

Abstract
Norovirus (NoV) is the leading cause of viral gastroenteritis globally. Since 1996, NoV variants of a single genetic lineage, GII.4, have been associated with at least six pandemics of acute gastroenteritis and caused between 62 and 80% of all NoV outbreaks. The emergence of these novel GII.4 variants has been attributed to rapid evolution and antigenic variation in response to herd immunity; however, the contribution of recombination as a mechanism facilitating emergence is increasingly evident. In this study, we sought to examine the role that intragenotype recombination has played in the emergence of GII.4 variants. Using a genome-wide approach including 25 complete genome sequences generated as part of this study, 11 breakpoints were identified within the NoV GII.4 lineage. The breakpoints were located at three recombination hot spots: near the open reading frame 1/2 (ORF1/2) and ORF2/3 overlaps, as well as within ORF2, which encodes the viral capsid, at the junction of the shell and protruding domains. Importantly, we show that recombination contributed to the emergence of the recent pandemic GII.4 variant, New Orleans 2009, and a newly identified GII.4 variant, termed Sydney 2012. Reconstructing the evolutionary history of the GII.4 lineage reveals the widespread impact of both inter- and intragenotype recombination on the emergence of many GII.4 variants. Lastly, this study highlights the many challenges in the identification of true recombination events and proposes that guidelines be applied for identifying NoV recombinants.
INTRODUCTION
Norovirus (NoV), a member of the Caliciviridae family, is the leading cause of acute viral gastroenteritis and is estimated to cause almost half of all cases of gastroenteritis globally (1). Although a common cause of sporadic disease, NoV is primarily associated with outbreaks of acute gastroenteritis in institutional settings, such as aged-care facilities, hospitals, cruise ships, and child-care centers (2, 3). A highly infectious pathogen, NoV is readily transmitted from person to person or through contamination of water and food sources (4–6). Furthermore, epidemics of acute gastroenteritis are associated with the emergence of antigenic variants from a specific genetic lineage, the genogroup II, genotype 4 (GII.4) viruses. These epidemics have occurred globally with increasing frequency since the mid-1990s (7–9). Consequently, NoV-associated gastroenteritis has become a major public health concern for which there is no available antiviral agent or preventative vaccine.
NoV possesses a single-stranded, positive-sense, polyadenylated RNA genome of approximately 7,600 nucleotides (nt), which is packaged within a naked icosahedral virion of 27 to 32 nm in diameter (10). The viral genome is organized into three open reading frames (ORFs) with short untranslated regions at both the 5′ and 3′ ends. ORF1 encodes a 200-kDa polyprotein that is cleaved by the viral protease into at least six nonstructural proteins, which includes an RNA-dependent RNA polymerase (RdRp) (11). Two structural capsid proteins, VP1 and VP2, are encoded by ORF2 and ORF3, respectively. VP1 is the major component of the viral capsid (90 dimers per virion) and is divided into three major structural domains. These include a conserved shell (S) domain connected by a flexible hinge to a protruding stem (P1) domain that leads to the hypervariable P2 domain, which forms the external surface of the viral capsid (12). VP2 is a small basic protein with an undefined function, although roles in capsid assembly (13) and RNA recruitment into the virion (14) have both been proposed.
Like most RNA viruses, NoV demonstrates extensive genetic diversity. It has been classified into six genogroups (GI to GVI) on the basis of the VP1 amino acid sequence (15, 16). Each genogroup can be further divided into genotypes; currently, more than 36 genotypes have been described (15, 17). Human NoVs include viruses from GI, GII, and GIV, with the GII.4 viruses being most commonly identified in both outbreak and sporadic settings (8). NoVs are also known to infect a wide range of mammals (18–23).
Since the mid-1990s, variants of the NoV GII.4 lineage have caused 62 to 80% of NoV outbreaks globally (8, 24). Furthermore, six distinct GII.4 variants have been associated with global epidemics of acute gastroenteritis from 1996 to the present and include US 1995/96 in 1996 (25, 26), Farmington Hills in 2002 (27, 28), Hunter in 2004 (29), 2006b virus in 2007 and 2008 (30), New Orleans virus from 2009 to 2012 (31), and most recently, Sydney 2012 (32). A number of additional GII.4 variants have been identified, including Henry 2001, Japan 2001, Asia 2003, 2006a, and Apeldoorn 2008; however, these viruses were associated with epidemics localized to a particular region rather than a global pandemic (7, 8, 33–38).
Numerous mechanisms are thought to drive the evolution of the GII.4 lineage (reviewed in reference 39). The GII.4 viruses have a larger susceptible population to infect than viruses from other genotypes as a result of binding to a wider range of histo-blood group antigens (HBGAs), which are proposed to be attachment factors (40, 41). Additionally, through a high rate of evolution, new antigenic variants emerge from the GII.4 lineage every 2 to 3 years and are often associated with widespread epidemics (9, 42). Antigenic change is most evident within the hypervariable P2 domain of VP1, which contains the host cell receptor binding and antigenic regions of the viral capsid and is therefore under the greatest selective pressure (42–44). Furthermore, recent work has confirmed the potential antigenic properties of these sites in the P2 domain through the isolation and characterization of neutralizing antibodies derived from human sera (45). In this regard, NoV capsid evolution is reminiscent of the evolution of influenza A virus hemagglutinin, where immune-driven selection leads to new antigenic variants that emerge to replace their predecessors (46, 47).
As well as evolution through antigenic drift, NoV undergoes homologous recombination with breakpoints most often identified at the ORF1/2 overlap (48, 49). Some studies, however, have reported recombination within ORF1 (50) and ORF2 (44, 51). Recombination at the ORF1/2 overlap is important, as it facilitates the exchange of nonstructural (ORF1) and structural (ORF2-3) genes between different NoV lineages, which provides another mechanism, in addition to mutation, for generating antigenically novel viruses. Therefore, analogous to reassortment in influenza virus, recombination is likely to be an important mechanism contributing to viral emergence (52).
NoV intergenotype recombinants are frequently identified in molecular epidemiological studies (30, 48, 53, 54); however, recent work suggests that intragenotype recombination may have also played a role in the emergence of some GII.4 variants (37, 55). Full-length genome sequencing is not common practice in NoV molecular epidemiological studies, and the lack of full-length GII.4 sequence data has hindered the search for bona fide intragenotype recombination within the NoV GII.4 lineage. Indeed, the lack of genome sequences has made it difficult to distinguish true intragenotype recombination from other processes shaping the evolution of the NoV GII.4 lineage, including rapid evolution and natural selection in the capsid gene, rate variation (among lineages and sites), or even artifacts created during PCR or errors introduced during sequence assembly (reviewed in reference 56).
Since 1996, NoV GII.4 variants have been associated with at least six pandemics of acute gastroenteritis and continue to cause millions of infections across the globe annually. Rapid evolution and antigenic variation are important evolutionary forces shaping the epidemiological success and persistence of the GII.4 viruses in the population; however, the ultimate source of their diversity and mechanisms driving the emergence of novel GII.4 viruses remain unknown. In this study, we investigated the role that recombination has played in the emergence of GII.4 variants. Using a genome-wide approach, we examined the evolutionary history of the GII.4 lineage and revealed the widespread impact of both inter- and intragenotype recombination on the emergence of many GII.4 variants, including the two most recent GII.4 variants, New Orleans 2009 and Sydney 2012.
MATERIALS AND METHODS
Specimen collection and preparation.
Stool samples were collected through the South Eastern Area Laboratory Services (SEALS) at the Prince of Wales Hospital (POWH) as part of routine surveillance and outbreak investigations; therefore, human ethics approval was not requested for this study. Furthermore, patient consent was not required, as all specimens were deidentified and our analysis did not include any patient demographic information. All stool specimens were prepared as 20% (vol/vol) suspensions, and viral RNA was extracted as previously described (3).
Amplification and sequencing of full-length NoV GII.4 genomes.
