

Abstract
Hepatitis C virus (HCV) genome replication is thought to occur in a membranous cellular compartment derived from the endoplasmic reticulum (ER). The molecular mechanisms by which these membrane-associated replication complexes are formed during HCV infection are only starting to be unraveled, and both viral and cellular factors contribute to their formation. In this study, we describe the discovery of nonopioid sigma-1 receptor (S1R) as a cellular factor that mediates the early steps of viral RNA replication. S1R is a cholesterol-binding protein that resides in lipid-rich areas of the ER and in mitochondrion-associated ER membranes (MAMs). Several functions have been ascribed to this ER-resident chaperone, many of which are related to Ca2+ signaling at the MAMs and lipid storage and trafficking. Downregulation of S1R expression by RNA interference (RNAi) in Huh-7 cells leads to a proportional decrease in susceptibility to HCV infection, as shown by reduced HCV RNA accumulation and intra- and extracellular infectivity in single-cycle infection experiments. Similar RNAi studies in persistently infected cells indicate that S1R expression is not rate limiting for persistent HCV RNA replication, as marked reduction in S1R in these cells does not lead to any decrease in HCV RNA or viral protein expression. However, subgenomic replicon transfection experiments indicate that S1R expression is rate limiting for HCV RNA replication without impairing primary translation. Overall, our data indicate that the initial steps of HCV infection are regulated by S1R, a key component of MAMs, suggesting that these structures could serve as platforms for initial RNA replication during HCV infection.
INTRODUCTION
It is estimated that 170 million humans are chronically infected with hepatitis C virus (HCV). Chronic HCV infection is associated with persistent liver inflammation, fibrosis, cirrhosis, and hepatocellular carcinoma (1). Recently, combination therapy, including pegylated alpha 2a interferon (IFN-α2a), ribavirin, and specific HCV protease inhibitors, has been approved for the treatment of HCV-infected patients, with high cure rates compared with pegylated IFN-α2a and ribavirin alone, the previous standard of care (2, 3). However, adverse effects and cost considerations limit the implementation of these new treatment regimens.
HCV is an enveloped RNA virus with a single 9.6-kb positive-strand RNA genome that encodes a single open reading frame of approximately 3,000 amino acids flanked by 5′ and 3′ untranslated regions (UTR) that regulate translation and replication of the viral genome. The 5′ UTR contains an internal ribosomal entry site (4) that cooperates with the 3′ UTR regions for efficient viral polyprotein translation and RNA replication. Individual viral proteins are produced as a result of the sequential proteolysis of the HCV polyprotein by cellular and viral proteases, which produce structural proteins (core, E1, and E2) that are major components of the viral particles and p7 and NS2, which mediate infectious-particle assembly in coordination with the nonstructural proteins (NS3, NS4A, NS4B, NS5A, and NS5B), which are sufficient for viral RNA replication (reviewed in references 5 and 6).
HCV enters target cells in a multistep process that involves several cellular receptors that recognize viral and cellular proteins, as well as lipid components present in the incoming infectious particles (reviewed in reference 7). Internalization of the infectious virions occurs by receptor-mediated endocytosis, followed by fusion of the viral and cellular membranes and release of the viral genome into the cytosol (reviewed in reference 8). Translation of the incoming HCV genomes (primary translation) leads to expression of the viral polyprotein and establishment of replication complexes, which produce progeny viral RNA that will be either translated or encapsidated to assemble infectious progeny virions (reviewed in reference 5). HCV assembly takes place in specialized areas of the endoplasmic reticulum (ER) in close proximity to cytoplasmic lipid droplets (9) and strongly depends, not only on the viral structural and nonstructural proteins, but also on cellular factors, including those required for the production of hepatic lipoproteins (10), some of which are incorporated into HCV virions (11–13).
A common feature of plus-strand RNA viruses is that viral replication occurs in specialized compartments derived from modified host membranes (14, 15). HCV RNA replication is thought to occur in modified ER membranes condensed in the form of a membranous web (16). It has been shown that HCV replication occurs at specific ER locations within detergent-resistant membrane subdomains rich in cholesterol and specific subsets of sphingolipids (17–20). While all the viral components of the replicase complex are directly or indirectly bound to the ER membrane, only NS4B (21) and NS5A (22) have been shown to be capable of modifying the architecture of the membranes where they are inserted, and this property is essential for efficient HCV RNA replication (21, 22). Other viral proteins promote membrane remodeling by recruiting specific lipids (20) or cellular enzymes capable of producing a specific lipid environment (23, 24). These modifications and others derived from the direct interaction of viral proteins with multiple cellular factors (25, 26) lead to the construction of specialized structures that bear the viral replicase (22).
HCV infection causes mitochondrial dysfunction and oxidative stress in patients (27) and in cell culture models (28). Expression of viral proteins triggers Ca2+ flux from the ER into the mitochondria, which leads to alterations in the mitochondrial metabolism and production of reactive oxygen species (ROS) (29). ER Ca2+ export is carried out in mitochondrion-associated ER membranes (MAMs) (30), which are areas of physical interaction that enable bidirectional communication between the two organelles and regulate physiological processes, like mitochondrial energy and lipid metabolism (reviewed in reference 31). A major player in the regulation of Ca2+ transition from the ER to the mitochondria is the intracellular nonopioid sigma-1 receptor (S1R), a chaperone that modulates Ca2+ transition and cell survival under stress conditions by interaction with inositol 1,4,5-phosphate receptors (IP3R) (32). S1R also binds cholesterol and sphingolipids, and this capacity determines its localization into detergent-resistant lipid rafts in the ER and MAMs (33) and its role in the redistribution of cellular lipids throughout intracellular compartments (34). Screening of a drug-like library for anti-HCV compounds revealed the antiviral activity of known S1R ligands (35). Thus, we set out to determine whether S1R plays a role in the HCV life cycle. In the present study, we describe the discovery of S1R as a cellular factor that mediates early steps of viral RNA replication downstream of translation of the incoming viral genomes, suggesting that HCV uses elements of the MAMs as platforms for the initial steps of HCV RNA replication.
MATERIALS AND METHODS
Viruses and cells.
JFH-1 and a cell culture-adapted JFH-1 variant, D183, viruses have been described previously (36, 37). Huh-7 (38) and Huh-7.5.1 (36) human hepatoma cells and HEK293T cells (39) were cultured in Dulbecco's modified Eagle's medium supplemented with 100 mM HEPES, nonessential amino acids (Gibco), penicillin/streptomycin (Gibco), and 10% fetal calf serum, as previously described (36). JFH-1-derived intergenotypic chimeric viruses were produced by electroporation of the in vitro-transcribed genomic RNA, as described previously (40). Influenza A virus (A/WSN/33) was obtained from Juan Ortín (Centro Nacional de Biotecnología [CNB], Madrid, Spain). Vesicular stomatitis virus (Indiana) was obtained from the ATCC (Manassas, VA).
Plasmids.
