

Abstract
Conventional CD4+ T cells play an important role in viral immunity. In most virus infections, they provide essential help for antiviral B and T cell responses. In chronic infections, including HIV infection, an expansion of regulatory T cells (Tregs) has been demonstrated, which can suppress virus-specific CD4+ T cell responses in vitro. However, the suppressive activity of Tregs on effector CD4+ T cells in retroviral infection is less well documented in vivo. We took advantage of a transgenic mouse in which Tregs can be selectively depleted to determine the influence of such cells on retrovirus-specific CD4+ T cell responses during an ongoing infection. Mice were infected with Friend retrovirus (FV), and Tregs were depleted during the acute phase of the infection. In nondepleted mice, activated CD4+ T cells produced Th1-type cytokines but did not exhibit any antiviral cytotoxicity as determined in a major histocompatibility complex (MHC) class II-restricted in vivo cytotoxic T lymphocyte (CTL) assay. Depletion of Tregs significantly increased the numbers of virus-specific CD4+ T cells and improved their cytokine production, whereas it induced only very little CD4+ T cell cytotoxicity. However, after dual depletion of Tregs and CD8+ T cells, conventional CD4+ T cells developed significant cytotoxic activity against FV epitope-labeled target cells in vivo and contributed to the control of virus replication. Thus, both Tregs and CD8+ T cells influence the cytotoxic activity of conventional CD4+ T cells during an acute retroviral infection.
INTRODUCTION
CD4+ helper T cells and cytotoxic CD8+ T cells are key players in adaptive immune responses against acute viral infections. However, during antiviral immune responses, T cells may become functionally exhausted, thereby allowing immune escape and the establishment of chronicity (1–4). Taking advantage of a transgenic mouse model, we have previously demonstrated that one mechanism contributing to the exhaustion of CD8+ T cells during an ongoing retroviral infection is suppression by regulatory T cells (Tregs) (5). Tregs expand in the late phase of the acute infection of mice with Friend virus (FV) and suppress the cytotoxic activity of effector CD8+ T cells in vivo (6, 55). Such functional suppression results in increased viral loads and contributes to viral immune escape. While these studies clearly document the inhibitory effect of Tregs on effector CD8+ T cells during retroviral infection, the suppressive activity of Tregs on CD4+ T cells in vivo is less well understood. In vitro studies show that Tregs suppress the proliferation and cytokine production of human immunodeficiency virus (HIV)-specific CD4+ T cells (7–9). In addition, a correlation between the number of Tregs, functional exhaustion of CD4+ T cells, and viral loads in lymph nodes of HIV-positive patients has been demonstrated (10), suggesting that Tregs may inhibit retrovirus-specific CD4+ helper T cell responses in infected individuals. In mouse models, Treg suppression of retrovirus-specific T cell receptor (TCR) transgenic (Tg) CD4+ T cells has been found (11, 12). Virus-specific CD4+ TCR Tg cells were adoptively transferred into FV-infected mice, and their proliferation and cytokine production were subsequently controlled in the recipient mice by Tregs. However, those experiments did not fully reflect the situation in a normal infection, because TCR Tg T cells are known to exhibit some artificial functions compared to endogenous T cells (13). To better analyze Treg effects on CD4+ T cells in a less contrived setting, we utilized transgenic DEREG mice, in which Foxp3-expressing Tregs can be selectively depleted by injecting diphtheria toxin (14, 15). The mice are on the C57/BL6 background and therefore develop a chronic infection but no acute leukemia after inoculation of FV (16, 17). The depletion of Tregs resulted in enhanced CD4+ T cell responses during acute retroviral infection. Interestingly, only dual depletion of Tregs and CD8+ T cells induced cytotoxic activity of virus-specific CD4+ T cells that was associated with the control of virus replication.
MATERIALS AND METHODS
Mice.
Inbred C57BL/6 (B6) and DEREG (15) mice were maintained under pathogen-free conditions. Experiments were done using mice (H-2b/b, Fv1b/b, Fv2r/r) or transgenic mice backcrossed on the C57BL/6 background that are resistant to FV-induced leukemia. All mice were females of 8 to 16 weeks of age at the beginning of the experiments. Mice were treated in accordance with institutional guidelines.
Virus and viral infection.
The FV stock used in these experiments was an FV complex containing B-tropic Friend murine leukemia helper virus and polycythemia-inducing spleen focus-forming virus (55). The stock was prepared as a 10% spleen cell homogenate from BALB/c mice infected 14 days previously with 3,000 spleen focus-forming units (SFFU) of noncloned virus stock. Experimental mice were injected intravenously with 0.5 ml phosphate-buffered saline (PBS) containing 20,000 SFFU of FV. The virus stock was free of lactate dehydrogenase-elevating virus.
IC assays.
The assay to determine levels of infection by infectious centers (ICs) has been previously described (18).
Cell surface and intracellular staining by flow cytometry.
