Ang-2 expression (16). However, significant increase in Ang-2 transcription occurs only 54 h postinfection (16) and requires the expression of several viral genes (40). Here, we report that a significant amount of Ang-2 is presynthesized and stored in the WPBs of endothelial cells and that it is rapidly released upon KSHV infection. This finding defines a novel mechanism by which KSHV infection contributes to increased levels of Ang-2 to promote KS tumor growth.
Unlike transcriptional upregulation, KSHV induction of rapid Ang-2 release does not require viral gene expression but viral binding to its cellular integrin receptors. As summarized in Fig. 7, blocking viral binding with soluble heparin sulfate, anti-integrin antibodies, or RGD peptide inhibits KSHV-induced Ang-2 release. KSHV binding to integrins enhances tyrosine phosphorylation of several downstream signaling proteins, including the kinases Src and FAK and the Calα2 subunit of the l-type Ca2+ channel. Our results suggest that this KSHV-integrin-transduced protein phosphorylation plays a key role in regulating Ang-2 release, since the protein tyrosine phosphorylation inhibitors genistein and PP2 abolish Ang-2 release. Consistent with previous reports (41), our data confirm that changes in the intracellular Ca2+ concentration mediate Ang-2 release and that Ca2+ chelators and Ca2+ channel blockers inhibit Ang-2 release. We also demonstrated that KSHV binding to HUVECs induces instantaneous Ca2+ influx, which can be inhibited by protein tyrosine kinase inhibitors. Such Ca2+ influx-transduced “outside-in” signaling appears to play a critical role in mediating Ang-2 release. This may explain why thapsigargin, an agent that raises cytosolic Ca2+ by blocking the ability of the cell to pump Ca2+ into the endoplasmic reticulum, has no effect on KSHV-induced Ang-2 release. Together, these results suggest that the opening of the Ca2+ channel is regulated through its own phosphorylation. Although integrins have been previously found to induce tyrosine phosphorylation-dependent Ca2+ influx in endothelial cells, our finding that KSHV binding to endothelial cells enhances phosphorylation of the Calα2 subunit of the l-type Ca2+ channel for induction of Ca2+ influx is new.
Fig 7.
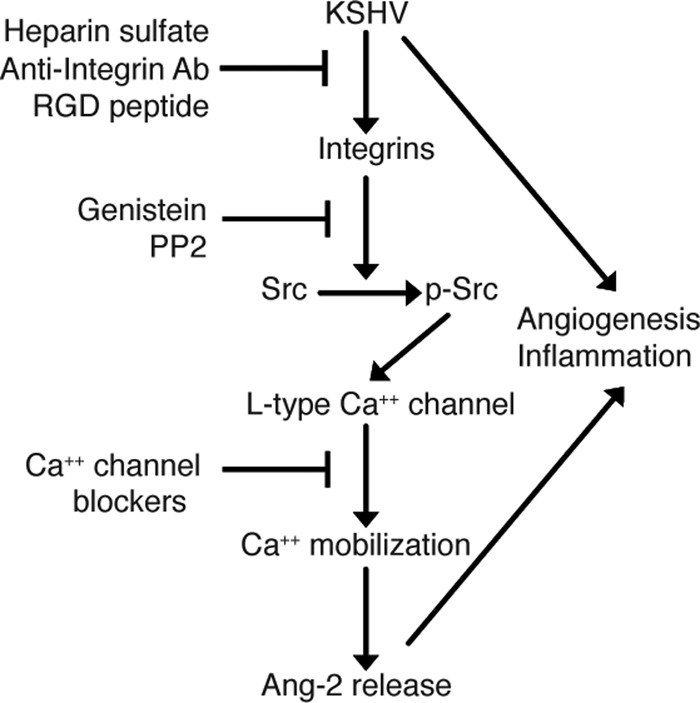
Summary of the mechanisms by which KSHV induces rapid Ang-2 release from endothelial cells. KSHV binding to its integrin receptors enhances phosphorylation of tyrosine kinases FAK and Src and the Calα2 subunit of the l-type calcium channel and results in Ca2+ influx. The Ca2+ influx ultimately mediates rapid Ang-2 release from the WPBs of endothelial cells through regulated exocytosis. Blocking viral binding with heparin sulfate, anti-integrin antibodies, or RGD peptide; inhibiting protein tyrosine phosphorylation with genistein or PP2; and treatment with Ca2+ channel blockers all inhibit Ang-2 release.