JFH-1 cDNA was obtained from Apath (Brooklyn, NY). Mission short hairpin RNA (shRNA) plasmids pLKO-Puro and those expressing shRNAs targeting SIGMAR1 (S1R) were purchased from Sigma-Aldrich (St. Louis, MO). Control shRNA plasmid expressing a sequence from Thermotoga sp. was generated by insertion of the adaptor primer pair (5′ CCGGTAATTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTTG and 5′ AATTCAAAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAATTA) immediately after the U6 promoter in the AgeI/EcoRI-digested pLKO-puro vector. A similar strategy was used for insertion of the HCV internal ribosome entry site (IRES) targeting shRNA (41) using the primer pair 5′ CCGGTACCTCAAAGAAAAACCAAATTCAAGAGATTTGGTTTTTCTTTGAGGTTTTTTG and 5′ AATTCAAAAAACCTCAAAGAAAAACCAAATCTCTTGAATTTGGTTTTTCTTTGAGGTA. Plasmids necessary for vesicular stomatitis virus G protein (VSV-G)-pseudotyped lentivirus production, pMDLg/pRRE (Addgene ID12251), pM2.G (Addgene ID12259), and pRSV-Rev (Addgene ID12253), were kindly provided by Didier Trono (Ecole Polytechnique Federale de Lausanne) (42). The genomic lentiviral plasmid sinPPTCMV-PREU3Nhe, kindly provided by Inder Verma (Salk Research Institute), was used to clone S1R cDNA produced by real-time (RT)-PCR from total RNA extracted from Huh-7 cells using the following primers: 5′ GCAGCAGGATCCATGCAGTGGGCCGTGGGC and 5′ GCAGCAGTCGACTCAAGGGTCCTGGCCAAAGAG). The product was digested and cloned into the BamHI and SalI sites of the lentiviral vector. The shRNA4-resistant S1R mutant was generated by introducing 4 silent mutations in the target sequence of S1R cDNA by PCR-mediated recombination using the mutagenic primers 5′ GTGGAGTGGGGCCCTAATACTTGGATGGTCGAGTACGGCCGG and 5′ CCGGCCGTACTCGACCATCCAAGTATTAGGGCCCCACTCCAC and the S1R primers described above. Plasmids required for murine leukemia virus-based hepatitis C virus pseudotype (HCVpp) production were kindly provided by Francois Loic Cosset (INSERM, Lyon, France) and have been previously described (43). Plasmids pFKi389Luc-EI/NS3-3′_JFH1_dg, pWPI-CE1-BSD, and pWPI-SpE2p7NS2-BSD, permitting the production of defective reporter HCV virions by transcomplementation (HCVtcp) (44), and the genotype 2a and 1a subgenomic replicons JFH-1 and Con1Luc, bearing adaptive mutations E1202G and T1280I, were kindly provided by Ralf Bartenschlager (University of Heidelberg) (45).
Antibodies.
Goat (S-18) and mouse monoclonal F5 anti-S1R antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal antibody against caveolin-2 was purchased from Epitomics (Burlingame, CA). Mouse monoclonal anti-NS3 and anti-NS5A were purchased from Biofront Technology (Tallahassee, FL). Mouse monoclonal antibody against human beta actin (ab8226) was purchased from Abcam (Cambridge, United Kingdom). Rabbit sera against NS3, NS4B, and NS5A were kindly provided by R. Bartenschlager and have been previously described (22). Human monoclonal anti-E2 (AR3A) was provided by Mansun Law (Scripps Research Institute). Antibodies against influenza virus NP and VSV N were kindly provided by Juan Ortín and Dolores Rodriguez (CNB-CSIC, Madrid, Spain), respectively. Rabbit antibodies against mitochondria (TOMM22) and endoplasmic reticulum (PDI) were purchased from Sigma (St. Louis, MO).
Lentiviral particle production and Huh-7 cell transduction.
Lentiviral particles were produced in HEK-293T cells by cotransfection of plasmids pMDLg/pRRE, pM2.G, and pRSV-Rev, together with each of the pLKO-based shRNA vectors or the genomic vector encoding S1R cDNA, as described previously (46). Supernatants were collected 36 to 48 h posttransfection, filtered through a 0.45-μm filter, and used to inoculate Huh-7 cells. For shRNA-expressing lentiviral vectors, which confer resistance to puromycin, the smallest amount of supernatant sufficient to confer resistance to puromycin (2.5 μg/ml) on 100% of the cells was used. Silencing was verified by Western blotting, typically at days 3 and 6 posttransduction for Huh-7 and subgenomic replicon cells and days 6 and 9 in persistently infected cultures, as the latter proliferate slowly compared with Huh-7 cells. Once S1R downregulation was verified, cell growth was monitored by counting control and S1R-silenced cell suspensions, plating equal cell numbers, and counting the cells again 3 and 5 days later. shRNA constructs that led to differences of more than 25% were discarded.
Western blot analysis.
Total protein samples were prepared by lysis of the cells in 1× Laemmli loading buffer. Samples were subjected to SDS-PAGE and transferred into a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with either 5% nonfat milk or 3% bovine serum albumin in PBS-0.25% Tween 20 for 1 h at room temperature and subsequently incubated with primary antibody dilutions in the same buffer for an additional hour (except for goat anti-S1R S-18, which was incubated for 5 h). The membranes were washed for 1 h in PBS-0.25% Tween 20, incubated with appropriate dilutions of the corresponding secondary antibody, washed for 1 h in PBS-0.25% Tween 20, and developed using enhanced chemiluminescence (ECL). Nonsaturated films were scanned, and relative density values were measured using Image J software (47).
Preparation of virus stocks.
JFH-1 and D183 viruses were propagated in Huh-7.5.1 clone 2 cells by inoculation at low multiplicity (multiplicity of infection [MOI] = 0.01) (11). Cell supernatants were collected at different times postinoculation (p.i.), and the titers were determined using endpoint dilution and immunofluorescence microscopy as previously described (46).
Infection experiments.
For single-cycle infection experiments, Huh-7 cells expressing different lentiviral constructs were plated at a density of 5 × 104 cells per well in 12-well plates (Corning, Lowell, MA). The next day, the cells were inoculated with D183 virus stocks at an MOI of 10 and incubated at 37°C. Five hours later, the inoculum was removed and the cells were washed twice with warm phosphate-buffered saline (PBS). Twenty-four and 48 h later, samples of the cells and the supernatants were collected for infectious-virus titer analysis (see above) and HCV RNA quantitation (by RT-quantitative PCR [qPCR]). Intra- and extracellular HCV infectivity titers were determined as previously described (46). Persistently infected cell cultures were generated by inoculation of Huh-7 cells with JFH-1 virus (MOI = 0.01). The cells were cultured until infection was propagated to nearly 100% of the population as revealed by immunofluorescence microscopy, typically at days 12 to 14 postinoculation (36).
Influenza virus and vesicular stomatitis virus infections were performed by inoculating Huh-7 cells at an MOI of 10 with a virus diluted in PBS with 5 μg/ml bovine serum albumin (BSA) for 30 min at room temperature. The inoculum was removed, and the cells were replenished with complete growth medium. Protein samples were collected at different time points and analyzed by Western blotting using specific antibodies.
Analysis of viral entry using HCVpp.
HCVpp bearing the JFH-1 envelope were produced in HEK293T cells as previously described (43). As a control, cells were transfected with the genomic plasmid but without the plasmid expressing the viral envelope (no env). Target Huh-7 cells (2 × 104 cells per well) were plated on a 96-well plate. The next day, the cells were inoculated with HCVpp and vesicular stomatitis virus pseudotype (VSVpp) preparations diluted to produce similar luciferase activities. Forty-eight hours later, the cells were lysed and the luciferase activity was detected and quantitated in a luminometer using a commercial kit (Luciferase Assay Kit; Promega, Madison, WI). Relative infection values were calculated as percentages of cells transduced with an empty vector. For most HCVpp experiments, parallel cultures were infected with HCVtcp (see below) to verify the reduced susceptibility of S1R-deficient cells to HCV infection.
In vitro transcription and HCV RNA transfection.
Plasmids containing the sequence corresponding to wild-type and replication-deficient mutant (D318N) subgenomic JFH-1 replicons bearing a luciferase reporter gene have been described previously (44, 48). After digestion with the restriction enzyme MluI or XbaI, respectively, the linearized plasmids were in vitro transcribed using a commercial kit (Megascript T7; Ambion, Paisley, United Kingdom). The resulting products were digested with DNase and precipitated with LiCl. The pelleted RNA was washed with 75% and 100% ethanol and resuspended in nuclease-free water. In vitro-transcribed RNA was transfected with Lipofectamine 2000 (Life Technologies, Carlsbad, CA) using the manufacturer's recommendations. Luciferase activities were measured in the sample using a commercial kit (Dual Luciferase Assay System; Promega, Madison, WI) at different times posttransfection.
Genotype 1b subgenomic replicon electroporation.