Cell surface staining was performed using T cell antibodies as follows: anti-CD4 (RM 4-5; eBioscience), anti-CD8 (53-6.7; BD Biosciences), anti-CD43 (1B11; BioLegend), anti-CD62L (MEL-14; eBioscience), anti-CD44 (IM7; eBioscience), and anti-CD11b (M1/70; BD Biosciences). In surface stainings, dead cells were excluded by propidium iodide (eBioscience) staining, while fixable viability dye (eBioscience) was applied in intracellular stainings. Intracellular granzyme B (GzmB) antibody (GB11; Invitrogen), gamma interferon (IFN-γ) (XMG1.2; eBioscience), interleukin-2 (IL-2) (JES6-5H4; eBioscience), and IL-4 (11B11; eBioscience) staining was performed using the Cytofix/Cytoperm intracellular staining kit (BD Biosciences) as described previously (19). For cytokine staining, splenocytes were isolated and restimulated with coated anti-CD3 and anti-CD28 for 4 h (20). For granzyme staining, splenocytes were directly fixed without previous stimulation. Data were acquired on an LSR II flow cytometer (BD Biosciences) from 250,000 to 1,000,000 lymphocyte-gated events per sample. Analyses were done using FlowJo 7.6 software (Tree Star Inc.).
Lymphocyte depletion and agonistic anti-CD137 antibody treatment.
Briefly, mice were inoculated 4 times intraperitoneally (i.p.) with 0.5 ml of supernatant fluid obtained from hybridoma cell culture 169.4 producing CD8a-specific monoclonal antibody (21). The treatment depleted more than 95% of the CD8+ cells in the spleen (at 10 days postinfection [dpi]). CD4+ T cells were depleted with the same efficacy by i.p. injection of 0.5 ml of supernatant fluid obtained from hybridoma cell culture YTS 191.1 producing CD4-specific monoclonal antibody (21). The CD4 depletion antibody was diluted 1:3 in endotoxin-free PBS. Depletion of CD8+ and CD4+ T cells was carried out four times, every other day starting 2 days after FV infection (22). To deplete Tregs, DEREG mice were injected with diphtheria toxin (DT) (Merck) diluted in endotoxin-free PBS. A total of 1 μg DT was inoculated on days 3, 6, and 9 after FV infection. The treatment depleted more than 97% of the CD4+ T cells expressing green fluorescent protein (GFP) in the spleens of DEREG mice (at 10 dpi). To characterize the role of the costimulatory receptor CD137, infected mice were treated with 100 μg of anti-CD137 (clone LOB12.3; Bioxcell) administered every other day from day four after FV infection by i.p. injection.
Tetramers and tetramer staining.
Major histocompatibility complex (MHC) class II tetramer (Tet II) Ab-restricted Friend murine leukemia virus (F-MuLV) envelope epitope (H19-Env) (EPLTSLTPRCNTAWNRLUL) staining was performed as previously described (12).
In vivo cytotoxicity assay.
The described in vivo cytotoxic T lymphocyte (CTL) assay (23) was modified to measure cytotoxicity in FV-infected mice. Lymph node cells and bone marrow cells from naive mice were loaded with 1 μM Ab-restricted F-MuLV envelope epitope (H19-Env) (EPLTSLTPRCNTAWNRLUL) (24). These loaded cells were then stained with 7.5 μM CellTrace Violet (Invitrogen). As a control, lymph node cells and bone marrow cells without peptide were stained with 1 μM PKH26 (Sigma-Aldrich). Cells were transferred intravenously (i.v.) (107 cells of each population) into naive or FV-infected and T cell-depleted mice. Two hours after the transfer of cells, spleens were harvested, and cell suspensions were prepared. Target cells were distinguished from recipient cells and from one another based on CellTrace Violet and PKH26 stainings. The percentage of killing was calculated as follows: 100 − {[(% peptide pulsed in infected cells/% unpulsed in infected cells)/(% peptide pulsed in uninfected cells/% unpulsed in uninfected cells)] × 100}.
Statistical analysis.
Statistical data were derived using the nonparametric Student t test (GraphPad Prism software).
RESULTS
Activation and proliferation of FV-specific CD4+ T cells.
The first aim of the current study was to determine the kinetics of the total CD4+ T cell response after FV infection. It was not possible to study the full repertoire of epitope specificities, since only one MHC II tetramer (Tet II) specific for the FV H19-Env epitope (24) is currently available. Thus, we analyzed the bulk population of virus-specific CD4+ T cells using a combination of CD43 and CD62L staining. It has previously been shown that all CD4+ T cells from FV-infected mice that upregulated the activation-induced glycoform of CD43 (25, 26) also expressed CD11a+ (27), indicating that they were activated by cognate antigen rather than nonspecific inflammatory modulators (28). We prefer CD43 as a marker for effector T cells because unlike CD11a, CD43 is downregulated upon differentiation into memory T cells (29, 30). Another step in the activation of T cells is downregulation of the L-selectin (CD62L) adhesion molecule, which allows egress from lymphatic tissues and migration to sites of inflammation (31). A comparison of these markers on Tet II− CD4+ T cells from naive mice versus Tet II+ CD4+ T cells from infected mice revealed upregulation of CD43 and downregulation of CD62L after FV infection (Fig. 1A). Multicolor analysis of Tet II+ CD4+ T cells showed that the majority had a mature effector phenotype with both upregulated expression of CD43 and downregulated CD62L (Fig. 1B).
Fig 1.