A long reverse transcription-PCR (RT-PCR) was employed to amplify the full-length genome of NoV GII.4 variants. First, cDNA was transcribed from viral RNA using a modified oligo(dT)30 primer (GV270 in Table 1) and a SuperScript III first-strand synthesis system (Invitrogen, Carlsbad, CA). Primer GV270 was designed to bind to the NoV poly(A) tail and generate full-length cDNA transcripts while incorporating a DNA tag sequence (5′-GCATGACTGACATAGCACAGCGGCCGCCC-3′). Following cDNA synthesis, a long RT-PCR was performed using primers GV207 (or GV17) and GV271 (Table 1) and a SequalPrep long PCR kit (Invitrogen). Primers GV207 and GV17 bind to the first 33 bases of the NoV GII genome, with GV207 also having a NotI restriction site. Primer GV271 was complementary to the DNA tag sequence present in cDNA primer GV270. The conditions for the long RT-PCR were as follows: initial denaturation at 94°C for 120 s, then 10 cycles of 94°C for 15 s and 68°C for 510 s, and then 30 cycles starting at 94°C for 15 s and 68°C for 510 s, with the extension time increasing by 20 s with every cycle. Following the RT-PCR, amplicons were analyzed by agarose gel electrophoresis, and bands corresponding to the full-length NoV genome (approximately 7.6 kb) were excised from the gel and purified using a QIAquick gel extraction kit (Qiagen, Hilden, Germany).
Table 1.
Primers used in this study
| Primer | Sequence (5′–3′)a | Orientation | Nucleotide positionb |
|---|---|---|---|
| G2F3 | TTGTGAATGAAGATGGCGTCGA | Sense | 5079 |
| G2SKR | CCRCCNGCATRHCCRTTRTACAT | Antisense | 5389 |
| GV5 | AACACTGTCATATGTGCCAC | Sense | 3521 |
| GV6 | TTRTTGACCTCTGGKACGAG | Antisense | 5155 |
| GV17 | GTGAATGAAGATGGCCTCTAACGACGCTTCCGC | Sense | 1 |
| GV63 | GGAAGAACCACTTCATGAC | Antisense | 1178 |
| GV86 | TTNGGCACGGTTGAGACTGT | Antisense | 7422 |
| GV132 | CCRGCRAAGAAAGCTCCAGCCAT | Antisense | 6729 |
| GV202 | GTCATGAAGTGGTTCTTCCC | Sense | 1160 |
| GV203 | CCWGGYCAACCTGACATGTGGAA | Sense | 1892 |
| GV205 | TTCTCAAGCAAAGGTCTBAGTGATGA | Sense | 2678 |
| GV206 | TTGGCCTCYTCYTCTTCACAGAA | Antisense | 2850 |
| GV207 | CGAGCGGCCGCGTGAATGAAGATGGCGTCTAACGACGCTTCCGC | Sense | 1 |
| GV220 | TGYTATGCCTTCTGYTGTTGGGT | Sense | 617 |
| GV221 | CGTGGAGGAGCCCTGATGCTCG | Antisense | 2066 |
| GV269 | CAACGAGACTTGGTTCTACAGCTGG | Sense | 7230 |
| GV270 | GCATGACTGACATAGCACAGCGGCCGCCC-(T30) | Antisense | Poly(A) tail |
| GV271 | GCATGACTGACATAGCACAGCGGCCGCCC | Antisense | TAG |
| GV272 | GCHGCTGGTGGTTTGTGCACACC | Antisense | 477 |
| GV287 | GTCCTTGCCACCAAGRTAGGC | Antisense | 3721 |
| GV288 | CCAGATGTGTCTGCGATGAT | Sense | 944 |
| GV305 | CAGRCAAGAGCCAATGTTCAGATGG | Sense | 4999 |
| GV306 | GGCCTCAATTTGTGCTTGGAGC | Antisense | 6916 |
| GV307 | CAGTYTCTTGTCGAGTYCTCACG | Sense | 5674 |
| GV308 | GCTTGGAGCATCTCTTTRTCATG | Antisense | 6903 |
Degenerate bases are N (A, C, T, or G), H (A, C, or T), B (C, G, or T), K (G or T), W (A or T), R (A or G), and Y (T or C).
Relative to the sequence of Farmington Hills (GenBank accession number AY502023).
All purified PCR products were sequenced with the primers listed in Table 1 using dye terminator chemistry on an ABI 3730 DNA analyzer (Applied Biosystems, Carlsbad, CA). The raw sequence reads were first edited with FinchTV v1.4 software (Geospiza, Seattle, WA) and then assembled with the MEGA5 (57) and Geneious v5.6 (58) programs.
Genomewide examination for recombination in the GII.4 lineage.
Full-length GII.4 genome sequences were obtained from GenBank and then combined with 25 new sequences generated as part of this study. In the combined data set (262 sequences), there was an overrepresentation of highly homogeneous 2006b sequences from two large Japanese data sets of full-length GII.4 genomes identified between 2006 and 2009 (37, 59). Other GII.4 clades, such as Farmington Hills 2002, were also found to contain highly homogeneous sequences. Therefore, those GII.4 clades overrepresented by homogeneous sequences were trimmed by randomly removing sequences of strains identified within the same year and location that clustered close together by phylogenetic analysis. This resulted in a reduced alignment of 91 sequences with representatives from all major GII.4 clades identified between 1974 and 2012 except Japan 2001, as no full-length genome representative was available from GenBank (Table 2).
Table 2.
GII.4 variants examined in this study
| GII.4 varianta | Period of circulation | Representative strain with a full-length genome b | GenBank accession no. | Reference(s) or sourcec |
|---|---|---|---|---|
| CHDC 1970s | 1974–1977 | CHDC5191/1974/US | FJ537134 | 71 |
| Lordsdale 1993 | 1987–1993 | Lorsdale/1993/GB | X86557 | 78 |
| Camberwell 1994 | 1987–1994 | Camberwell/1994/AU | AF145896 | 79, 80 |
| US 1995/96* | 1995–2002 | Dresden174/1997/DE | AY741811 | 25 |
| Henry 2001 | 2000–2004 | TCH186/2002/US | EU310927 | 81 |
| Japan 2001 | 2002–2003 | Kaiso/2003/JPd | AB294779 | 82 |
| Farmington Hills 2002* | 2002–2004 | FarmingtonHills/2002/US | AY502023 | 15, 27 |
| Asia 2003 | 2003–2006 | Guangzhou/2003/CN | DQ369797 | 83 |
| Hunter 2004* | 2004–2006 | Hunter-NSW504D/2004/AU | DQ078814 | 29 |
| 2006a | 2006–2008 | Kenepuru-NZ327/2006/NZ | EF187497 | 3 |
| 2006b* | 2006–2012 | Shellharbour-NSW696T/2006/AU | EF684915 | 3 |
| Osaka 2007 | 2007–2008 | Osaka1/2007/JP | AB541319 | 37 |
| Cairo 2007 | 2007 | Sutherland-NSW505G/2007/AU | GQ845368 | 84 |
| Apeldoorn 2008 | 2007–2011 | JB-15/2008/KR | HQ009513 | 33 |
| New Orleans 2009* | 2008–2012 | Orange-NSW001P/2008/AU | GQ845367 | 30, 31, 85 |
| Sydney 2012* | 2012 | Sydney-NSW0514/2012/AU | JX459908 | This study |
GII.4 variants marked with an asterisk were associated with a pandemic of acute gastroenteritis and demonstrated a global distribution.
Prototype strains are presented as strain name/year/country. US, United Staes; GB, Great Britain; AU, Australia; DE, Germany; JP, Japan; CN, China; NZ, New Zealand; KR, South Korea.
References refer to the first report or full epidemiological description for each variant.
The strain Kaiso/2003/JP had no full-length genome sequence available in GenBank.
The alignment was then examined for strains that were positioned outside the major GII.4 clades by phylogenetic analysis of the full-length genome using the neighbor-joining method in MEGA5 (57). This led to exclusion of two strains, Iwate5/2007/JP and PC51/2007/IN, as they represented isolated recombination events without independent identification of similar strains (or similar breakpoints) by other groups. The remaining 89 sequences were then analyzed using the 3SEQ program (60), which considers each triplet of sequences to identify potential recombination breakpoints. The significance threshold was a P value of 0.05, and Dunn-Sidak correction was applied to correct for multiple testing. Maximum likelihood phylogenies were then produced using the PhyML program (61), based on sequences between recombination breakpoint regions identified by 3SEQ. The best-fit model of molecular evolution was chosen using the JModelTest tool (62) according to the Akaike information criterion with correction (AICc). In order to obtain an optimal tree topology, a heuristic search was performed with five initial random trees using the best (highest likelihood) of the methods: subtree pruning and regrafting (SPR) and nearest-neighbor interchange (NNI). The support for each node was determined from 1,000 bootstrap replicates. Putative recombinants were also examined by comparisons of genetic similarity to potential parental strains using the SimPlot program (63).
Bayesian coalescent analysis of the NoV GII.4 recombinant regions using time-stamped sequences.