S1R-deficient Huh-7 cells and control cells expressing an irrelevant shRNA were electroporated with in vitro-transcribed HCV genotype 1b (Con1-ET)-luc replicon RNA as previously described (45). Twenty thousand cells per well were plated in 96-well plates and cultured for 4, 24, and 48 h before collection for luciferase activity analysis. Parallel cultures were treated with high doses (10 μM) of the specific NS5B polymerase inhibitor 2′-C-methyladenosine (2mAde) (BOC Sciences, Shirley, NY) to determine the luciferase activity derived from primary translation of the transfected viral genome in the absence of replication. Luciferase activity values in the presence of 2mAde were subtracted at different time points, and replication was calculated as the ratio of the luciferase values at the given time point and the values 4 h postelectroporation, which derive exclusively from primary translation (45). Comparable transfection efficiencies were monitored by RT-qPCR of HCV RNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA on samples collected from the different cell lines 4 h postelectroporation.
HCVtcp production and infection.
Infectious replication-defective HCV virions (HCVtcp) were produced by trans-encapsidation using using Huh-7.5.1 clone 2 cells and the methodology described previously (44). HCVtcp are infectious HCV particles that bear genomes lacking the coding region proteins required for viral assembly. Thus, these viruses can only deliver subgenomic replicons into target cells and cannot produce infectious particles, unless they infect cells expressing in trans the viral proteins required for virus assembly. Packaging cells were produced by transducing Huh-7.5.1 clone 2 cells with lentiviral vectors bearing genomic constructs that express HCV genotype 2a (J6CF) core-E1 and E2-NS2 regions. This cell line was subsequently electroporated with an in vitro-transcribed dicistronic subgenomic JFH-1 replicon bearing the luciferase gene. Supernatants of the electroporated packaging cells were collected after 24, 48, and 72 h and used to inoculate naive Huh-7 cells to determine the presence of infectious HCVtcp using luciferase as the readout. For infection experiments, target cells (2 × 104 cells per well) were plated onto a 96-well plate. The next day, HCVtcp preparations (typically 200 μl) were used to inoculate the cells. Luciferase activity was measured 24, 48, or 72 h postinfection as described above.
RNA extraction and real-time quantitative PCR.
RNA was extracted using the GTC extraction method (49). Reverse transcription of total RNA was performed using random hexamers and the Multiscribe Reverse Transcription Kit (Life Technologies, Carlsbad, CA). cDNA was quantified by qPCR using previously described primers for HCV and GAPDH (50) and Power SYBR Green Mix (Life Technologies, Carlsbad, CA).
Confocal immunofluorescence microscopy.
Huh-7 cells were grown on glass coverslips and infected at a high MOI (MOI = 10) with D183 virus. Sixteen, 48, and 72 h postinfection, the cells were fixed for 20 min at −20°C with methanol, washed twice with PBS, and incubated with a buffer containing 2% BSA in PBS for 1 h. Antibodies were diluted in 0.1% BSA in PBS and incubated with the cells for 1 h, after which the cells were washed with PBS and subsequently incubated with a 1:500 dilution of a goat anti-mouse antibody conjugated to Alexa 488 or Alexa 594 (Invitrogen, Carlsbad, CA). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole). The cells were washed with PBS and mounted on glass slides with Prolong (Invitrogen, Carlsbad, CA).
Confocal microscopy was performed with a Leica TCS SP5 laser scanning system (Leica Microsystems). Images of 1,024 by 1,024 pixels at eight bit gray scale depth were acquired sequentially every 0.13 to 0.3 μm through a 63×/1.40-numerical-aperture (NA) immersion oil lens, employing LAS AF v 2.6.0 software (Leica Microsystems). Image analysis was performed using Image J (47) and the Jacop software plug-in (51) to determine Pearson's and Mander's coefficients of at least 15 individual cells (8 to 10 stacks). The statistical significance of the colocalization was analyzed for NS3/S1R, NS4B/S1R, and NS5A/S1R using the Costes et al. randomization (52) and the methods of Van Steensel et al. (53), which confirmed that colocalization occurred in a nonrandom manner. Colocalized pixel maps were generated using the “colocalization threshold” tool of the Image J package.
Detergent-resistant membrane flotation assay.
The protocol of the detergent-resistant membrane flotation assay is similar to that described by Hayashi and Fujimoto (54). HCV-infected and naive cells (2 × 106) were lysed by adding 1 ml of TNE buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 2 mM EDTA) containing 0.5% Triton X-114 and protease inhibitors (Complete; Roche, Basel, Switzerland). The lysates were incubated for 2 h on ice before applying them on top of a 10 to 40% discontinuous sucrose-TNE gradient. Samples were spun for 16 h at 120,000 rpm. Fourteen fractions were collected from the top and analyzed by SDS-PAGE and Western blotting for the presence of S1R, NS3, caveolin-2, and beta-actin.
RESULTS
Sigma-1 receptor is required for efficient HCV infection in cell culture.
Since we have previously identified putative S1R ligands (haloperidol, rimcazole, lofepramine, methyl paroxetine, prochlorperazine, fluphenazine, cyproheptadine, trifluoperazine, azelastine, desloratadine, salmeterol, carvedilol, amiodarone, and benproperine) (35, 55) as HCV infection inhibitors, we postulated that S1R might be involved in HCV infection. In order to investigate the potential role of S1R in the HCV life cycle, we generated S1R-deficient cell populations by transducing human hepatoma (Huh-7) cells with lentiviral vectors expressing four different shRNAs against S1R mRNA or an irrelevant sequence, as described previously (46). Analysis of total cell extracts by Western blotting at day 6 posttransduction revealed reduced expression of the S1R to different extents in the S1R shRNA-transduced cells compared with the control population (Fig. 1A). It is noteworthy that S1R protein silencing was maximal 5 to 6 days after lentiviral transduction, probably due to its relatively long half-life (approximately 48 h) (32). Once S1R downregulation was verified, the different S1R-deficient cell lines were subsequently inoculated at a high multiplicity of infection (MOI = 10) with a cell-culture-adapted HCV (D183v) (37), and their susceptibilities to infection were evaluated by measuring progeny virus production, as well as intracellular viral RNA accumulation in a single cycle of infection. Analysis of intra- and extracellular infectivity 24 and 48 h postinfection revealed a reduction in the progeny viral titer that was proportional to that of the intracellular S1R protein expression in all cases (Fig. 1B and C), suggesting that intracellular S1R levels are rate limiting for HCV infection. Intracellular HCV RNA levels measured by RT-qPCR at 5 h p.i. were comparable in control and S1R-deficient cells, indicating that the inoculum size and virus adsorption to the target cells were similar in all cell lines (Fig. 1D). Remarkably, at 24 and 48 h p.i., S1R-deficient cells displayed reduced intracellular HCV RNA content compared to the controls (Fig. 1D), in parallel with their infectivity titers (Fig. 1B and C) and intracellular S1R expression levels (Fig. 1A), suggesting that a step in HCV infection that leads to accumulation of intracellular HCV RNA is dependent on the sigma-1 receptor. Similar results were obtained with defective HCV virions produced by trans-encapsidation (HCVtcp) (44), which can produce only a single round of infection (Fig. 2A) (44). Using this system, extended kinetic studies revealed that luciferase levels in S1R-deficient cells were proportionally reduced 24 and 48 h postinfection but that they were partially (S1R-2) or completely (S1R-4) restored to control levels 72 h postinfection (Fig. 2B), supporting the notion that early steps of HCV infection leading to HCV RNA accumulation are limited by S1R expression. For the sake of simplicity, subsequent experiments aimed at determining which step of the infection is S1R dependent were carried out using shRNA4 (S1R-4) and -2 (S1R-2), because they provided consistent and prolonged S1R downregulation without any overt impact on cell viability (data not shown).
Fig 1.

Cellular levels of S1R are limiting for HCV infection. Huh-7 cells were transduced with lentiviral vectors expressing an irrelevant sequence or shRNAs targeting S1R mRNA and infected at an MOI of 10 with D183v. (A) Western blot analysis performed on total cell extracts showing reduced S1R expression in shRNA-expressing cell lines at the time of inoculation (6 days posttransduction) and a loading control (β-actin). (B and C) Intracellular (B) and extracellular (C) infectivity titers (FFU/ml) in HCV-infected cells expressed as averages and standard deviations (n = 3). (D) Normalized HCV RNA levels were determined at 5, 24, and 48 h postinfection by RT-qPCR as described in Materials and Methods and are expressed as averages and standard deviations (n = 3). Normalized S1R expression in panels B and C was quantified from panel A. The statistical significance of the differences with the control data set was determined using Student's t test (*, P < 0.05; **, P < 0.01).