Activation profile of FV-specific CD4+ T cells and kinetic analysis of the expansion of activated CD4+ T cells during acute FV infection. (A) Splenocytes from naive and 10-day-infected B6 mice were isolated, and CD4+ Tet II+ T cells reactive with I-Ab MHC class II tetramers specific for the FV H19-Env epitope were analyzed by flow cytometry for their expression of activation markers CD43 and CD62L. Expression levels were compared between the whole CD4+ T cell population of naive mice and the Tet II+ population of infected mice. The background staining of Tet II+ in CD4+ T cells from uninfected mice was around 0.1%. This staining is nonspecific, because similar results are obtained for uninfected MHC class II-deficient mice. Numbers in the gates represent percentages. (B) Representative density plot showing the combination of the markers CD43 and CD62L on FV H19-Env tetramer-positive CD4+ T cells from 10-day-infected mice. (C) Kinetic analysis of the expansion of activated CD4+ T cells during acute FV infection. (D and E) Frequency of CD4+ T cells specific for the FV epitope H19-Env (D) and frequency of total activated CD4+ T cells (E). Each dot represents a single mouse, and mean values are indicated by a line. Statistically significant differences between the time points were determined by the unpaired t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0001.
Proportions of activated CD4+ T cells peaked at 10 dpi (mean of ∼15% of the total CD4+ T cells) and slowly but significantly decreased thereafter to ∼11% at 21 dpi (Fig. 1C). This was the same time point at which the peak expansion of Tet-II+ CD4+ T cells was found in a previous study (12). At 10 days, the Tet II+ CD4+ T cells represented about 6% of the total activated (CD43+ CD62L−) CD4+ T cells (Fig. 1D and E). At 21 dpi, the proportion of activated CD4+ T cells was still significantly increased compared to that in naive mice (Fig. 1C), most likely due to the chronic infection that starts to develop at 21 dpi. Based on these findings, all subsequent functional experiments with CD4+ T cells were performed by analyzing CD4+ CD43+ CD62L cells at 10 dpi.
CD4+ T cells produce Th1 cytokines but do not mediate cytotoxic effects during acute FV infection.
In a previous study we showed that FV-specific CD4+ T cells isolated from CD4+ TCR-β Tg mice expanded, produced IFN-γ, and suppressed viral replication in FV-infected recipient mice (12). However, the antiviral effects of endogenous CD4+ T cells in FV infection have not been studied so far. We therefore performed functional studies on FV-induced CD4+ T cells by analyzing the key cytokines IFN-γ, IL-2, and IL-4 (Fig. 2A). Compared to cells from naive animals, activated CD4+ T cells expressed significantly more IFN-γ and IL-2. No significant increase was observed for the Th2-like cytokine IL-4. Activated CD4+ T cells also produced significantly more granzyme B (GzmB) (Fig. 2B) and showed elevated levels of perforin mRNA (data not shown), molecules that can induce apoptosis in infected target cells. To analyze whether or not these activated CD4+ T cells mediate any FV-specific killing of MHC class II H19-Env peptide-presenting target cells, an in vivo cytotoxicity assay was performed. However, fewer than 5% of the peptide-loaded cells were killed in the recipient mice (Fig. 2C). Since values of at least 10% are considered to indicate specific killing in this assay, we did not detect any significant CD4+ T cell-mediated FV-specific cytotoxicity in acutely infected mice.
Fig 2.
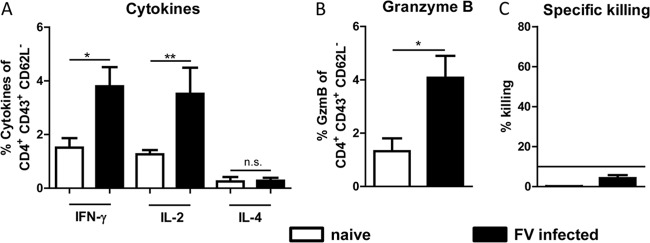
Functional analysis of CD4+ effector T cells in acute FV infection. Splenocytes from 10-day-infected B6 mice were isolated, and CD4+ CD43+ CD62L− T cells were stained for intracellular expression of cytokines and granzyme B. (A) Percentage of activated CD4+ T cells that express the cytokines IFN-γ, IL-2, and IL-4. (B) Percentage of activated CD4+ T cells that express the cytotoxic molecule GzmB. (C) In vivo killing of FV-specific CD4+ T cells measured in an in vivo cytotoxicity assay. Lymph node cells and bone marrow cells from naive mice were loaded with the FV H19-Env epitope and labeled with cell trace violet. Target cells were injected intravenously into naive and FV-infected mice. The figure shows the percentage of the target cell killing in the spleen. Statistically significant differences between the groups were determined by the unpaired t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0001. n.s., not significant. Data were pooled from two independent experiments, and the mean values with standard errors of the means (SEM) are shown.
Tregs modulate the activation of and cytokine production by CD4+ T cells during acute FV infection.
To analyze the effect of Tregs on the endogenous CD4+ T cell response during acute FV infection, we used DEREG mice, in which Tregs can be efficiently and selectively depleted during FV infection (6, 55). Following depletion of Tregs in acutely infected DEREG mice, we observed a significant increase in the frequency of total activated (CD43+ CD62L−) (Fig. 3A) as well as FV-specific (Tet II+) CD4+ T cells (Fig. 3B). To examine the functional properties of CD4+ T cells after Treg depletion, intracellular cytokine staining was performed. The expression of IFN-γ, IL-2, and IL-4 increased significantly following Treg depletion in FV-infected mice (Fig. 3C to E). The most striking increase was found for IL-4-producing CD4+ T cells, which were virtually absent in infected nondepleted mice. As a negative control, we depleted Tregs by DT injection into uninfected mice, which did not result in activation of CD4+ cells or production of cytokines or granzyme B (data not shown). The results show that both Th1 and Th2 cytokine responses of endogenous CD4+ T cells were tightly regulated by Tregs during acute FV infection.