Our study highlights the importance of the dynamic interaction between KSHV and its integrin receptors in triggering Ang-2 release. In contrast to KSHV, adenovirus, used as a control in our study, does not induce Ang-2 release. Previous studies suggested that integrins facilitate adenovirus entry and internalization (42). However, mutant adenovirus without an RGD motif can enter cells efficiently (43), suggesting that the interaction between adenovirus and integrins is RGD independent. This different mechanism may explain why adenovirus does not induce Ang-2 release.
Ang-2 is best known for its role in blood vessel remodeling (44, 45). It is an antagonist of the endothelial cell-specific tyrosine kinase receptor Tie-2 (44). In the presence of VEGF, Ang-2 destabilizes existing blood vessels to promote angiogenesis (44). Ang-2 is highly expressed in most cancers and is a prognosticator of cancer progression. A high level of Ang-2 is associated with aggressive tumor cell migration, invasion, and cancer metastasis (46, 47, 48). Ang-2 is also a proinflammatory cytokine that plays an essential role in eliciting the host inflammatory response against infection by sensitizing endothelial cells to TNF-α (49). In addition, Ang-2 promotes infiltration of inflammatory cells, such as monocytes and neutrophils (50, 51, 52, 53). Previously, we reported that KSHV-induced Ang-2 enhances blood vessel growth in a Matrigel-based in vivo angiogenesis assay (16). More recently, we found that the rapidly released Ang-2 induced by KSHV enhances the adhesion of monocytes to endothelial cells (data not shown). The fact that this enhanced monocyte adhesion to endothelial cells can be abolished by soluble Tie-2 suggests that the Ang-2 rapidly released from KSHV-infected HUVECs also plays a role in promoting inflammation.
In summary, we have found that KSHV stimulates rapid release of the proangiogenic and proinflammatory cytokine Ang-2 from endothelial cells through dynamic interaction with its integrin receptors. This finding reveals a novel mechanism of KSHV induction of angiogenesis and inflammation, which might play important roles in the early stages of KS tumor development.
ACKNOWLEDGMENTS
This study was supported by start-up funds from Case Western Reserve University to Feng-Chun Ye; grants DE017333, CA096512, CA124332, and CA119889 from the National Institutes of Health to Shou-Jiang Gao; and Public Health Service grant CA075911 to Bala Chandran.
We thank Zhang Xinwen in the Department of Periodontics, School of Dental Medicine, Case Western Reserve University, for technical assistance in measuring intracellular calcium concentrations.
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 2. Ensoli B, Sgadari C, Barillari G, Sirianni MC, Sturzl M, Monini P. 2001. Biology of Kaposi's sarcoma. Eur. J. Cancer 37:1251–1269 [DOI] [PubMed] [Google Scholar]
- 3. Ensoli B, Sirianni MC. 1998. Kaposi's sarcoma pathogenesis: a link between immunology and tumor biology. Crit. Rev. Oncogenesis 9:107–124 [DOI] [PubMed] [Google Scholar]
- 4. Sivakumar R, Sharma-Walia N, Raghu H, Veettil MV, Sadagopan S, Bottero V, Varga L, Levine R, Chandran B. 2008. Kaposi's sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications. J. Virol. 82:1759–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60:4873–4880 [PubMed] [Google Scholar]
- 6. Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S, Chandran B. 2004. Host gene induction and transcriptional reprogramming in Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection. Cancer Res. 64:72–84 [DOI] [PubMed] [Google Scholar]
- 7. Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G, Samaniego F. 2002. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J. Natl. Cancer Inst. 94:926–935 [DOI] [PubMed] [Google Scholar]
- 8. Lane BR, Liu J, Bock PJ, Schols D, Coffey MJ, Strieter RM, Polverini PJ, Markovitz DM. 2002. Interleukin-8 and growth-regulated oncogene alpha mediate angiogenesis in Kaposi's sarcoma. J. Virol. 76:11570–11583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwarz M, Murphy PM. 2001. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J. Immunol. 167:505–513 [DOI] [PubMed] [Google Scholar]