Fig 2.
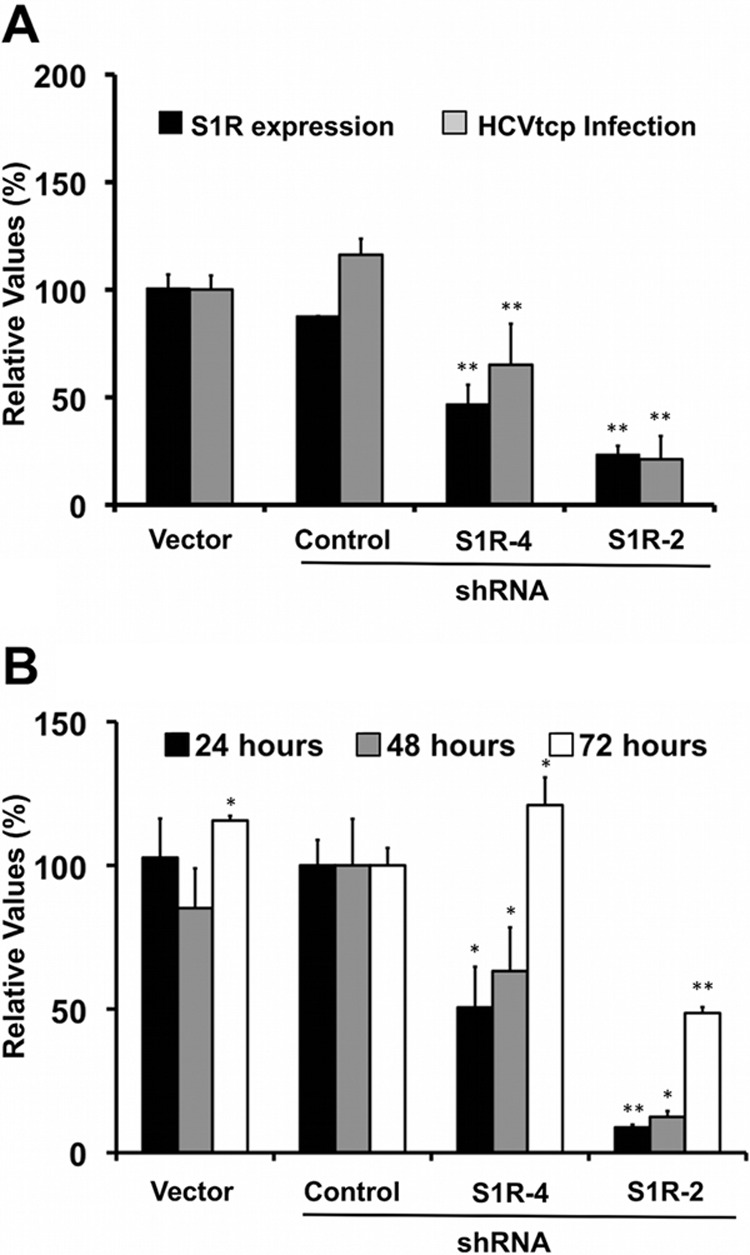
Cellular levels of S1R are limiting for HCVtcp infection. Huh-7 cells were transduced with lentiviral vectors expressing irrelevant sequences or shRNAs targeting S1R mRNA and infected with HCVtcp. (A) Twenty-four hours postinfection, cell lysates were used to measure luciferase activity and S1R expression. The data are shown as average percentages of the empty vector and standard deviations (n = 9). (B) Extended kinetics of HCVtcp infection at 24, 48, and 72 h postinfection. The data are shown as average percentages of the control vector and standard deviations for a representative experiment (n = 3). The statistical significance of the differences with the control data set was determined using Student's t test (*, P < 0.05; **, P < 0.01).
In order to demonstrate that S1R expression downregulation is responsible for the reduced susceptibility to HCV infection, an S1R cDNA carrying silent mutations conferring resistance to shRNA4 was overexpressed in shRNA4-expressing cells and Huh-7 cells using lentiviral vectors. As expected, nontransduced S1R-deficient cells displayed reduced susceptibility to HCV infection compared with cells expressing an irrelevant shRNA or the parental Huh-7 cells, as shown by the reduced extracellular infectivity titers after single-cycle HCV infection (MOI = 10) (Fig. 3). Remarkably, increasing doses of S1R cDNA rescued susceptibility to HCV infection to normal levels but marginally increased the infectivity titers in parental Huh-7 cells (Fig. 3). These results confirm that reduced S1R expression is indeed responsible for the reduced susceptibility of S1R-deficient cells to HCV infection. In order to extend our observations to other HCV genotypes, we infected S1R-deficient cells, expressing shRNA2 and -4 with JFH-1 and chimeric recombinant viruses bearing the structural region from genotypes 1a (H77) and 1b (Con1) (40). Infection with JFH-1 and the chimeric viruses paralleled S1R expression levels (data not shown), indicating that HCV S1R dependence is not restricted to JFH-1. In order to determine if S1R-deficient cells were refractory to infection by other RNA viruses, we performed single-cycle infection studies (MOI = 10) of S1R-deficient Huh-7 cells with unrelated viruses, such as influenza A virus and VSV. As shown in Fig. 4, analysis of viral nucleoprotein (VSV N and influenza virus NP) accumulation at different times postinfection did not reveal any significant difference between control and S1R-deficient cells, indicating that S1R expression is not limiting for influenza A virus or VSV infection.
Fig 3.
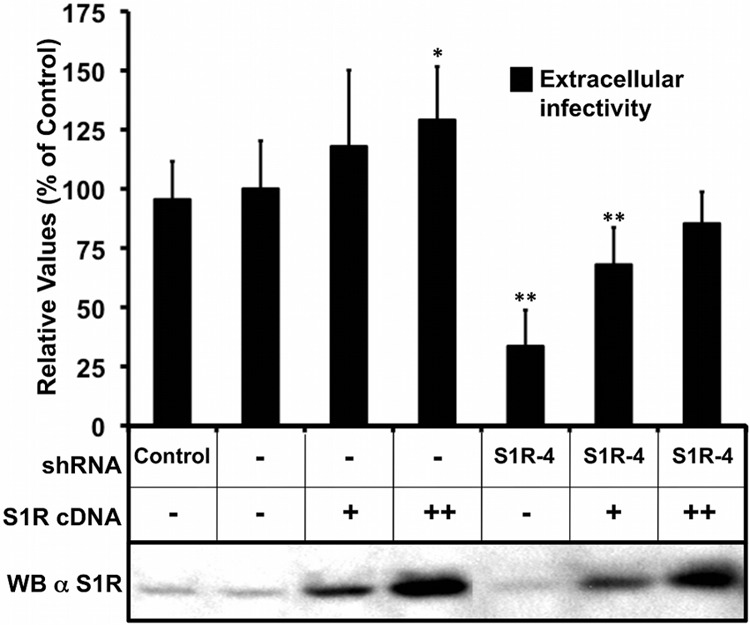
Expression of S1R cDNA restores susceptibility to infection of S1R-deficient cells. Mock-treated Huh7 cells and S1R shRNA4-expressing cells were transduced with a lentiviral vector expressing an shRNA-resistant S1R cDNA. The cells were subsequently infected at an MOI of 10 with D183v. Extracellular infectivity titers were determined at 24 h postinfection. The data are shown as averages and standard deviations of two independent experiments performed in triplicate (n = 6) and are represented as percentages of the mock-treated cells. The statistical significance of the differences with the control data set was determined using Student's t test (*, P < 0.05; **, P < 0.01). WB, Western blot; α S1R, anti-S1R; +, transduction with lentiviral vector; ++, transduction with higher dose of vector; −, no transduction.
Fig 4.