In order to determine the timing of possible recombination events, the alignments of regions between breakpoints were then analyzed using the Bayesian coalescent methods of BEAST (64). For each analysis, the SRD06 codon-based model of evolution was used (65). The substitution rate parameters and base frequencies were unlinked across the 1st plus 2nd and the 3rd codon partitions to allow separate model estimates, and the Bayesian Skyline demographic model (66) with 15 groups in the piecewise-constant function size was used to account for the complex population structure. Three different molecular clocks were compared using Bayes tests: a strict clock, an uncorrelated exponential relaxed clock, and an uncorrelated lognormal relaxed clock. Across all partitions, an uncorrelated exponential relaxed clock was favored over the other clock models (Bayes factor, ≥32.837 for all partitions when comparing the uncorrelated exponential relaxed clock against the strict clock). For each analysis, at least three independent chains were run for between 30 million and 50 million steps to ensure convergence before the runs were combined after the removal of appropriate burn-in. The maximum clade credibility tree was then inferred with node heights scaled to mean values using the Tree Annotator program and visualized with the FigTree program (available from http://tree.bio.ed.ac.uk/software). The BEAST software package is available from http://beast.bio.ed.ac.uk.
Nucleotide sequence accession numbers.
The GenBank accession numbers for strains sequenced in this study are as follows: GQ845367 to GQ845369, HM748971 to HM748973, JQ613570 to JQ613573, JX459900 to JX459908, EF187497, EF684915, GQ845024, GQ845366, and DQ078814. The sequence alignments (including GenBank accession numbers for all strains) used for each analysis are available by request.
RESULTS AND DISCUSSION
Preparation of full-length NoV GII.4 genome data sets.
In order to examine the evidence for inter- and intragenotype recombination as an evolutionary process facilitating the emergence of novel GII.4 variants, we developed a two-step RT-PCR, to amplify the full-length NoV genome as a single amplicon (approximately 7.6 kb) (Fig. 1). Using this method, we amplified and sequenced 25 new full-length genomes from representative GII.4 strains identified between 2004 and 2012 in New South Wales, Australia (except for one New Zealand isolate collected in 2006). This included representative strains from the following GII.4 variants: Hunter 2004, 2006a, 2006b, Osaka 2007, Cairo 2007, Apeldoorn 2008, New Orleans 2009, and Sydney 2012 (Table 2). These 25 sequences were aligned against all available full-length genome sequences from GII.4 strains in GenBank (n = 237 as of 4 October 2011). Following the removal of overrepresented sequences, a refined alignment of 91 sequences was obtained with representatives from all major GII.4 clades identified between 1974 and 2012, except Japan 2001 (Table 2).
Fig 1.

Representative results from the RT-PCR amplification of the NoV GII full-length genome. Amplicons were analyzed by 0.8% (wt/vol) agarose gel electrophoresis and stained with ethidium bromide. Lane 1, a 1-kb DNA ladder (Promega) with selected molecular sizes marked; lane 2, unpurified RT-PCR product for NoV strain NSW217I/2010/AU; lane 3, NoV genome amplicon (approximately 7.6 kb) from NSW217I/2010/AU following gel extraction and purification; lanes 4 and 5, similar results for NoV strain NSW817L/2010/AU showing the unpurified and gel-extracted amplicons, respectively.
To identify and remove possible artificial recombinant genomes, the alignment was then examined for individual sequences that were not positioned within a major cluster by phylogenetic analysis of the full-length genome. This identified two recombinants strains which were represented by only a single sequence (Fig. 2). Strain Iwate5/2007/JP (GenBank accession number AB541275), referred to as Japan 2007b by Motomura et al. (37), was found to have three breakpoints at nucleotide positions 352, 2765, and 5085. This produced a mosaic genome from the Apeldoorn 2008 and 2006b parental strains (Fig. 2A). The GII.4 strain PC51/2007/IN (GenBank accession number EU921388) was another virus with only one representative and recombination breakpoints within ORF1 (Fig. 2B). Across the majority of the genome, PC51/2007/IN was related to Osaka 2007; however, between nucleotide positions 3744 and 4320, the sequence showed high similarity to GII.b viruses, including strain PC52/2007/IN (GenBank accession number EU921389), which was characterized in the same study (67). Therefore, the detection of recombination here may be an artifact, especially since the authors used 10 separate RT-PCRs to amplify the full-length genome and the breakpoints were adjacent to primer binding sites (67). These two strains were excluded from further analysis, as they represented only single examples and were not verified by the independent identification of a similar virus showing similar recombination breakpoints.
Fig 2.

SimPlot analysis of suspect putative recombinant NoV strains. (A) SimPlot for recombinant strain Iwate5/2007/JP (GenBank accession number AB541275). Three breakpoints were identified at nucleotide positions 352, 2765, and 5085. This produced a mosaic genome from Apeldoorn 2008 and 2006b parental strains. (B) SimPlot for recombinant strain PC51/2007/IN (GenBank accession number EU921388). This recombinant had an Osaka 2007 backbone with a mosaic insertion between nucleotide positions 3744 and 4320 from a GII.b parental strain. For each panel, the vertical axis represents the percent nucleotide sequence similarity between the putative recombinant and each parent strain, and the horizontal axis for panel A shows the nucleotide position along the full-length genome, while that for panel B shows the nucleotide position only in the ORF1 region. The breakpoint positions are shown and marked with dashed lines. Each analysis used a window size of 300 nt and a step size of 5 nt. The parental strains were as follows: 2006b for Hiroshima3/2008/JP (GenBank accession number AB541256), Apeldoorn 2008 for Hokkaido5/2008/JP (GenBank accession number AB541268), Osaka 2007 for Osaka1/2007/JP (GenBank accession number AB541319), and GII.b for PC52/2007/IN (GenBank accession number EU921389).
Genome-wide examination of recombination in GII.4 variants from 1974 to 2012.
For the remaining strains (n = 89), the complete genome sequences were examined for recombination using 3SEQ (60). This program provides nonparametric statistical tests for detecting mosaic structures in sequences by comparing relationships among sequence triplets. We used this method as it is resilient to false positives from rate variation among sites, it is well-suited to large data sets, and it is able to calculate breakpoints with corresponding statistical significance. In this analysis, a total of 11 potential recombination events were detected within the 89 genome sequences examined (Table 3). The breakpoints and their range of positions along the genome are plotted in Fig. 3. Most breakpoints (n = 6/11) were located near the ORF1/2 overlap (between nucleotide positions 5085 to 5104), a site of recombination commonly identified in NoV (48, 49). Two breakpoints were identified near the ORF2/3 overlap, and three breakpoints within ORF2 were identified. To examine the putative recombination events, the same alignment was used to produce maximum likelihood phylogenies of the regions between the recombination breakpoints identified by 3SEQ. The regions of analysis were ORF1 (nucleotide positions 5 to 5104), ORF2a (nucleotide positions 5085 to 5603), ORF2b (nucleotide positions 5604 to 6706), and ORF3 (nucleotide positions 6706 to 7513) (Fig. 4).
Table 3.