- 10. Hensler HR, Rappocciolo G, Rinaldo CR, Jenkins FJ. 2009. Cytokine production by human herpesvirus 8-infected dendritic cells. J. Gen. Virol. 90:79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qin Z, Kearney P, Plaisance K, Parsons CH. 2010. Pivotal advance: Kaposi's sarcoma-associated herpesvirus (KSHV)-encoded microRNA specifically induce IL-6 and IL-10 secretion by macrophages and monocytes. J. Leukoc. Biol. 87:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin D, Gutkind JS. 2009. Kaposi's sarcoma virally encoded, G-protein-coupled receptor: a paradigm for paracrine transformation. Methods Enzymol. 460:125–150 [DOI] [PubMed] [Google Scholar]
- 13. Li X, Liang D, Lin X, Robertson ES, Lan K. 2011. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen reduces interleukin-8 expression in endothelial cells and impairs neutrophil chemotaxis by degrading nuclear p65. J. Virol. 85:8606–8615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma-Walia N, Paul AG, Bottero V, Sadagopan S, Veettil MV, Kerur N, Chandran B. 2010. Kaposi's sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 6:e1000777 doi:10.1371/journal.ppat.1000777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sin SH, Dittmer DP. 2012. Cytokine homologs of human gammaherpesviruses. J. Interferon Cytokine Res. 32:53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye FC, Blackbourn DJ, Mengel M, Xie JP, Qian LW, Greene W, Yeh IT, Graham D, Gao SJ. 2007. Kaposi's sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J. Virol. 81:3980–3991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ye FC, Lattif AA, Xie JP, Weinberg A, Gao SJ. 2012. Nutlin-3 induces apoptosis, disrupts viral latency and inhibits expression of angiopoietin-2 in Kaposi's sarcoma tumor cells. Cell Cycle 11:1393–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. 2004. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36:687–693 [DOI] [PubMed] [Google Scholar]
- 19. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, Kriz W, Thurston G, Augustin HG. 2004. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood 103:4150–4156 [DOI] [PubMed] [Google Scholar]
- 20. Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang FZ, Akula SM, Sharma-Walia N, Zeng L, Chandran B. 2003. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 77:3131–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ye FC, Zhou FC, Xie JP, Kang T, Greene W, Kuhne K, Lei XF, Li QH, Gao SJ. 2008. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 82:4235–4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greene W, Zhang W, He M, Witt C, Ye F, Gao SJ. 2012. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi's sarcoma-associated herpesvirus in endothelial cells. PLoS Pathog. 8:e1002703 doi:10.1371/journal.ppat.1002703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Collins PW, Macey MG, Cahill MR, Newland AC. 1993. Von Willebrand factor release and P-selectin expression is stimulated by thrombin and trypsin but not IL-1 in cultured human endothelial cells. Thromb. Haemost. 70:346–350 [PubMed] [Google Scholar]
- 25. Romani de Wit T, de Leeuw HP, Rondaij MG, de Laaf RT, Sellink E, Brinkman HJ, Voorberg J, van Mourik JA. 2003. Von Willebrand factor targets IL-8 to Weibel-Palade bodies in an endothelial cell line. Exp. Cell Res. 286:67–74 [DOI] [PubMed] [Google Scholar]
- 26. Oynebraten I, Bakke O, Brandtzaeg P, Johansen FE, Haraldsen G. 2004. Rapid chemokine secretion from endothelial cells originates from 2 distinct compartments. Blood 104:314–320 [DOI] [PubMed] [Google Scholar]
- 27. Mesri EA, Cesarman E, Arvanitakis L, Rafii S, Moore MA, Posnett DN, Knowles DM, Asch AS. 1996. Human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med. 183:2385–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Akula SM, Wang FZ, Vieira J, Chandran B. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282:245–255 [DOI] [PubMed] [Google Scholar]
- 29. Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419 [DOI] [PubMed] [Google Scholar]
- 30. Garrigues HJ, Rubinchikova YE, Dipersio CM, Rose TM. 2008. Integrin alphaVbeta3 binds to the RGD motif of glycoprotein B of Kaposi's sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J. Virol. 82:1570–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stouffer GA, Smyth SS. 2003. Effects of thrombin on interactions between beta3-integrins and extracellular matrix in platelets and vascular cells. Arterioscler. Thromb. Vasc. Biol. 23:1971–1978 [DOI] [PubMed] [Google Scholar]