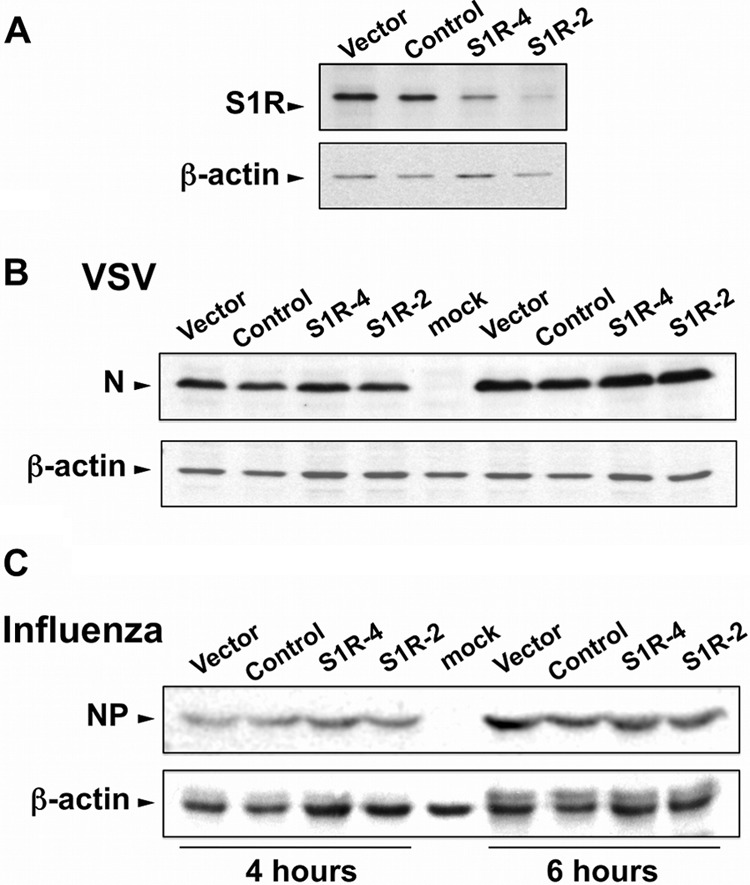
S1R is not rate limiting for other RNA virus infections. Huh-7 cells were transduced with lentiviral vectors expressing irrelevant sequences or shRNAs targeting S1R mRNA. (A) Western blot analysis against S1R and a loading control (β-actin). (B and C) The cells were subsequently infected (MOI = 10) with VSV (B) or influenza virus (C). Protein samples were collected at 4 and 6 h postinfection and analyzed by Western blotting against VSV N (B) or influenza virus NP (C).
Persistent HCV infection is not dependent on S1R expression levels.
In order to further characterize the role of S1R in HCV infection, we set out to determine the impact of S1R expression downregulation in persistently infected cells. In order to generate persistently infected cultures, Huh-7 cells were inoculated at low multiplicity with JFH-1 virus and cultured for 2 weeks, at which time the infection had spread to virtually all the cells (36). Persistently infected cell cultures are characterized by constant viral RNA replication, protein expression, and progeny virus production (37). In order to investigate the role of S1R during persistent HCV infection, lentiviral vectors expressing a control shRNA and two shRNAs directed against the S1R mRNA were used to transduce persistently infected cultures. Maximum silencing was observed typically at day 9 posttransduction in these cells (data not shown). Figure 5 shows that S1R expression was effectively reduced by approximately 60% (S1R-4) and 80% (S1R-2) compared with the controls (Fig. 5A and B). Despite this notable reduction in S1R expression, these cells expressed NS3 levels comparable to those of the control cells (Fig. 5A and B). These results suggest that intracellular S1R levels are not rate limiting for steady-state HCV RNA replication. In order to test this hypothesis, we performed similar silencing experiments using two additional controls: a potent and specific polymerase inhibitor (2mAde) and an shRNA targeting the HCV IRES, to be used as positive controls of agents that actively perturb HCV replication. As expected, treatment of persistently infected cells with 10 μM 2mAde resulted in a profound reduction of intracellular HCV RNA levels and undetectable extracellular-infectivity titers (Fig. 5C and D, 2mAde). In addition, transduction of persistently infected cells with a lentiviral vector expressing an shRNA targeting the viral RNA resulted in a marked parallel reduction of intracellular HCV RNA levels and extracellular-infectivity titers (Fig. 5C and D, HCV shRNA). However, S1R-deficient cells displayed intracellular HCV RNA levels that were comparable to those in the control cells, albeit slightly higher (Fig. 5C, S1R-4 and -2 shRNAs), as were the extracellular-infectivity titers (Fig. 5D, S1R-4 and -2 shRNAs). These results reinforce the notion that intracellular S1R levels are not rate limiting for steady-state HCV RNA replication or infectious-particle assembly and secretion. These results also rule out the possibility that the deficient HCV infection observed in S1R-deficient cells is due to a general, nonspecific effect on cell viability and proliferation, as persistent HCV RNA replication is very sensitive to these changes (56).
Fig 5.

S1R downregulation does not interfere with persistent HCV infection. Persistently infected Huh-7 cells were untreated (Mock) or transduced with lentiviral vectors expressing an irrelevant shRNA (Control), an HCV-targeting shRNA (shRNA-HCV), or S1R-targeting (S1R-4 and S1R-2) shRNAs. Parallel cultures were treated with a specific HCV polymerase inhibitor (2mAde; 10 μM). Samples of the cells and supernatants were collected at day 9 posttransduction. (A) Western blotting against HCV NS3 protein (NS3), S1R, and loading control (β-actin) from one representative experiment of two. (B) Quantitation of S1R and NS3 protein expression normalized to the loading control. (C) Normalized intracellular HCV RNA levels determined by RT-qPCR. (D) Extracellular HCV infectivity titers. The data in panels C and D are shown as averages and standard deviations for two independent experiments performed in triplicate (n = 6) and are represented as percentages of the mock-treated cells. The statistical significance of the differences with the control data set was determined using Student's t test (**, P < 0.01).
Identical results were obtained in Huh-7 cells harboring a subgenomic JFH-1 replicon (Fig. 6). Marked S1R downregulation by shRNA-induced silencing did not significantly affect viral protein or RNA accumulation (Fig. 6). As expected, lentivirus-mediated HCV shRNA expression and treatment with the polymerase inhibitor 2mAde led to a marked reduction in HCV NS3 protein abundance, as determined by Western blotting (Fig. 6A), as well as to a reduction in HCV RNA accumulation compared with the control cells measured by RT-qPCR (Fig. 6B). These results, together with those obtained in persistently infected cells (Fig. 5) suggest that HCV infection can persist in S1R-deficient cells and indicate that S1R is likely rate limiting at an early step of the infection that leads to viral RNA accumulation.
Fig 6.