Summary of recombination events within the GII.4 lineage identified in this study
| GII.4 varianta | Genomic representativeb | GenBank accession | 3SEQ breakpoint |
SimPlot breakpoint position(s) (nt) | Classificationc |
||||
|---|---|---|---|---|---|---|---|---|---|
| Position (nt) | P value | ORF1 (5–5104) | ORF2a (5085–5603) | ORF2b (5604–6707) | ORF3 (6707–7513) | ||||
| Japan 2008b | Toyama2/2008/JP | AB541356 | 4981–5109 | <1.00 × 10−40 | 5094 | GII.4 Apeldoorn 2008 | GII.4 2006b | GII.4 2006b | GII.4 2006b |
| ∼ | Randwick-NSW882J/2011/AU | JX459900 | 5628–5647 | <1.00 × 10−40 | 5637 | GII.4 2006b | GII.4 New Orleans 2009 | GII.4 New Orleans 2009 | GII.4 New Orleans 2009 |
| ∼ | Toyama5/2008/JP | AB541362 | 5591–5605 | <1.00 × 10−40 | 5624 | GII.4 2006b | GII.4 Apeldoorn 2008 | GII.4 Apeldoorn 2008 | GII.4 Apeldoorn 2008 |
| ∼ | CHDC2094/1974/US | FJ537135 | 6820–6822 | <1.00 × 10−40 | 6723 | GII.4 CHDC 1970s | GII.4 CHDC 1970s | GII.4 CHDC 1970s | Novel GII |
| Osaka 2007 | Osaka1/2007/JP | AB541319 | 5002–5100, 6438–6646 | 6.35 × 10−28, <1.00 × 10−40 | 5124, 6655 | GII.ed | GII.4 Osaka 2007 | GII.4 Osaka 2007 | GII.4 2006b |
| New Orleans 2009 | Orange-NSW001P/2008/AU | GQ845367 | 4972–5016 | 5.06 × 10−38 | 5154 | GII.4 2006a | GII.4 Apeldoorn 2008-like | GII.4 Apeldoorn 2008-like | GII.4 Apeldoorn 2008-like |
| Sydney 2012 | Sydney-NSW0514/2012/AU | JX459908 | 4972–5100 | 3.25 × 10−38 | 5073 | GII.4 Osaka 2007 | GII.4 Apeldoorn 2008-like | GII.4 Apeldoorn 2008-like | GII.4 Apeldoorn 2008-like |
| Asia 2003 | Guangzhou/2003/CN | DQ369797 | 5110–5126 | 6.11 × 10−25 | 5072 | GII.12d | GII.4 Asia 2003 | GII.4 Asia 2003 | GII.4 Asia 2003 |
| CHDC 1970s | CHDC5191/1974/US | FJ537134 | 4967–5275 | 4.19 × 10−20 | 5118 | Novel GII | GII.4 CHDC 1970s | GII.4 CHDC 1970s | GII.4 CHDC 1970s |
| Cairo 2007 | Sutherland-NSW505G/2007/AU | GQ845368 | 5591–5602 | 4.82 × 10−18 | 5608 | GII.4 Japan 2001 | GII.4 Japan 2001 | GII.4 Osaka 2007-like | GII.4 Osaka 2007-like |
Those variants identified by a tilde represent sporadic recombination events that do not form part of a major GII.4 clade.
Genomic representative strains are presented as the strain identifier/year/country.
Classified to the level of genotype with the GII.4 variant assignment also shown. Numbers in parentheses represent nucleotide positions.
The NoroNet classification is shown, but the strains but may in fact be ancestral GII.4 lineages.
Fig 3.
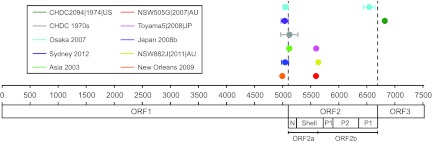
Recombination breakpoints identified across the full-length genome in the NoV GII.4 lineage. An alignment of 89 full-length genome sequences that contained representatives from all major GII.4 variant clades (except Japan 2001) was analyzed using the program 3SEQ. The summarized results identified 11 recombination breakpoints along the entire genome. The range and mean value for each breakpoint are shown along with its relative genome nucleotide position (horizontal axis). The putative recombinants are listed in the key, with the colored lines matching each corresponding breakpoint. The three ORFs are shown along with the structural domains of the capsid protein (ORF2), which are the N-terminal (N), shell (S), P1, and P2 domains. The ORF2a and ORF2b regions analyzed in this study are also indicated. Most breakpoints localized to the ORF1/2 and ORF2/3 overlaps; however, three breakpoints were also identified within ORF2.
Fig 4.

Maximum likelihood phylogenies of the NoV GII.4 genome. Maximum likelihood phylogenies of the ORF1 (A), ORF2a (B), ORF2b (C), and ORF3 (D) regions from the alignment of the NoV GII.4 strains used in the 3SEQ recombination analysis (n = 89) are shown. The genomic position for each region of analysis is shown. Bootstrap values for key nodes are shown as the percentages of 1,000 replicates. All major GII.4 clades are represented and labeled. The branches of recombinant clades/strains are colored as follows; dark green, CHDC2094/1974/US; gray, CHDC 1970s; purple, Sydney 2012; light blue, Osaka 2007; light green, Asia 2003; red, NSW505G/2007/AU; pink, Toyama5/2008/JP; yellow, NSW882J/2011/AU; orange, New Orleans 2009; and dark blue, Japan 2008b. The scale bars represent the number of substitutions per site.
ORF1/2 recombination in the GII.4 lineage.
The phylogenetic analysis revealed that the two most recently identified GII.4 variants, New Orleans 2009 and Sydney 2012, are both ORF1/2 recombinants with breakpoints located in the regions from nucleotide positions 4972 to 5016 and 4972 to 5100, respectively. The GII.4 variant New Orleans 2009 has been the predominant NoV strain in circulation globally since 2009 (31) and has been associated with three consecutive epidemics of acute gastroenteritis in NSW, Australia (2009 to 2011). New Orleans 2009 has an ORF1 region derived from a 2006a parent and an ORF2-3 region from Apeldoorn 2008 (Fig. 4 and 5A; Table 3). The recently emerged GII.4 variant, Sydney 2012, was first identified in NSW, Australia, during early 2012 and has since replaced New Orleans 2009 as the predominant GII.4 variant in circulation. It caused approximately 30% of acute gastroenteritis outbreaks in Sydney that year (data not shown); however, it was also associated with atypical increases in gastroenteritis in other regions, including New Zealand, France, Japan, Hong Kong, and the United States (32, 68, 69). The ORF1 region of Sydney 2012 was derived from an Osaka 2007 virus, while the ORF2/3 region appears to be related to the most recent common ancestor of Apeldoorn 2008 and New Orleans 2009 (Fig. 4 and 5B). Since Apeldoorn 2008 is ancestral to New Orleans 2009 in the ORF2/3 region, the likely parent for Sydney 2012 in the ORF2/3 region was an early Apeldoorn 2008 strain (Table 3).
Fig 5.

SimPlots for all putative NoV GII.4 recombinants analyzed in this study. Comparisons of genetic similarity between recombinant and possible parental strains were made using SimPlot. The results are shown for the GII.4 variants New Orleans 2009 (A), Sydney 2012 (B), Japan 2008b (C), Asia 2003 (D), Osaka 2007 (E), and CDHC 1970s (F) and the GII.4 strains CDHC2094/1974/US (G), NSW882J/2011/AU (H), Toyama5/2008/JP (I), and NSW505G/2007/AU (J). The vertical axis represents the percent nucleotide sequence similarity between the putative recombinant and each strain used for comparison, and the horizontal axis shows the relative nucleotide position along the full-length genome. The relative positions of ORFs 1, 2, and 3 are also provided, with colors matching the likely parent strain in that region. Furthermore, gray color indicates that the region was relatively novel and no suitable parent was identified; light or dark shading highlights distinct novel regions. The breakpoint positions are shown and marked with dashed lines. Each analysis used a window size of 300 nt and a step size of 5 nt. The strains used in this analysis, both recombinants and parents, were selected from the full-length genome representatives shown in Table 2.
The Japan 2008b clade also presented with evidence of ORF1/2 intragenotype recombination, with breakpoints located between nucleotide positions 4981 and 5109 (Fig. 4 and 5C), as previously reported by Motomura et al. (37). The parent for the Japan 2008b ORF1 was Apeldoorn 2008, and the parent for ORF2-3 was 2006b (Table 3). The Japan 2008b recombinant strains have not been reported outside Japan.
Breakpoints were also identified at the ORF1/2 overlap for the GII.4 variants Asia 2003, Osaka 2007, and CHDC 1970s (Fig. 3). Phylogenetic analysis revealed that each had novel ORF1 sequences that were positioned away from the remaining GII.4 clades (Fig. 4A and 5). The mean intergroup nucleotide distances from the Osaka 2007 and Asia 2003 clades to the main GII.4 lineage were 12.47% and 11.82%, respectively. The CHDC 1970s viruses are the oldest GII.4 strains sequenced and have a 14.19% mean intergroup nucleotide distance in ORF1 to the main GII.4 lineage. In comparison, the mean intragroup distance within the main GII.4 lineage was only 5.61%. Therefore, it is possible that Asia 2003, Osaka 2007, and CHDC 1970s are intergenotype recombinants with an ORF1/2 breakpoint (Table 3).