Fig 3.

CD4+ T cell responses following FV infection and Treg depletion. FV-infected DEREG mice were treated with diphtheria toxin (DT) to deplete Foxp3+ Tregs during the first 10 days of infection. Control mice were FV infected but received PBS instead of DT. (A and B) Flow cytometry of spleen cells was used to determine the percentage of activated CD4+ T cells (A) as well as the percentage of FV-specific tetramer-positive CD4+ T cells (B). (C to E) Activated CD4+ T cells were analyzed for IFN-γ (C), IL-2 (D), and IL-4 (E) cytokine expression. Each dot represents a single mouse, and mean values are indicated by a line. Data were pooled from at least two independent experiments. Differences between the two groups were analyzed by the unpaired t test. Statistically significant differences between the groups are indicated.
Both CD8+ T cells and Tregs influence the cytotoxicity of CD4+ T cells during acute FV infection.
Our finding of a Treg-mediated inhibition of the CD4+ T cell cytokine production during acute FV infection raised the question of whether the cytotoxic function of CD4+ T cells was also controlled by Tregs. Following Treg depletion, we found a significant increase in GzmB expression of up to 10% of the activated CD4+ T cells (Fig. 4A). In contrast, no significant difference was found for perforin mRNA levels in CD4+ T cells from FV-infected versus FV-infected Treg-depleted mice (data not shown). However, the direct cytotoxic activity of FV-specific CD4+ T cells measured by the in vivo cytotoxicity assay increased slightly but significantly in infected and Treg-depleted mice (Fig. 4B). Thus, Treg depletion enhanced the effector functions of FV-specific CD4+ T cells, including their ability to directly kill MHC class II FV-specific-peptide-loaded target cells in vivo.
Fig 4.
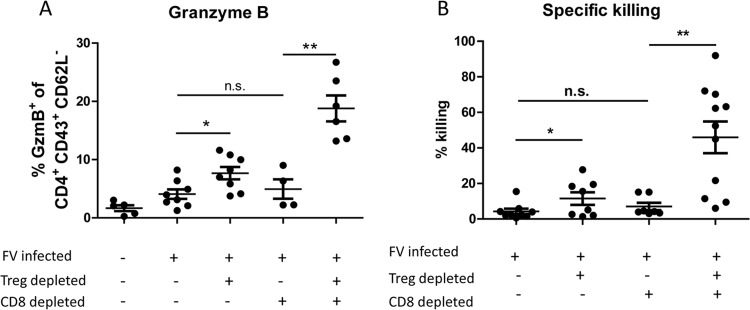
Cytotoxic function of CD4+ T cells after T cell depletion. FV-infected B6 or DEREG mice were treated with DT to deplete Foxp3+ Tregs and/or with CD8-depleting antibodies during the first 10 days of infection. (A) Flow cytometry was used to determine the percentage of activated (CD43+ CD62L−) CD4+ cells isolated from the spleen that express the cytotoxic molecule GzmB at 10 dpi. (B) In vivo cytotoxicity of CD4+ T cells. The figure shows the percentage of the target cell killing in the spleens of mice depleted for CD8+ T cells and/or Tregs. Each dot represents a single mouse, and mean values are indicated by a line. Data were pooled from at least two independent experiments. Statistically significant differences between the groups were determined by the unpaired t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0001.
It is well known that compensatory mechanisms can sometimes rescue deficient immunological functions, as reported previously (32–35). Thus, we were interested in whether CD4+ T cells might be able to compensate for the lack of cytotoxic T cells in CD8-depleted mice, especially when no Tregs were present to modulate their activity. Depletion of CD8+ T cells alone did not result in a significant increase of GzmB-expressing CD4+ T cells compared to that in the acutely infected but nondepleted group (Fig. 4A). This was supported by our findings from the in vivo cytotoxicity assay, where we did not detect any significant CD4+ T cell killing in CD8+ T cell-depleted mice. In contrast, dual depletion of both Treg and CD8+ T cells significantly increased the proportion of GzmB-expressing activated CD4+ T cells (Fig. 4A). In addition, a mean of around 50% MHC II-restricted target cell killing was observed in this group of mice, which was more than 5 times higher than that in the group of FV-infected nondepleted mice (Fig. 4B).
Cytotoxic CD4+ T cells are associated with control of virus replication.
As shown previously, CD8+ T cell depletion resulted in significantly increased viral loads, whereas Treg depletion produced significant reductions in viral loads due to enhanced CD8+ T cell responses (Fig. 5) (6, 55). Interestingly, promoting in vivo CD4+ T cell cytotoxicity by Treg depletion allowed CD8-depleted mice to control viral loads as efficiently as nondepleted mice (Fig. 5). This control over viral loads was lost when CD4+ T cells were also depleted in addition to CD8+ T cells and Tregs. These results indicate that virus control in Treg/CD8-depleted mice was dependent on CD4+ T cells and associated with in vivo CD4+ T cell cytotoxicity.
Fig 5.