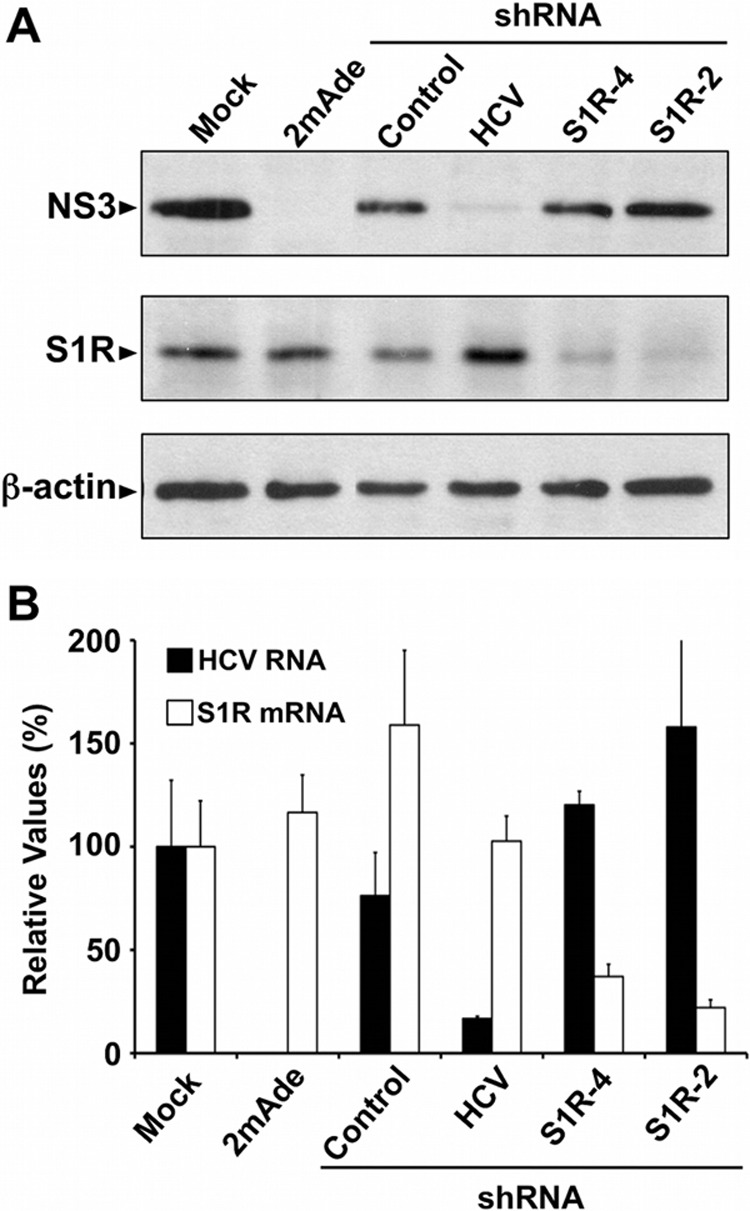
S1R downregulation does not interfere with steady-state subgenomic JFH-1 replication. Huh-7 cells bearing a subgenomic JFH-1 replicon were transduced with an empty vector or with lentiviral vectors expressing an irrelevant shRNA (Control), an HCV-targeting shRNA (HCV), or S1R-targeting (S1R-4 and S1R-2) shRNAs. Parallel cultures were treated with a specific HCV polymerase inhibitor (2mAde; 10 μM). Samples of the cells were collected at day 6 posttransduction. (A) Western blotting against HCV NS3 protein (NS3), S1R, and loading control (β-actin). (B) Normalized intracellular HCV RNA and S1R mRNA levels determined by RT-qPCR. The data are shown as averages and standard deviations of triplicate (n = 3) samples from a representative experiment and are represented as percentages of the cells treated with an empty vector.
S1R downregulation does not affect viral entry.
The results obtained in single-cycle infection experiments indicate that HCV RNA accumulation is reduced in S1R-deficient cells (Fig. 1C). Since HCV RNA replication per se does not appear to be dependent on S1R expression (Fig. 5 and 6), it is possible that the reduced HCV RNA accumulation observed after infection of S1R-deficient cells is due to deficient viral entry. Many aspects of HCV entry have been studied using HCV-pseudotyped retroviral vectors (HCVpp) (43). Therefore, we used retroviral particles pseudotyped with the JFH-1 envelope glycoproteins (HCVpp) or with the vesicular stomatitis virus G protein as a control to test their abilities to infect S1R-deficient cells. HCVpp and VSVpp infection efficiencies were comparable in all cell lines despite the differential expression of S1R (Fig. 7), suggesting that S1R is not rate limiting for viral entry and that either primary translation (translation of the incoming genomes) or establishment of HCV RNA replication is dependent on S1R expression. Thus, we set out to determine if initial steps of viral infection downstream of viral entry are affected by reduced S1R expression.
Fig 7.
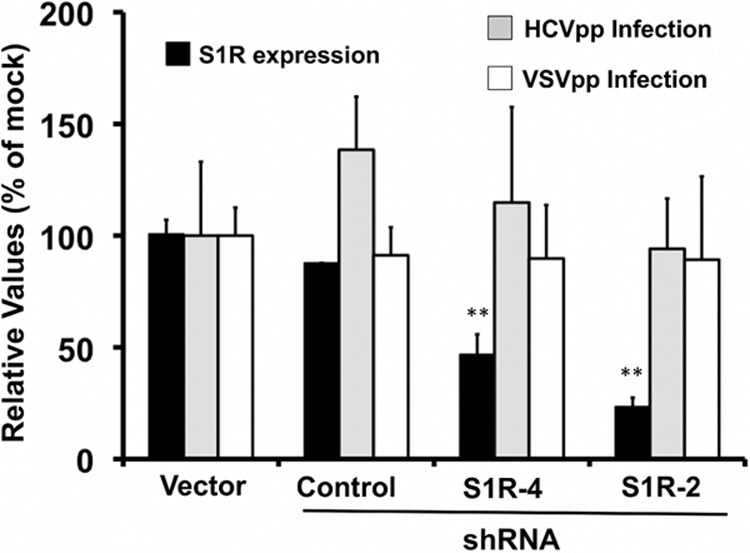
HCV entry is not altered by S1R downregulation. Huh-7 cells were transduced with lentiviral vectors expressing an empty vector (Vector), an irrelevant shRNA (Control) or S1R-targeting (shRNA4 and shRNA2) shRNAs. Once downregulation was verified by Western blotting (black bars), the cells were inoculated with pseudotyped retroviruses bearing JFH-1 (HCVpp) or VSV (VSVpp) envelope glycoproteins. Luciferase activity levels were determined at 48 h in total cell extracts. Normalized S1R expression levels were determined by Western blotting against S1R and β-actin. The data are shown as averages and standard deviations (n = 12) for four independent experiments performed in triplicate and are represented as percentages of cells transduced with an empty vector. The statistical significance of the differences with the control data set was determined using Student's t test (**, P < 0.01).
S1R protein expression downregulation impairs initiation of HCV RNA replication without affecting primary translation.
In order to study primary translation and establishment of viral replication complexes, we bypassed the entry step by transfection of an in vitro-transcribed HCV subgenomic replicon (genotype 2a; JFH-1) RNA bearing a luciferase reporter into S1R-deficient and control Huh-7 cells. Luciferase accumulation was measured at different times posttransfection to determine the ability of S1R-deficient cells to support HCV RNA translation and replication. In order to specifically study primary translation, a point mutation was introduced in the catalytic site of the NS5B polymerase (57). Thus, all the signal detected 5 h posttransfection reflects the translation efficiency in the different cell lines. Translations of the replication-deficient subgenomic replicon were comparable in control and S1R cells (Fig. 8), except for the unexpected increase in S1R shRNA2-expressing cells and the expected decrease in the cells expressing the shRNA targeting viral RNA (Fig. 8). Overall, these results suggest that S1R expression levels are not limiting for the translation of incoming HCV genomes.
Fig 8.
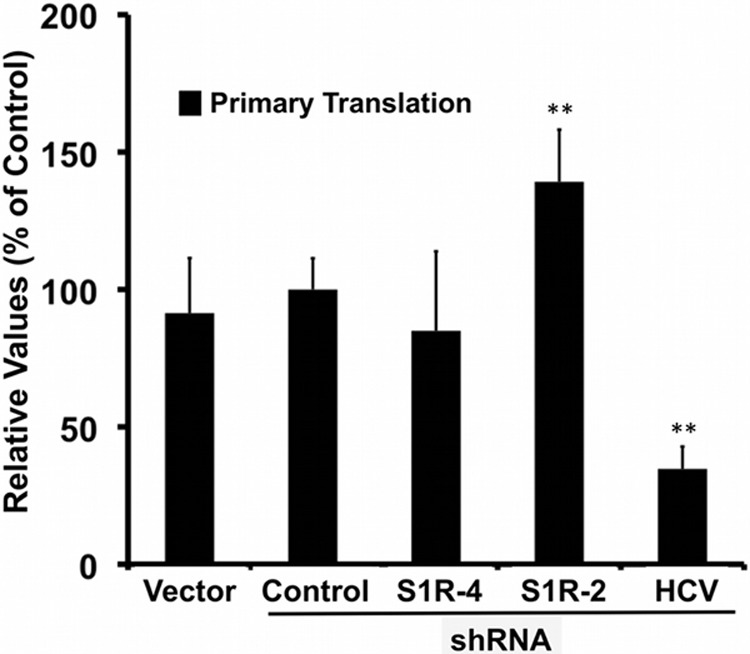
S1R expression levels are not limiting for primary translation. Huh-7 cells were transduced with lentiviral vectors expressing an irrelevant shRNA (Control), an HCV-targeting shRNA (HCV), or S1R-targeting (S1R-4 and S1R-2) shRNAs. S1R-deficient and control cells were transfected with a subgenomic JFH-1 replicon bearing a luciferase reporter gene and a polymerase activity-inactivating (GND) mutation. Luciferase activity was measured 5 h posttransfection. The data are shown as averages and standard deviations (n = 6) for two independent experiments performed in triplicate and are represented as percentages of cells transduced with the control vector. The statistical significance of the differences with the control data set was determined using Student's t test (**, P < 0.01).