The most recent NoroNet nomenclature system has reclassified ORF1 of Asia 2003 as a GII.12 ORF1 (available from http://www.rivm.nl/en/Topics/Topics/N/NoroNet). The Asia 2003 ORF1 was previously referred to as “recombinant GII.4” (9, 48) or “Lordsdale-like” (70), since the ORF1 was related to the main GII.4 lineage yet was associated with numerous other ORF2/3 genotypes, including GII.3, GII.10, and GII.12 (9, 48, 70). Since the phylogenetic relationship between the GII.12 ORF1 and ORF2 shows little congruence, assigning Asia 2003 as an intergenotype recombinant may be misleading. Likewise, the ORF1 region of Osaka 2007 has been classified as GII.e by NoroNet and may also be an intragenotype recombinant. However, the relative distance of Asia 2003 and Osaka 2007 in ORF1 compared to that of the ORF1s of the CHDC 1970s variants, which are the earliest GII.4 strains described (71), suggests that these novel ORF1s may not represent different genotypes but may have evolved from distinct ancestral GII.4 lineages. Therefore, clarification with regard to the classification of the ORF1 regions of both Asia 2003 (GII.12) and Osaka 2007 (GII.e) is required, as the confusion will impact the classification of other viruses, such as Sydney 2012, which has an Osaka 2007-like ORF1. These issues make classifying these recombination events as either intergenotype or intragenotype difficult.
ORF2/3 recombination in the GII.4 lineage.
Two of the 11 recombination breakpoints were identified near the ORF2/3 overlap (a single-nucleotide overlap). The Osaka 2007 variants had a second breakpoint between nucleotide positions 6438 and 6646, which is located near the ORF2/3 overlap (Fig. 3 and 5E). The phylogenetic analysis shows that Osaka 2007 viruses have a distinct GII.4 ORF2 region; however, their ORF3s clustered closely with the ORF3s of 2006b variants (Fig. 4D; Table 3). The ancestral GII.4 strain, CHDC2094/1974/US (GenBank accession number FJ537135), was also found to have a breakpoint near the ORF2/3 overlap at nucleotide positions 6820 to 6822 (Fig. 3; Table 3). Across both ORF1 and ORF2, CHDC2094/1974/US was related to the other ancestral GII.4 CHDC 1970s variants; however, the CHDC2094/1974/US ORF3 sequence was novel and demonstrated only 83.88% nucleotide similarity to the ORF3 sequences of its closet GII.4 relatives, the CHDC 1970s variants CHDC5191/1974/US and CHDC4871/1977/US (Fig. 4D and 5G). Importantly, this ORF3 region also did not demonstrate any close relationship to that of any other NoV GII genotype. Therefore, similar to the difficulties proving ORF1/2 recombination for the ancestral GII.4s, we cannot confirm the recombination event in CHDC2094/1974/US without more NoV sequences for comparison.
Recombination within the ORF2 region.
In NoV, recombination typically occurs at the ORF1/2 overlap; however, a number of studies have suggested that recombination may occur at breakpoints within ORF2, which encodes the major capsid protein VP1, near the P2 domain boundaries (44, 51, 55). This scenario could lead to the creation of antigenically novel variants and contribute to the emergence of GII.4 variants. In our analysis of full-length genome sequences, three viruses were found to have breakpoints near the capsid shell/P1 domain boundary (near genome nucleotide position 5600): the sporadic strains NSW882J/2011/AU and Toyama5/2008/JP and the Cairo 2007 variant NSW505G/2007/AU (Fig. 3; Table 3).
The recombination within ORF2 for the sporadic strains NSW882J/2011/AU and Toyama5/2008/JP was evident by both comparisons of genetic similarity and phylogenetic analysis (Fig. 4B and C and 5H and I). The breakpoints for NSW882J/2011/AU and Toyama5/2008/AU were located at nucleotide positions 5628 to 5647 and 5591 to 5605, respectively (Fig. 3; Table 3). The ORF1/partial ORF2 parent for both viruses was 2006b, while for NSW882J/2011/AU, the partial ORF2/3 region was derived from a New Orleans 2009 parent (Fig. 5H); the same partial ORF2/3 region for Toyama5/2008/JP was derived from Apeldoorn 2008 (Fig. 5I).
The breakpoint for Cairo 2007 strain NSW505G/2007/AU was also found between nucleotide positions 5591 and 5602 (Fig. 3; Table 3); however, the parents for this recombination event were difficult to establish. In ORF1/partial ORF2, Cairo 2007 was distantly related to the Lordsdale 1993 variants (Fig. 4A and B), whereas in the remaining ORF2 region (ORF2b), it clustered closer to the Osaka 2007 variant (Fig. 4C). The Japan 2001 GII.4 variant was not included in our 3SEQ analysis, as no full-length genome sequence was available. In order to determine if the Japan 2001 variant was a possible parent, the region of analysis was shortened to a partial ORF1/complete ORF2 sequence (genome nucleotide positions 4286 to 6707). Using similarity plots, the Cairo 2007 strain NSW505G/2007/AU was then compared to Japan 2001, Lordsdale 1993, US 1995/96, and Osaka 2007 to identify possible parental strains (Fig. 5J). This revealed that the likely ORF1 parent of Cairo 2007 was Japan 2001 and not Lordsdale 1993. Following a sharp drop in similarity to Japan 2001 at the proposed ORF2 breakpoint (near nucleotide position 5608), the Cairo 2007 sequence was more closely related to the Osaka 2007 and US 1995/96 variant sequences than to the Japan 2001 sequence (Fig. 5J). However, for both these possible parents, the similarity to the Cairo 2007 sequence fell below 90% in the P2 domain, indicating that if this were a true recombination event, it took place in the past and antigenic drift within the P2 domain subsequently occurred. The identity of the parental strains for the recombinant Cairo 2007 strain is further complicated by the fact that the Japan 2001 variant was also previously suggested to be recombinant with a breakpoint position at nucleotide 5879 (44).
Origin of the Apeldoorn-like capsid P2 domain.
It has been suggested that the most recent pandemic GII.4 variant, New Orleans 2009, was a recombinant based on a 2006a backbone with a mosaic P2 domain derived from a 2006b-like ancestral virus (55). In our analysis of full-length genome sequences using 3SEQ, we did not detect any mosaic signal within ORF2 for New Orleans 2009; instead, we identified a single recombination breakpoint at the ORF1/2 overlap (Fig. 3), which suggested that New Orleans 2009 was a 2006a (ORF1)/Apeldoorn 2008 (ORF2/3) intragenotype recombinant (Fig. 3). Despite this, the phylogenies of the ORF2b region presented in this study (Fig. 4C and 6C), which are approximately equivalent to those for the mosaic P2 domain region in the study by Lam et al. (55), suggest that members of the entire clade, which includes Apeldoorn 2008, New Orleans 2009, and Sydney 2012 (referred to here as Apeldoorn-like), share a recent common ancestor with 2006b. For the maximum likelihood phylogenies (Fig. 4C), the bootstrap support for this relationship was 49%, and from the Bayesian analysis using BEAST, the estimated posterior probability of the equivalent node was 1 (Fig. 6C); therefore, support for this relationship was relatively strong. Furthermore, according to the coalescent estimates from BEAST, the time to this most recent common ancestor was 10.13 years (8.41 to 11.95 years; 95% highest probability density [HPD]), which would place this Apeldoorn 2008/2006b ancestor in circulation in about 2002. The possible 2006b origin of the ORF2b (P2 domain) region for Apeldoorn-like viruses contrasts against the origins for the remaining regions of the genome (ORF1, ORF2a, and ORF3): it was shown that the Apeldoorn 2008, New Orleans 2009, and Sydney 2012 viruses have distinct, non-2006b-like origins (Fig. 6A, B, and D). Therefore, if the P2 domain was of a recombinant origin from a 2006b-like virus, then the recombination event likely took place in the early 2000s and will be difficult to confirm due to the strong effects of selection and rapid evolution within this region of the NoV capsid. It is also important to consider the possibility that strong selective pressure from herd immunity and host-driven adaptation may have directed the early Apeldoorn-like and 2006b-like viruses toward similar evolutionary paths, analogous to convergent evolution, thereby mimicking recombination. Lastly, the different rates of evolution between the relatively conserved shell domain and the rapidly evolving protruding domain may also influence the analysis of recombination in the capsid gene. This phenomenon contributed to the misidentification of recombination in the hemagglutinin gene of the 1918 Spanish flu virus (72).
Fig 6.

Bayesian time-scaled phylogenies of the NoV GII.4 genome. Bayesian time-scaled phylogenies of the ORF1 (A), ORF2a (B), ORF2b (C), and ORF3 (D) regions from the alignment of NoV GII.4 strains used in the 3SEQ recombination analysis (n = 89) are shown. Support for key nodes is shown as estimated posterior probabilities. All major GII.4 clades are represented and labeled. The branches of recombinant clades/strains are colored as follows; dark green, CHDC2094/1974/US; gray, CHDC 1970s; purple, Sydney 2012; light blue, Osaka 2007; light green, Asia 2003; red, NSW505G/2007/AU; pink, Toyama5/2008/JP; yellow, NSW882J/2011/AU; orange, New Orleans 2009; and dark blue, Japan 2008b. The phylogenies are scaled to time (x axis) and are shown as years.