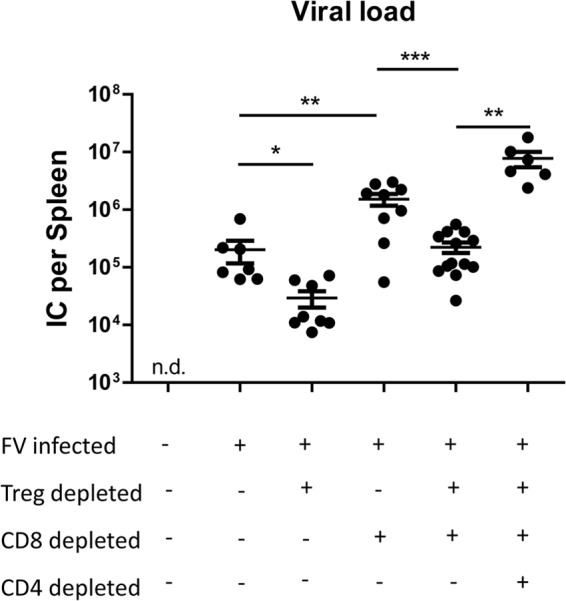
FV loads after T cell depletion. Splenocytes from differentially depleted animals were assayed at 10 dpi. Numbers of infected cells per spleen are given. For three naive control animals tested, we did not detect any virus in this assay. Each dot represents a single mouse, and mean values are indicated by a line. Data were pooled from at least two independent experiments. Statistically significant differences between the groups were determined by the unpaired t test: *, P < 0.05; **, P < 0.005; ***, P < 0.0001.
DISCUSSION
CD4+ helper T cells are a critical component of a functional immune system, but their direct antiviral effector functions in vivo are not very well understood. Previous studies have shown that effector CD4+ T cells can mediate killing of virus-infected target cells (36), a functional property that is usually attributed to CD8+ T cells or NK cells. For retroviral infections, only limited knowledge on CD4+ T cell cytotoxicity has been generated so far. In HIV-infected patients, CD4+ T cells that could kill HIV-infected target cells in vitro were found (37, 38). An in vitro characterization of an FV-specific CD4+ T cell clone demonstrated its MHC class II-restricted cytotoxic potential against virus-infected cells (56), suggesting that CD4+ T cell cytotoxicity might play a role in retroviral immunity. However, we previously showed in CD4+ T cell depletion experiments that CD4+ T cells have no direct anti-FV activity in the presence of CD8+ T cells (39). The current results indicate that Tregs and CD8+ T cells influence the cytotoxic activity of CD4+ T cells during an ongoing retroviral infection. This may be a reason why such cells are not frequently found in many viral infections.
We and others previously reported that CD8+ T cells are essential in controlling FV infection (40–42). After administration of anti-CD8 monoclonal antibodies, resistant mice can no longer control acute virus replication and develop severe splenomegaly and subsequent leukemia. These studies indicate that cytotoxicity against virus-infected target cells is critical for retroviral immunity. In contrast, the depletion of CD4+ T cells during acute FV infection results mainly in reduced antibody responses and in earlier decline of the CD8+ T cell responses (12). In FV vaccine studies, CD4+ T cells had no direct antiviral activity, but they were required in addition to CD8+ T cells and B cells for complete protection against challenge infection (43, 44). Thus, the most important function of CD4+ T cells during acute retrovirus infection seems to be their helper function for other effector cell populations of the adaptive immune system (45). This helper function, which is mediated by the production of immunostimulatory cytokines (46), was negatively regulated by Tregs. Depletion of Tregs significantly increased the production of cytokines (Fig. 3C to E) and also of the cytotoxic molecule GzmB (Fig. 4A) during acute FV infection. However, only very weak CD4+ T cell-mediated MHC class II-restricted cytotoxicity was measured in the absence of Tregs (Fig. 4B). Most likely, a direct antiviral effect of CD4+ T cells is not required in a system in which the cytotoxic function of CD8+ T cells is sufficient to mediate successful killing of virus-infected cells. This might be the reason why CD4+ T cell cytotoxicity was found only when CD8+ T cells were depleted in addition to Treg ablation. At first glance, such a dual depletion seems to be a very artificial experimental model. However, during chronic infections, e.g., with viruses such as HIV, hepatitis C virus (HCV), or FV, cytotoxic CD8+ T cells become functionally exhausted (47), a situation that might be similar to experimental CD8+ T cell depletion. In that scenario, the cytotoxicity of CD4+ T cells might be controlled exclusively by Tregs, since the inhibitory function of CD8+ T cells is lost. In fact, cytotoxic activity of CD4+ T cells against human pathogens has been described mainly for chronic viral infections (36). Furthermore, we have previously shown that CD4+ T cells control chronic FV infection when cytotoxic CD8+ T cells have developed functional exhaustion (22). Cytotoxic CD4+ T cells might therefore be especially important during chronic infections to keep virus replication in check, but these responses may be at least partially controlled by Tregs.
How can CD4+ T cells mediate killing of FV-infected cells, since they can recognize antigens only in the context of MHC class II? The main target cells of FV are erythroid precursor cells that do not express MHC class II. However, B cells and monocytes are also efficiently infected by FV (27). These cell types express MHC class II and can be recognized as possible target cells by cytotoxic CD4+ T cells. Interestingly, during chronic FV infection, the main virus reservoir is B cells (22), which would explain why cytotoxic CD4+ T cells can keep persistent virus in check. However, complete virus elimination might be hampered by the partial suppression of the CD4+ T cell response by Tregs.
The question remains how CD8+ T cells and Tregs control CD4+ T cell cytotoxicity mechanistically. Several different mechanisms for Treg suppression of conventional CD4+ T cells have been described (48). In HIV infection, a cyclic AMP (cAMP)-dependent mechanism involving the enzyme CD39 was recently described (49). This effect requires cell-to-cell contact, which is in line with data from in vitro Treg suppression assays in the FV model (50). However, the precise mechanism of Treg inhibition of effector T cell responses during FV infection in vivo has not been identified so far.