In order to study HCV RNA replication, we transfected a replication-competent subgenomic JFH-1 replicon into control and S1R-deficient cell lines. As expected from the results in Fig. 8, we found no significant differences in luciferase accumulation at 5 h posttransfection (Fig. 9), since early luciferase accumulation derives predominantly from primary translation (48, 58). When measured at 24 and 48 h, luciferase levels were reduced in the two S1R-deficient cell lines (Fig. 9A and B). It is noteworthy that the degree to which viral replication is reduced parallels the reduction of S1R expression in the target cells, with cells expressing shRNA S1R-4 displaying a moderate decrease and those expressing shRNA S1R-2 a more pronounced reduction both in S1R expression and in luciferase accumulation. Similar results were obtained after electroporation of a genotype 1b (Con1) subgenomic replicon RNA bearing a luciferase reporter into S1R-deficient Huh-7 cells (Fig. 10), indicating that HCV S1R dependence is not restricted to the JFH-1 replicase. Overall, these results suggest that the reduced accumulation of HCV RNA in S1R-deficient cells in single-cycle infection experiments (Fig. 1) is due to a defect in the establishment of HCV RNA replication during HCV infection, downstream of primary translation.
Fig 9.

S1R expression levels are limiting for initiation of HCV RNA replication. S1R-deficient cells were transfected with a subgenomic JFH-1 replicon bearing a luciferase reporter gene. Luciferase activity was measured at the indicated time points. S1R expression was quantitated by Western blotting. (A) Representative transfection experiment. Data are shown as averages and standard deviations (n = 3) of the measured light units. (B) Averages and standard deviations for three independent transfection experiments performed in triplicate (n = 9) represented as percentages of the cells transduced with an empty vector. (C and D) Similar experiments were conducted in Huh-7.5.1. cells (n = 6). The statistical significance of the differences with the control data set was determined using Student's t test (**, P < 0.01).
Fig 10.
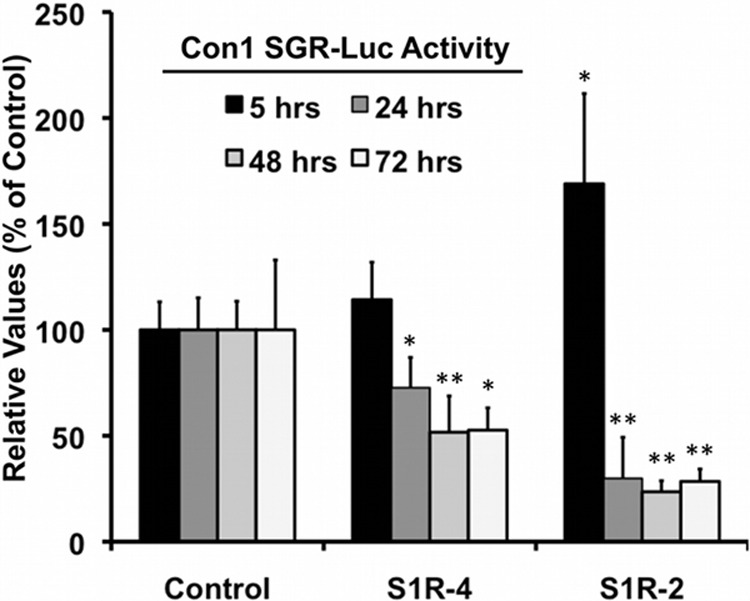
S1R expression is rate limiting for early genotype 1b HCV RNA replication. Control and S1R-deficient Huh-7 cells were prepared by lentiviral transduction of Huh-7 cells. S1R-deficient and control cells were electroporated with an in vitro-transcribed dicistronic HCV subgenomic replicon RNA from genotype 1b (Con1) bearing a luciferase reporter gene. Samples were collected at the indicated time points, and relative replication was determined as described in Materials and Methods. The data are shown as averages and standard deviations for two independent experiments performed in triplicate (n = 6). The statistical significance of the differences with the control data set was determined using Student's t test (*, P < 0.05; **, P < 0.01).
Similar experiments were carried out in Huh-7.5.1 cells, which have been shown to be highly permissive for HCV infection and RNA replication (36). S1R-deficient Huh-7.5.1 cells show comparable primary translation, measured by luciferase accumulation 5 h posttransfection, compared with control cells (Fig. 9C and D). As expected, Huh-7.5.1. cells display more rapid and efficient HCV RNA replication than Huh-7 cells, with a clear increase in luciferase accumulation 24 h after transfection in the control cells. During this initial phase, S1R expression levels appear to be rate limiting for HCV RNA replication (Fig. 9C and D), while once luciferase accumulation stalls after 24 h in control Huh-7.5.1 cells, replication in S1R-deficient cells appears to reach levels that are comparable to those in control cells at 48 h, supporting the notion that S1R expression is rate limiting early after primary translation but is dispensable once replication reaches steady-state levels, as observed in persistently infected cells (Fig. 5).
S1R colocalizes with nonstructural HCV proteins.
Since S1R appears to modulate the establishment of viral replication, we set out to determine whether S1R and components of the viral replicase colocalize during HCV infection. Huh-7 cells were infected at high multiplicity (MOI = 10) with D183 virus and incubated for 16, 48, and 72 h, after which the cells were fixed and processed for immunofluorescence microscopy. In mock-infected Huh-7 cells, immunofluorescence staining against S1R reveals two distinct patterns of subcellular distribution. Most cells display discrete cytoplasmic punctae that are juxtaposed to mitochondria when stained with antibodies against TOMM22, an outer mitochondrial membrane protein (Fig. 11). The rest of the cells display a diffuse cytoplasmic pattern that colocalizes with endoplasmic reticulum markers, such as protein disulfide isomerase (PDI) (Fig. 11). This dual distribution has been described previously (59) and is due to the fact that S1R shuttles between MAMs and peripheral ER areas, even under normal culture conditions. In HCV-infected cells, viral antigens were only reliably detectable 48 h postinfection using this fixation procedure (Fig. 11B), at which time more than 70% (72 ± 14; n = 430) of the infected cells display a diffuse perinuclear pattern (32, 33). In order to determine whether S1R and HCV replication complexes colocalize, infected cells were stained with antibodies directed against components of the viral replicase, NS3, NS4B, and NS5A (22). Interestingly, our data indicate that NS3, NS4B, and NS5A display partial perinuclear colocalization with endogenous S1R 48 h postinfection (Fig. 12A). The calculated averages (± standard deviations; n > 25) of the Mander's overlap coefficient for S1R is 60 ± 18 with NS3, 69 ± 18 with NS4B, and 65 ± 14 with NS5A 48 h postinfection. These results suggest that S1R is relocalized to ER-derived membranes that are also targeted by HCV NS proteins during early steps of viral infection (Fig. 11B). Interestingly, later during infection (72 h), more than 60% of the infected cells (63 ± 10; n = 320) display discrete cytoplasmic punctae that do not clearly colocalize with the bulk NS protein signal (Fig. 12B), suggesting that a fraction of S1R shuttles back to the original punctate pattern and that the general S1R perinuclear relocalization observed at 48 h is transient. Overall, these results suggest that S1R is recruited to areas of the ER where HCV proteins accumulate at early stages of viral infection.
Fig 11.