Influence of recombination on evolution of the NoV GII.4 lineage.
Using the data from both the 3SEQ breakpoints (Fig. 3; Table 3) and the ORF1, ORF2a, ORF2b, and ORF3 phylogenies (Fig. 4 and 6), the evolution of the NoV GII.4 lineage through time was reconstructed, showing the influence of recombination (Fig. 7). This model highlights the expansion in genetic diversity among the GII.4 viruses, initially through antigenic drift and more recently due to widespread inter- and intragenotype recombination. In fact, since the emergence of Farmington Hills 2002, all subsequently identified GII.4 variants have been influenced by recombination as the parent of a recombinant or as a recombinant themselves (Fig. 7). This may be partly explained by an increase in the number of cocirculating GII.4 variants. Since the early 1990s, at most three variants have been in circulation at one time; however, between 2007 and 2008, up to six different GII.4 variants were in circulation (Table 2). Furthermore, these six variants each formed four distinct sublineages: Hunter 2004 and 2006a, Cairo 2007 and Osaka 2007, 2006b, and Apeldoorn 2008 and New Orleans 2009. Intragenotype recombination provides a mechanism for mixing between these distinct sublineages and will increase the genetic repertoire of the major GII.4 lineage. This genetic mixing has culminated in the emergence of the GII.4 variant Sydney 2012, which contains sequence derived through recombination from all four of the aforementioned sublineages.
Fig 7.

Model for the emergence and origin of the NoV GII.4 variants. On the basis of evidence collected from the phylogenetic and recombination analyses, the evolution of the NoV GII.4 lineage through time was reconstructed, showing the influence of recombination. Each of the following major GII.4 variants is shown as a labeled box: CHDC 1970s (CH70s), Lordsdale 1993 (Lrd93), Camberwell 1994 (Cm94), US 1995/96 (US96), Japan 2001 (Jpn01), Farmington Hills 2002 (FH02), Asia 2003 (Asi03), Hunter 2004 (Hnt04), 2006a, 2006b, Osaka 2007 (Osk07), Cairo 2007 (Cai07), Apeldoorn 2008 (Apl08), New Orleans 2009 (NO09), and Sydney 2012 (Syd12). Black circles represent recombination events, with the dashed lines showing the direction of the nonvertical evolution from parental strains. The locations of the breakpoints are shown next to each recombination event in red text. The mosaic P2 domain recombination leading to the emergence of the Apeldoorn 2008-like viruses was not confirmed in this study; therefore, it has been shown with dotted lines and a question mark in the recombination node. Together, these results highlight the widespread recombination in the GII.4 lineage and its impact on the emergence of novel variants.
Most of the recombination events described in this study have involved recombination at the ORF1/2 overlap (n = 6/11) and have coincided with the emergence of a novel GII.4 variant. This is not surprising, as most of the reported intergenotype recombination in NoV occurs at the ORF1/2 overlap (48, 49) and would facilitate the exchange of nonstructural and structural genes. Clearly, this would affect the antigenic properties of the virus and therefore contribute to the emergence of new epidemic viruses. However, the acquisition of novel ORF1 regions may also confer a selective advantage that contributes to viral emergence, for example, by altering the balance of replication and mutation rates that have been shown to be important determinants of viral fitness (73–75).
A framework for identifying recombination in the NoV GII.4 lineage.
As a consequence of the potential problems in identifying true recombination events, Boni et al. have proposed guidelines for the identification of homologous recombination in influenza virus (76). Currently, no such guidelines exist for NoV; however, the principles of the guidelines for influenza virus could easily be applied. For example, the guidelines include limiting sources of contamination, determining if a breakpoint occurs near a primer binding site, checking alignments for errors, assessing the statistical significance of recombination events by using both mosaic-based methods such as 3SEQ and phylogenetic methods (including those available in the RDP package) (77), and then confirming proposed recombinants by independent identification by another laboratory. Our group has also highlighted previously that, in order to account for artificial recombination events from the amplification of sequences from two coinfecting viruses, it is necessary to generate a single amplicon across the region of analysis (48, 49). In this study, we avoided a number of these problems by employing a method of amplifying whole genomes in a single RT-PCR; furthermore, we tested for recombination using 3SEQ with a statistical threshold of a P value of <0.05. 3SEQ has the highest sensitivity and the lowest false-positive rate compared to other mosaic methods. We then assessed the support for any recombination event using a comprehensive phylogenetic approach. We also removed from our analysis a number of suspect putative recombinant sequences that could be artificial (those in Fig. 2). Lastly, in order to determine the source and mechanisms facilitating the emergence of novel GII.4 variants, molecular epidemiological studies that ensure thorough sampling coverage across time and different regions of the world are needed. By adopting a broader surveillance strategy with improved sequence coverage, we are more likely to identify novel GII.4 variants closer to the point of emergence as well as identify the intermediate transitional strains that may have facilitated their emergence.
In conclusion, this study has demonstrated that both intergenotype recombination and intragenotype recombination are widespread within the pandemic NoV GII.4 lineage and are likely to be important forces driving the evolution and emergence of novel GII.4 viruses.
ACKNOWLEDGMENTS
This work was supported in part by the Australian Research Council through Discovery Projects DP120104073 and DP110100465. M.F.B. is supported by the Wellcome Trust (098511/Z/12/Z).
We thank Rowena Bull of the School of Medical Sciences, Faculty of Medicine, University of New South Wales, for support in assay development and Juan Merif of the Department of Microbiology, South Eastern Sydney Laboratory Service, Prince of Wales Hospital, for the provision of samples.
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Patel MM, Hall AJ, Vinje J, Parashar UD. 2009. Noroviruses: a comprehensive review. J. Clin. Virol. 44:1–8 [DOI] [PubMed] [Google Scholar]
- 2. Blanton LH, Adams SM, Beard RS, Wei G, Bulens SN, Widdowson MA, Glass RI, Monroe SS. 2006. Molecular and epidemiologic trends of caliciviruses associated with outbreaks of acute gastroenteritis in the United States, 2000-2004. J. Infect. Dis. 193:413–421 [DOI] [PubMed] [Google Scholar]
- 3. Tu ET, Bull RA, Greening GE, Hewitt J, Lyon MJ, Marshall JA, McIver CJ, Rawlinson WD, White PA. 2008. Epidemics of gastroenteritis during 2006 were associated with the spread of norovirus GII.4 variants 2006a and 2006b. Clin. Infect. Dis. 46:413–420 [DOI] [PubMed] [Google Scholar]
- 4. Gallimore CI, Cubitt D, du Plessis N, Gray JJ. 2004. Asymptomatic and symptomatic excretion of noroviruses during a hospital outbreak of gastroenteritis. J. Clin. Microbiol. 42:2271–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teunis PF, Moe CL, Liu P, Miller SE, Lindesmith L, Baric RS, Le Pendu J, Calderon RL. 2008. Norwalk virus: how infectious is it? J. Med. Virol. 80:1468–1476 [DOI] [PubMed] [Google Scholar]
- 6. Tu ET, Bull RA, Kim MJ, McIver CJ, Heron L, Rawlinson WD, White PA. 2008. Norovirus excretion in an aged-care setting. J. Clin. Microbiol. 46:2119–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siebenga JJ, Lemey P, Kosakovsky Pond SL, Rambaut A, Vennema H, Koopmans M. 2010. Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 6:e1000884 doi:10.1371/journal.ppat.1000884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siebenga JJ, Vennema H, Zheng DP, Vinje J, Lee BE, Pang XL, Ho EC, Lim W, Choudekar A, Broor S, Halperin T, Rasool NB, Hewitt J, Greening GE, Jin M, Duan ZJ, Lucero Y, O'Ryan M, Hoehne M, Schreier E, Ratcliff RM, White PA, Iritani N, Reuter G, Koopmans M. 2009. Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001-2007. J. Infect. Dis. 200:802–812 [DOI] [PubMed] [Google Scholar]