The inhibitory mechanism of CD8+ T cells on CD4+ T cell cytotoxicity is also not understood, but similar findings were made in CD8-depleted, simian immunodeficiency virus (SIV)-infected macaques, where GzmB-expressing CD4+ T cells were shown to expand (51). One possible explanation is that CD8+ Tregs might play a role. CD8+ Foxp3− T cells were reported to mediate immunosuppression in cancer patients and in patients with autoimmune diseases (52). Depletion of such cells with anti-CD8 antibody in addition to Treg ablation might allow for antiviral cytotoxicity of CD4+ T cells. However, it is more likely that molecular mechanisms of the induction of T cell cytotoxicity play an important role. It has been shown in in vitro experiments that the transcription factor eomesodermin can trigger the expression of GzmB and cytotoxicity (53). Costimulatory receptors such as CD134 and CD137 (54) induce this transcription factor after binding of their ligands. These receptors are highly expressed on effector CD8+ T cells, possibly resulting in consumption of the ligands and subsequent induction of CD8+ T cell cytotoxicity. However, stimulation of these receptors on CD4+ T cells also induces granzyme expression and cytotoxicity (54), but in vivo CD8+ T cells may outcompete CD4+ T cells due to higher receptor expression levels. To provide some evidence for this hypothesis, we performed preliminary in vivo costimulation experiments with a signaling antibody against CD137 (54). Injection of this antibody only slightly enhanced the numbers of virus-specific CD4+ T cells in FV-infected mice, but their GzmB expression was greatly augmented (Fig. 6). This effect was even more pronounced when CD8+ T cells could not consume most of the antibody because they were experimentally depleted. In that experiment, up to 10% of all CD4+ T cells produced the cytotoxic molecule granzyme B. These results strongly suggest that costimulatory receptors may play a critical role in the induction of CD4+ T cell cytotoxicity. Unraveling these molecular mechanisms will certainly help to define the cytotoxic potential of CD4+ T cells in virus infections. This area of research is of high importance because cytotoxic CD4+ T cells may be a powerful new tool for the treatment of chronic infectious diseases.
Fig 6.

Role of CD137 in CD4+ T cell cytotoxicity. Mice were infected with FV and treated with anti-CD137 antibody or a combination of anti-CD137 and depleting anti-CD8 antibody. Ten days after infection, mice were sacrificed and spleen cells were analyzed by flow cytometry. Gated CD4+ T cells are shown in dot plots representative for 3 mice per group. Numbers in the gates represent percentages of total activated CD4+ T cells (CD43+ CD62L− (A), CD4+ T cells binding class II tetramers specific for the FV H19 Env epitope (A), and activated (CD43+ CD62L−) CD4+ T cells producing the cytotoxic molecule granzyme B (GzmB) (C).
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health tetramer facility for providing MHC II tetramers and also to Tim Sparwasser for providing us with DEREG transgenic mice.
This work was supported by a grant to U.D. and G.Z. from the Deutsche Forschungsgesellschaft (TRR60 Project B4) and by a grant to U.D. and I.A. (Di714/11-1).
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Welsh RM. 2001. Assessing CD8 T cell number and dysfunction in the presence of antigen. J. Exp. Med. 193:F19–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lieberman J, Shankar P, Manjunath N, Andersson J. 2001. Dressed to kill? A review of why antiviral CD8 T lymphocytes fail to prevent progressive immunodeficiency in HIV-1 infection. Blood 98:1667–1677 [DOI] [PubMed] [Google Scholar]
- 4. Yi JS, Cox MA, Zajac AJ. 2010. T-cell exhaustion: characteristics, causes and conversion. Immunology 129:474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, Evans LH, Peterson KE, Yang G, Hasenkrug KJ. 2004. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity 20:293–303 [DOI] [PubMed] [Google Scholar]
- 6. Zelinskyy G, Dietze K, Sparwasser T, Dittmer U. 2009. Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog. 5:e1000406 doi:10.1371/journal.ppat.1000406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kinter AL, Hennessey M, Bell A, Kern S, Lin Y, Daucher M, Planta M, McGlaughlin M, Jackson R, Ziegler SF, Fauci AS. 2004. CD25(+)CD4(+) regulatory T cells from the peripheral blood of asymptomatic HIV-infected individuals regulate CD4(+) and CD8(+) HIV-specific T cell immune responses in vitro and are associated with favorable clinical markers of disease status. J. Exp. Med. 200:331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weiss L, Donkova-Petrini V, Caccavelli L, Balbo M, Carbonneil C, Levy Y. 2004. Human immunodeficiency virus-driven expansion of CD4+CD25+ regulatory T cells, which suppress HIV-specific CD4 T-cell responses in HIV-infected patients. Blood 104:3249–3256 [DOI] [PubMed] [Google Scholar]
- 9. Aandahl EM, Michaelsson J, Moretto WJ, Hecht FM, Nixon DF. 2004. Human CD4+ CD25+ regulatory T cells control T-cell responses to human immunodeficiency virus and cytomegalovirus antigens. J. Virol. 78:2454–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andersson J, Boasso A, Nilsson J, Zhang R, Shire NJ, Lindback S, Shearer GM, Chougnet CA. 2005. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J. Immunol. 174:3143–3147 [DOI] [PubMed] [Google Scholar]