- 32. Kaleeba JA, Berger EA. 2006. Kaposi's sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science 311:1921–1924 [DOI] [PubMed] [Google Scholar]
- 33. Chakraborty S, Veettil MV, Chandran B. 2012. Kaposi's sarcoma associated herpesvirus entry into target cells. Front. Microbiol. 3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lowenstein CJ, Morrell CN, Yamakuchi M. 2005. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc. Med. 15:302–308 [DOI] [PubMed] [Google Scholar]
- 35. Bhattacharya S, Ying X, Fu C, Patel R, Kuebler W, Greenberg S, Bhattacharya J. 2000. Alpha(v) beta(3) integrin induces tyrosine phosphorylation-dependent Ca(2+) influx in pulmonary endothelial cells. Circ. Res. 86:456–462 [DOI] [PubMed] [Google Scholar]
- 36. Ross GR, Kang M, Akbarali HI. 2010. Colonic inflammation alters Src kinase-dependent gating properties of single Ca2+ channels via tyrosine nitration. Am. J. Physiol. Gastrointest. Liver Physiol. 298:G976–G984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Treinys R, Jurevicius J. 2008. L-type Ca2+ channels in the heart: structure and regulation. Medicina (Kaunas) 44:491–499 [PubMed] [Google Scholar]
- 38. Ensoli B, Sturzl M. 1998. Kaposi's sarcoma: a result of the interplay among inflammatory cytokines, angiogenic factors and viral agents. Cytokine Growth Factor Rev. 9:63–83 [DOI] [PubMed] [Google Scholar]
- 39. Brown LF, Dezube BJ, Tognazzi K, Dvorak HF, Yancopoulos GD. 2000. Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi's sarcoma and cutaneous angiosarcoma. Am. J. Pathol. 156:2179–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vart RJ, Nikitenko LL, Lagos D, Trotter MW, Cannon M, Bourboulia D, Gratrix F, Takeuchi Y, Boshoff C. 2007. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 67:4042–4051 [DOI] [PubMed] [Google Scholar]
- 41. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. 2006. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26:1002–1007 [DOI] [PubMed] [Google Scholar]
- 42. Nemerow GR, Cheresh DA, Wickham TJ. 1994. Adenovirus entry into host cells: a role for alpha(v) integrins. Trends Cell Biol. 4:52–55 [DOI] [PubMed] [Google Scholar]
- 43. Soudais C, Boutin S, Hong SS, Chillon M, Danos O, Bergelson JM, Boulanger P, Kremer EJ. 2000. Canine adenovirus type 2 attachment and internalization: coxsackievirus-adenovirus receptor, alternative receptors, and an RGD-independent pathway. J. Virol. 74:10639–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. 1997. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60 [DOI] [PubMed] [Google Scholar]
- 45. Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, Suri C, Campochiaro PA, Wiegand SJ, Yancopoulos GD. 2002. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 3:411–423 [DOI] [PubMed] [Google Scholar]
- 46. Lewis CE, De Palma M, Naldini L. 2007. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 67:8429–8432 [DOI] [PubMed] [Google Scholar]
- 47. Imanishi Y, Hu B, Jarzynka MJ, Guo P, Elishaev E, Bar-Joseph I, Cheng SY. 2007. Angiopoietin-2 stimulates breast cancer metastasis through the alpha(5)beta(1) integrin-mediated pathway. Cancer Res. 67:4254–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY. 2006. Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res. 66:775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fiedler U, Reiss Y, Scharpfenecker M, Grunow V, Koidl S, Thurston G, Gale NW, Witzenrath M, Rosseau S, Suttorp N, Sobke A, Herrmann M, Preissner KT, Vajkoczy P, Augustin HG. 2006. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat. Med. 12:235–239 [DOI] [PubMed] [Google Scholar]
- 50. Maliba R, Brkovic A, Neagoe PE, Villeneuve LR, Sirois MG. 2008. Angiopoietin-mediated endothelial P-selectin translocation: cell signaling mechanisms. J. Leukoc. Biol. 83:352–360 [DOI] [PubMed] [Google Scholar]
- 51. Orfanos SE, Kotanidou A, Glynos C, Athanasiou C, Tsigkos S, Dimopoulou I, Sotiropoulou C, Zakynthinos S, Armaganidis A, Papapetropoulos A, Roussos C. 2007. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit. Care Med. 35:199–206 [DOI] [PubMed] [Google Scholar]
- 52. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, Mazzieri R, Doglioni C, Naldini L. 2007. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 109:5276–5285 [DOI] [PubMed] [Google Scholar]
- 53. De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. 2007. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 28:519–524 [DOI] [PubMed] [Google Scholar]