- 9. Bull RA, Eden JS, Rawlinson WD, White PA. 2010. Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 6:e1000831 doi:10.1371/journal.ppat.1000831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kapikian AZ, Wyatt RG, Dolin R, Thornhill TS, Kalica AR, Chanock RM. 1972. Visualization by immune electron microscopy of a 27-nm particle associated with acute infectious nonbacterial gastroenteritis. J. Virol. 10:1075–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belliot G, Sosnovtsev SV, Mitra T, Hammer C, Garfield M, Green KY. 2003. In vitro proteolytic processing of the MD145 norovirus ORF1 nonstructural polyprotein yields stable precursors and products similar to those detected in calicivirus-infected cells. J. Virol. 77:10957–10974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290 [DOI] [PubMed] [Google Scholar]
- 13. Bertolotti-Ciarlet A, Crawford SE, Hutson AM, Estes MK. 2003. The 3′ end of Norwalk virus mRNA contains determinants that regulate the expression and stability of the viral capsid protein VP1: a novel function for the VP2 protein. J. Virol. 77:11603–11615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glass PJ, White LJ, Ball JM, Leparc-Goffart I, Hardy ME, Estes MK. 2000. Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol. 74:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323 [DOI] [PubMed] [Google Scholar]
- 16. Mesquita JR, Barclay L, Nascimento MS, Vinje J. 2010. Novel norovirus in dogs with diarrhea. Emerg. Infect. Dis. 16:980–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kroneman A, Vennema H, Deforche K, v d Avoort H, Penaranda S, Oberste MS, Vinje J, Koopmans M. 2011. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51:121–125 [DOI] [PubMed] [Google Scholar]
- 18. Liu BL, Lambden PR, Gunther H, Otto P, Elschner M, Clarke IN. 1999. Molecular characterization of a bovine enteric calicivirus: relationship to the Norwalk-like viruses. J. Virol. 73:819–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martella V, Campolo M, Lorusso E, Cavicchio P, Camero M, Bellacicco AL, Decaro N, Elia G, Greco G, Corrente M, Desario C, Arista S, Banyai K, Koopmans M, Buonavoglia C. 2007. Norovirus in captive lion cub (Panthera leo). Emerg. Infect. Dis. 13:1071–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martella V, Lorusso E, Decaro N, Elia G, Radogna A, D'Abramo M, Desario C, Cavalli A, Corrente M, Camero M, Germinario CA, Banyai K, Di Martino B, Marsilio F, Carmichael LE, Buonavoglia C. 2008. Detection and molecular characterization of a canine norovirus. Emerg. Infect. Dis. 14:1306–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang QH, Han MG, Cheetham S, Souza M, Funk JA, Saif LJ. 2005. Porcine noroviruses related to human noroviruses. Emerg. Infect. Dis. 11:1874–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wolf S, Williamson W, Hewitt J, Lin S, Rivera-Aban M, Ball A, Scholes P, Savill M, Greening GE. 2009. Molecular detection of norovirus in sheep and pigs in New Zealand farms. Vet. Microbiol. 133:184–189 [DOI] [PubMed] [Google Scholar]
- 23. Pinto P, Wang Q, Chen N, Dubovi EJ, Daniels JB, Millward LM, Buonavoglia C, Martella V, Saif LJ. 2012. Discovery and genomic characterization of noroviruses from a gastroenteritis outbreak in domestic cats in the US. PLoS One 7:e32739 doi:10.1371/journal.pone.0032739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2010. Viral shape-shifting: norovirus evasion of the human immune system. Nat. Rev. Microbiol. 8:231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noel JS, Fankhauser RL, Ando T, Monroe SS, Glass RI. 1999. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J. Infect. Dis. 179:1334–1344 [DOI] [PubMed] [Google Scholar]
- 26. White PA, Hansman GS, Li A, Dable J, Isaacs M, Ferson M, McIver CJ, Rawlinson WD. 2002. Norwalk-like virus 95/96-US strain is a major cause of gastroenteritis outbreaks in Australia. J. Med. Virol. 68:113–118 [DOI] [PubMed] [Google Scholar]
- 27. Lopman B, Vennema H, Kohli E, Pothier P, Sanchez A, Negredo A, Buesa J, Schreier E, Reacher M, Brown D, Gray J, Iturriza M, Gallimore C, Bottiger B, Hedlund KO, Torven M, von Bonsdorff CH, Maunula L, Poljsak-Prijatelj M, Zimsek J, Reuter G, Szucs G, Melegh B, Svennson L, van Duijnhoven Y, Koopmans M. 2004. Increase in viral gastroenteritis outbreaks in Europe and epidemic spread of new norovirus variant. Lancet 363:682–688 [DOI] [PubMed] [Google Scholar]
- 28. Widdowson MA, Cramer EH, Hadley L, Bresee JS, Beard RS, Bulens SN, Charles M, Chege W, Isakbaeva E, Wright JG, Mintz E, Forney D, Massey J, Glass RI, Monroe SS. 2004. Outbreaks of acute gastroenteritis on cruise ships and on land: identification of a predominant circulating strain of norovirus—United States, 2002. J. Infect. Dis. 190:27–36 [DOI] [PubMed] [Google Scholar]
- 29. Bull RA, Tu ET, McIver CJ, Rawlinson WD, White PA. 2006. Emergence of a new norovirus genotype II.4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 44:327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eden JS, Bull RA, Tu E, McIver CJ, Lyon MJ, Marshall JA, Smith DW, Musto J, Rawlinson WD, White PA. 2010. Norovirus GII.4 variant 2006b caused epidemics of acute gastroenteritis in Australia during 2007 and 2008. J. Clin. Virol. 49:265–271 [DOI] [PubMed] [Google Scholar]
- 31. Yen C, Wikswo ME, Lopman BA, Vinje J, Parashar UD, Hall AJ. 2011. Impact of an emergent norovirus variant in 2009 on norovirus outbreak activity in the United States. Clin. Infect. Dis. 53:568–571 [DOI] [PubMed] [Google Scholar]
- 32. van Beek J, Ambert-Balay K, Botteldoorn N, Eden JS, Fonager J, Hewitt J, Iritani N, Kroneman A, Vennema H, Vinje J, White PA, Koopmans M, NoroNet 2013. Indications for worldwide increased norovirus activity associated with emergence of a new variant of genotype II. 4, late 2012. Euro Surveill. 18(1):pii=20345.http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20345 [PubMed] [Google Scholar]
- 33. Belliot G, Kamel AH, Estienney M, Ambert-Balay K, Pothier P. 2010. Evidence of emergence of new GGII.4 norovirus variants from gastroenteritis outbreak survey in France during the 2007-to-2008 and 2008-to-2009 winter seasons. J. Clin. Microbiol. 48:994–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han TH, Kim CH, Chung JY, Park SH, Hwang ES. 2011. Emergence of norovirus GII-4/2008 variant and recombinant strains in Seoul, Korea. Arch. Virol. 156:323–329 [DOI] [PubMed] [Google Scholar]
- 35. Mans J, de Villiers JC, du Plessis NM, Avenant T, Taylor MB. 2010. Emerging norovirus GII.4 2008 variant detected in hospitalised paediatric patients in South Africa. J. Clin. Virol. 49:258–264 [DOI] [PubMed] [Google Scholar]
- 36. Mattison K, Sebunya TK, Shukla A, Noliwe LN, Bidawid S. 2010. Molecular detection and characterization of noroviruses from children in Botswana. J. Med. Virol. 82:321–324 [DOI] [PubMed] [Google Scholar]
- 37. Motomura K, Yokoyama M, Ode H, Nakamura H, Mori H, Kanda T, Oka T, Katayama K, Noda M, Tanaka T, Takeda N, Sato H. 2010. Divergent evolution of norovirus GII/4 by genome recombination from May 2006 to February 2009 in Japan. J. Virol. 84:8085–8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pang XL, Preiksaitis JK, Wong S, Li V, Lee BE. 2010. Influence of novel norovirus GII.4 variants on gastroenteritis outbreak dynamics in Alberta and the Northern Territories, Canada between 2000 and 2008. PLoS One 5:e11599 doi:10.1371/journal.pone.0011599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bull RA, White PA. 2011. Mechanisms of GII.4 norovirus evolution. Trends Microbiol. 19:233–240 [DOI] [PubMed] [Google Scholar]