- 11. Antunes I, Tolaini M, Kissenpfennig A, Iwashiro M, Kuribayashi K, Malissen B, Hasenkrug K, Kassiotis G. 2008. Retrovirus-specificity of regulatory T cells is neither present nor required in preventing retrovirus-induced bone marrow immune pathology. Immunity 29:782–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nair SR, Zelinskyy G, Schimmer S, Gerlach N, Kassiotis G, Dittmer U. 2010. Mechanisms of control of acute Friend virus infection by CD4+ T helper cells and their functional impairment by regulatory T cells. J. Gen. Virol. 91:440–451 [DOI] [PubMed] [Google Scholar]
- 13. Badovinac VP, Haring JS, Harty JT. 2007. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8(+) T cell response to infection. Immunity 26:827–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, Rudensky A, Sparwasser T. 2009. Depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J. Immunol. 183:7631–7634 [DOI] [PubMed] [Google Scholar]
- 15. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. 2007. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 204:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hasenkrug KJ, Dittmer U. 2000. The role of CD4 and CD8 T cells in recovery and protection from retroviral infection: lessons from the Friend virus model. Virology 272:244–249 [DOI] [PubMed] [Google Scholar]
- 17. Hasenkrug KJ, Dittmer U. 2007. Immune control and prevention of chronic Friend retrovirus infection. Frontiers in bioscience 12:1544–1551 [DOI] [PubMed] [Google Scholar]
- 18. Dittmer U, Brooks DM, Hasenkrug KJ. 1998. Characterization of a live-attenuated retroviral vaccine demonstrates protection via immune mechanisms. J. Virol. 72:6554–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zelinskyy G, Kraft AR, Schimmer S, Arndt T, Dittmer U. 2006. Kinetics of CD8+ effector T cell responses and induced CD4+ regulatory T cell responses during Friend retrovirus infection. Eur. J. Immunol. 36:2658–2670 [DOI] [PubMed] [Google Scholar]
- 20. He H, Messer RJ, Sakaguchi S, Yang G, Robertson SJ, Hasenkrug KJ. 2004. Reduction of retrovirus-induced immunosuppression by in vivo modulation of T cells during acute infection. J. Virol. 78:11641–11647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cobbold SP, Waldmann H. 1984. Therapeutic potential of monovalent monoclonal antibodies. Nature 308:460–462 [DOI] [PubMed] [Google Scholar]
- 22. Hasenkrug KJ, Brooks DM, Dittmer U. 1998. Critical role for CD4(+) T cells in controlling retrovirus replication and spread in persistently infected mice. J. Virol. 72:6559–6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barber DL, Wherry EJ, Ahmed R. 2003. Rapid in vivo killing by memory CD8 T cells. J. Immunol. 171:27–31 [DOI] [PubMed] [Google Scholar]
- 24. Schepers K, Toebes M, Sotthewes G, Vyth-Dreese FA, Dellemijn TA, Melief CJ, Ossendorp F, Schumacher TN. 2002. Differential kinetics of antigen-specific CD4+ and CD8+ T cell responses in the regression of retrovirus-induced sarcomas. J. Immunol. 169:3191–3199 [DOI] [PubMed] [Google Scholar]
- 25. Jones AT, Federsppiel B, Ellies LG, Williams MJ, Burgener R, Duronio V, Smith CA, Takei F, Ziltener HJ. 1994. Characterization of the activation-associated isoform of CD43 on murine T lymphocytes. J. Immunol. 153:3426–3439 [PubMed] [Google Scholar]
- 26. Onami TM, Harrington LE, Williams MA, Galvan M, Larsen CP, Pearson TC, Manjunath N, Baum LG, Pearce BD, Ahmed R. 2002. Dynamic regulation of T cell immunity by CD43. J. Immunol. 168:6022–6031 [DOI] [PubMed] [Google Scholar]
- 27. Duley AK, Ploquin MJ, Eksmond U, Ammann CG, Messer RJ, Myers L, Hasenkrug KJ, Kassiotis G. 2012. Negative impact of IFN-gamma on early host immune responses to retroviral infection. J. Immunol. 189:2521–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McDermott DS, Varga SM. 2011. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J. Immunol. 187:5568–5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Galvan M, Murali-Krishna K, Ming LL, Baum L, Ahmed R. 1998. Alterations in cell surface carbohydrates on T cells from virally infected mice can distinguish effector/memory CD8+ T cells from naive cells. J. Immunol. 161:641–648 [PubMed] [Google Scholar]
- 30. Harrington LE, Galvan M, Baum LG, Altman JD, Ahmed R. 2000. Differentiating between memory and effector CD8 T cells by altered expression of cell surface O-glycans. J. Exp. Med. 191:1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tedder TF, Steeber DA, Chen A, Engel P. 1995. The selectins: vascular adhesion molecules. FASEB J. 9:866–873 [PubMed] [Google Scholar]