- 40. de Rougemont A, Ruvoen-Clouet N, Simon B, Estienney M, Elie-Caille C, Aho S, Pothier P, Le Pendu J, Boireau W, Belliot G. 2011. Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 85:4057–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shanker S, Choi JM, Sankaran B, Atmar RL, Estes MK, Prasad BV. 2011. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: implications for epochal evolution. J. Virol. 85:8635–8645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lindesmith LC, Donaldson EF, Baric RS. 2011. Norovirus GII.4 strain antigenic variation. J. Virol. 85:231–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allen DJ, Noad R, Samuel D, Gray JJ, Roy P, Iturriza-Gomara M. 2009. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol. J. 6:150 doi:10.1186/1743-422X-6-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindesmith LC, Donaldson EF, Lobue AD, Cannon JL, Zheng DP, Vinje J, Baric RS. 2008. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5:e31 doi:10.1371/journal.pmed.0050031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lindesmith LC, Beltramello M, Donaldson EF, Corti D, Swanstrom J, Debbink K, Lanzavecchia A, Baric RS. 2012. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705 doi:10.1371/journal.ppat.1002705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Earn DJD, Dushoff J, Levin SA. 2002. Ecology and evolution of the flu. Trends Ecol. Evol. 17:334–340 [Google Scholar]
- 47. Koelle K, Cobey S, Grenfell B, Pascual M. 2006. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 314:1898–1903 [DOI] [PubMed] [Google Scholar]
- 48. Bull RA, Tanaka MM, White PA. 2007. Norovirus recombination. J. Gen. Virol. 88:3347–3359 [DOI] [PubMed] [Google Scholar]
- 49. Bull RA, Hansman GS, Clancy LE, Tanaka MM, Rawlinson WD, White PA. 2005. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 11:1079–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waters A, Coughlan S, Hall WW. 2007. Characterisation of a novel recombination event in the norovirus polymerase gene. Virology 363:11–14 [DOI] [PubMed] [Google Scholar]
- 51. Rohayem J, Munch J, Rethwilm A. 2005. Evidence of recombination in the norovirus capsid gene. J. Virol. 79:4977–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. 2008. The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bon F, Ambert-Balay K, Giraudon H, Kaplon J, Le Guyader S, Pommepuy M, Gallay A, Vaillant V, de Valk H, Chikhi-Brachet R, Flahaut A, Pothier P, Kohli E. 2005. Molecular epidemiology of caliciviruses detected in sporadic and outbreak cases of gastroenteritis in France from December 1998 to February 2004. J. Clin. Microbiol. 43:4659–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nayak MK, Chatterjee D, Nataraju SM, Pativada M, Mitra U, Chatterjee MK, Saha TK, Sarkar U, Krishnan T. 2009. A new variant of norovirus GII.4/2007 and inter-genotype recombinant strains of NVGII causing acute watery diarrhoea among children in Kolkata, India. J. Clin. Virol. 45:223–229 [DOI] [PubMed] [Google Scholar]
- 55. Lam TT-Y, Zhu H, Smith DK, Guan Y, Holmes EC, Pybus OG. 2012. The recombinant origin of emerging human norovirus GII.4/2008: intra-genotypic exchange of the capsid P2 domain. J. Gen. Virol. 93:817–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martin DP, Lemey P, Posada D. 2011. Analysing recombination in nucleotide sequences. Mol. Ecol. Resour. 11:943–955 [DOI] [PubMed] [Google Scholar]
- 57. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Swidan F, Thierer T, Wilson A. 2012. Geneious v5.6. Biomatters, Auckland, New Zealand [Google Scholar]
- 59. Motomura K, Oka T, Yokoyama M, Nakamura H, Mori H, Ode H, Hansman GS, Katayama K, Kanda T, Tanaka T, Takeda N, Sato H. 2008. Identification of monomorphic and divergent haplotypes in the 2006-2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J. Virol. 82:11247–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Boni MF, Posada D, Feldman MW. 2007. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176:1035–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321 [DOI] [PubMed] [Google Scholar]
- 62. Posada D. 2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25:1253–1256 [DOI] [PubMed] [Google Scholar]
- 63. Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214 doi:10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shapiro B, Rambaut A, Drummond AJ. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23:7–9 [DOI] [PubMed] [Google Scholar]
- 66. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185–1192 [DOI] [PubMed] [Google Scholar]
- 67. Chhabra P, Walimbe AM, Chitambar SD. 2010. Complete genome characterization of genogroup II norovirus strains from India: evidence of recombination in ORF2/3 overlap. Infect. Genet. Evol. 10:1101–1109 [DOI] [PubMed] [Google Scholar]
- 68. Chan MC, Chan PK. 2013. Complete genome sequence of a novel recombinant human norovirus genogroup II genotype 4 strain associated with an epidemic during summer of 2012 in Hong Kong. Genome Announc. 1(1):e00140–12 doi:10.1128/genomeA.00140-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Centers for Disease Control and Prevention 2013. Emergence of new norovirus strain GII.4 Sydney—United States, 2012. MMWR Morb. Mortal. Wkly. Rep. 62:55. [PMC free article] [PubMed] [Google Scholar]
- 70. Vinje J, Green J, Lewis DC, Gallimore CI, Brown DW, Koopmans MP. 2000. Genetic polymorphism across regions of the three open reading frames of “Norwalk-like viruses.” Arch. Virol. 145:223–241 [DOI] [PubMed] [Google Scholar]
- 71. Bok K, Abente EJ, Realpe-Quintero M, Mitra T, Sosnovtsev SV, Kapikian AZ, Green KY. 2009. Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 83:11890–11901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Worobey M, Rambaut A, Pybus OG, Robertson DL. 2002. Questioning the evidence for genetic recombination in the 1918 “Spanish flu” virus. Science 296:211. [DOI] [PubMed] [Google Scholar]
- 73. Sanjuan R, Cuevas JM, Furio V, Holmes EC, Moya A. 2007. Selection for robustness in mutagenized RNA viruses. PLoS Genet. 3:e93 doi:10.1371/journal.pgen.0030093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Domingo E, Holland JJ. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151–178 [DOI] [PubMed] [Google Scholar]
- 75. Regoes RR, Hamblin S, Tanaka MM. 2013. Viral mutation rates: modelling the roles of within-host viral dynamics and the trade-off between replication fidelity and speed. Proc. Biol. Sci. 280:20122047 doi:10.1098/rspb.2012.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boni MF, de Jong MD, van Doorn HR, Holmes EC. 2010. Guidelines for identifying homologous recombination events in influenza A virus. PLoS One 5:e10434 doi:10.1371/journal.pone.0010434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ando T, Mulders MN, Lewis DC, Estes MK, Monroe SS, Glass RI. 1994. Comparison of the polymerase region of small round structured virus strains previously classified in three antigenic types by solid-phase immune electron microscopy. Arch. Virol. 135:217–226 [DOI] [PubMed] [Google Scholar]
- 79. Cauchi MR, Doultree JC, Marshall JA, Wright PJ. 1996. Molecular characterization of Camberwell virus and sequence variation in ORF3 of small round-structured (Norwalk-like) viruses. J. Med. Virol. 49:70–76 [DOI] [PubMed] [Google Scholar]
- 80. Dingle KE, Lambden PR, Caul EO, Clarke IN. 1995. Human enteric Caliciviridae: the complete genome sequence and expression of virus-like particles from a genetic group II small round structured virus. J. Gen. Virol. 76(Pt 9):2349–2355 [DOI] [PubMed] [Google Scholar]
- 81. Parker TD, Kitamoto N, Tanaka T, Hutson AM, Estes MK. 2005. Identification of genogroup I and genogroup II broadly reactive epitopes on the norovirus capsid. J. Virol. 79:7402–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Seto Y, Iritani N, Kubo H, Kaida A, Murakami T, Haruki K, Nishio O, Ayata M, Ogura H. 2005. Genotyping of norovirus strains detected in outbreaks between April 2002 and March 2003 in Osaka City, Japan. Microbiol. Immunol. 49:275–283 [DOI] [PubMed] [Google Scholar]
- 83. Okada M, Tanaka T, Oseto M, Takeda N, Shinozaki K. 2006. Genetic analysis of noroviruses associated with fatalities in healthcare facilities. Arch. Virol. 151:1635–1641 [DOI] [PubMed] [Google Scholar]
- 84. Kamel AH, Ali MA, El-Nady HG, de Rougemont A, Pothier P, Belliot G. 2009. Predominance and circulation of enteric viruses in the region of greater Cairo, Egypt. J. Clin. Microbiol. 47:1037–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vega E, Barclay L, Gregoricus N, Williams K, Lee D, Vinje J. 2011. Novel surveillance network for norovirus gastroenteritis outbreaks, United States. Emerg. Infect. Dis. 17:1389–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]