- 32. Tyznik AJ, Sun JC, Bevan MJ. 2004. The CD8 population in CD4-deficient mice is heavily contaminated with MHC class II-restricted T cells. J. Exp. Med. 199:559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coers J, Gondek DC, Olive AJ, Rohlfing A, Taylor GA, Starnbach MN. 2011. Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog. 7:e1001346 doi:10.1371/journal.ppat.1001346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khounlotham M, Kim W, Peatman E, Nava P, Medina-Contreras O, Addis C, Koch S, Fournier B, Nusrat A, Denning TL, Parkos CA. 2012. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity 37:563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberts AD, Ordway DJ, Orme IM. 1993. Listeria monocytogenes infection in beta 2 microglobulin-deficient mice. Infect. Immun. 61:1113–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marshall NB, Swain SL. 2011. Cytotoxic CD4 T cells in antiviral immunity. J. Biomed. Biotechnol. 2011:954602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, Cooper DA, Rowland-Jones SL, Kelleher AD. 2002. Characterization of CD4(+) CTLs ex vivo. J. Immunol. 168:5954–5958 [DOI] [PubMed] [Google Scholar]
- 38. Zheng N, Fujiwara M, Ueno T, Oka S, Takiguchi M. 2009. Strong ability of Nef-specific CD4+ cytotoxic T cells to suppress human immunodeficiency virus type 1 (HIV-1) replication in HIV-1-infected CD4+ T cells and macrophages. J. Virol. 83:7668–7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zelinskyy G, Balkow S, Schimmer S, Werner T, Simon MM, Dittmer U. 2007. The level of friend retrovirus replication determines the cytolytic pathway of CD8+ T-cell-mediated pathogen control. J. Virol. 81:11881–11890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dittmer U, Brooks DM, Hasenkrug KJ. 1999. Protection against establishment of retroviral persistence by vaccination with a live attenuated virus. J. Virol. 73:3753–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robertson MN, Spangrude GJ, Hasenkrug K, Perry L, Nishio J, Wehrly K, Chesebro B. 1992. Role and specificity of T-cell subsets in spontaneous recovery from Friend virus-induced leukemia in mice. J. Virol. 66:3271–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zelinskyy G, Balkow S, Schimmer S, Schepers K, Simon MM, Dittmer U. 2004. Independent roles of perforin, granzymes, and Fas in the control of Friend retrovirus infection. Virology 330:365–374 [DOI] [PubMed] [Google Scholar]
- 43. Dittmer U, Brooks DM, Hasenkrug KJ. 1999. Requirement for multiple lymphocyte subsets in protection by a live attenuated vaccine against retroviral infection. Nat. Med. 5:189–193 [DOI] [PubMed] [Google Scholar]
- 44. Dittmer U, Hasenkrug KJ. 2000. Different immunological requirements for protection against acute versus persistent Friend retrovirus infections. Virology 272:177–182 [DOI] [PubMed] [Google Scholar]
- 45. Nair S, Bayer W, Ploquin MJ, Kassiotis G, Hasenkrug KJ, Dittmer U. 2011. Distinct roles of CD4+ T cell subpopulations in retroviral immunity: lessons from the Friend virus mouse model. Retrovirology 8:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kedzierska K, Crowe SM. 2001. Cytokines and HIV-1: interactions and clinical implications. Antivir. Chem. Chemother. 12:133–150 [DOI] [PubMed] [Google Scholar]
- 47. Li S, Gowans EJ, Chougnet C, Plebanski M, Dittmer U. 2008. Natural regulatory T cells and persistent viral infection. J. Virol. 82:21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt A, Oberle N, Krammer PH. 2012. Molecular mechanisms of Treg-mediated T cell suppression. Front. Immunol. 3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moreno-Fernandez ME, Rueda CM, Rusie LK, Chougnet CA. 2011. Regulatory T cells control HIV replication in activated T cells through a cAMP-dependent mechanism. Blood 117:5372–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robertson SJ, Messer RJ, Carmody AB, Hasenkrug KJ. 2006. In vitro suppression of CD8+ T cell function by Friend virus-induced regulatory T cells. J. Immunol. 176:3342–3349 [DOI] [PubMed] [Google Scholar]
- 51. von Gegerfelt A, Valentin A, Alicea C, Van Rompay KK, Marthas ML, Montefiori DC, Pavlakis GN, Felber BK. 2010. Emergence of simian immunodeficiency virus-specific cytotoxic CD4+ T cells and increased humoral responses correlate with control of rebounding viremia in CD8-depleted macaques infected with Rev-independent live-attenuated simian immunodeficiency virus. J. Immunol. 185:3348–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu L, Cantor H. 2008. Generation and regulation of CD8(+) regulatory T cells. Cell Mol. Immunol. 5:401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eshima K, Chiba S, Suzuki H, Kokubo K, Kobayashi H, Iizuka M, Iwabuchi K, Shinohara N. 2012. Ectopic expression of a T-box transcription factor, eomesodermin, renders CD4(+) Th cells cytotoxic by activating both perforin- and FasL-pathways. Immunol. Lett. 144:7–15 [DOI] [PubMed] [Google Scholar]
- 54. Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Menoret A, Mittler RS, Gordon SM, Reiner SL, Vella AT, Adler AJ. 2011. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J. Immunol. 187:3555–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zelinskyy G, Dietze KK, Hüsecken YP, Schimmer S, Nair S, Werner T, Gibbert K, Kershaw O, Gruber AD, Sparwasser T, Dittmer U. 2009. The regulatory T-cell response during acute retroviral infection is locally defined and controls the magnitude and duration of the virus-specific cytotoxic T-cell response. Blood 114:3199–3207 [DOI] [PubMed] [Google Scholar]
- 56. Iwashiro M, Peterson K, Messer RJ, Stromnes IM, Hasenkrug KJ. 2001. CD4+ T cells and gamma interferon in the long-term control of persistent Friend retrovirus infection. J. Virol. 75:52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]