S1R is associated with MAMs. (A) Confocal immunofluorescence microscopy of S1R in Huh-7 cells displaying two different subcellular patterns. (B and C) Higher-magnification images showing S1R (green) colocalization with mitochondria (TOMM22; red) in discrete cytoplasmic punctae (B) (the boxed area in the merged image is shown enlarged in the inset) and with ER markers (PDI; red) in a diffuse cytoplasmic staining (C). Nuclei were stained with DAPI and are shown in blue.
Fig 12.

S1R colocalizes with HCV NS proteins. Confocal immunofluorescence microscopy of HCV-infected Huh-7 cells (MOI = 10) at 48 (A) and 72 (B) h postinfection. S1R is shown in green, and NS3, NS4B, and NS5A are shown in red. Representative images from three independent experiments are shown. Colocalization masks (overlap) are shown in grayscale.
In HCV replicons, most of the HCV RNA and HCV replicase activity is associated with detergent-resistant, cholesterol- and sphingolipid-rich intracellular membranes that display a characteristic low density in isopycnic sucrose gradients (60). Since S1R is also associated with lipid-rich detergent-resistant ER membrane (DRM) structures that are particularly resistant to Triton X-114 treatment, we set out to study whether S1R-containing and viral replicase DRMs have similar biophysical properties after treatment with this detergent. JFH-1-infected and control Huh-7 cells were lysed on ice in a buffer containing 0.5% Triton X-114. Clear cell lysates were subjected to flotation using discontinuous sucrose (10 to 40%) gradients as described previously (54). Gradient fractions were then subjected to Western blot analysis for S1R, NS3, NS5A, and beta-actin. In addition, we used caveolin-2, a lipid raft-associated protein, as a marker of DRMs, because it had been previously reported to cofractionate with isolated HCV replicase complexes (60) and S1R (54). Western blot analysis of the sucrose gradient fractions indicated that caveolin-2 floated in the low-density fractions (Fig. 13A and B, fractions 1 to 5), as expected. In contrast, beta-actin, which was used as a marker of detergent-soluble proteins, migrated to higher-density fractions (Fig. 13A and B, fractions 9 to 11). S1R-specific Western blot analysis in mock- and HCV-infected cells showed that most S1R protein is associated with DRMs in fractions 1 to 5 (Fig. 13A and B), cofractionating with caveolin-2, as previously reported (54), suggesting that in HCV-infected cells, S1R remains associated with DRMs. In lysates from HCV-infected cells, most NS3 and NS5A is associated with DRMs and cofractionates with S1R and caveolin-2 (Fig. 13B). These results suggest that, during HCV infection, a substantial fraction of the NS proteins target intracellular Triton X-114-resistant membranes, similar to previously characterized S1R-containing DRMs (54). These results reinforce the notion that S1R and components of the HCV replicase target similar cholesterol- and sphingolipid-rich membrane environments during HCV infection, where S1R likely exerts its proviral functions at the onset of the infection.
Fig 13.
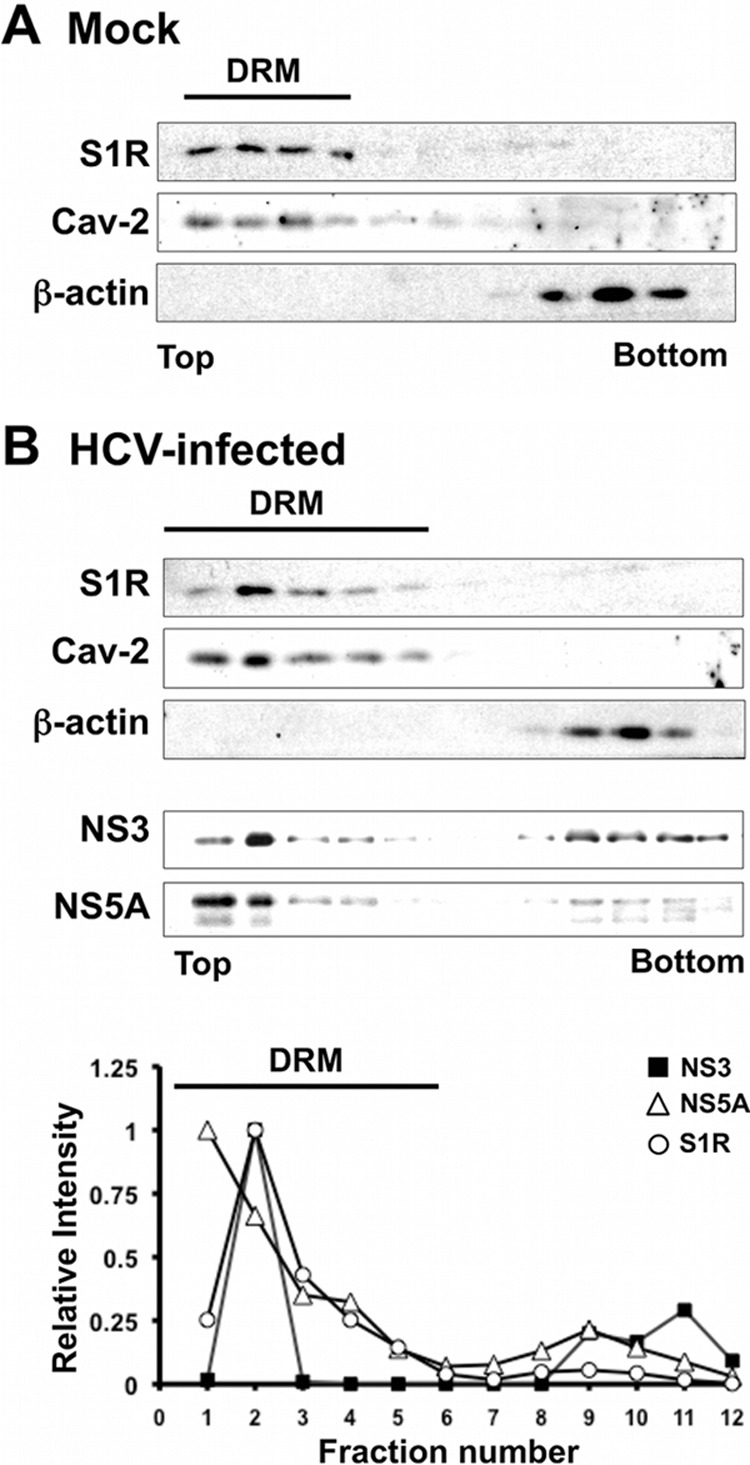
S1R cofractionates with HCV NS proteins in detergent-resistant membranes. Cell lysates were produced by incubating HCV-infected Huh-7 cells in TNE-0.5% Triton X-114. The cleared lysates were placed onto a discontinuous 10 to 40% sucrose-TNE gradient and centrifuged at 120,000 × g for 16 h. Gradient fractions were collected from the top and analyzed by Western blotting for the presence of NS3, NS5A, S1R, β-actin, and caveolin-2. (A) Western blot analysis of gradient fractions in mock-infected cells. (B) (Top) Western blot analysis of the gradient fractions in HCV-infected cells. (Bottom) Quantitation of S1R, NS3, and NS5A in the top blot. The values are shown as relative abundance per fraction relative to the maximum signal. Detergent-resistant membrane fractions containing caveolin-2 are marked as DRM.
DISCUSSION
The data presented in this study suggest a role for S1R in the hepatitis C virus life cycle. S1R silencing led to a proportional reduction of HCV infection efficiency, as shown by the reduced progeny infectious-virus production in single-cycle infection experiments (Fig. 1). This reduction is due to the decreased accumulation of viral RNA observed after infection of S1R-deficient cells (Fig. 1). Mechanistic studies using surrogate models for HCV entry and RNA replication indicate that S1R is rate limiting for the initial steps of HCV RNA replication immediately after translation of the incoming genomes, since viral entry (Fig. 7) and primary translation (Fig. 8) appear to be normal in S1R-deficient cells. These results could be demonstrated for subgenomic replicons from genotype 2a (Fig. 9) and 1b (Fig. 10), indicating that S1R dependence is not restricted to the genotype 2a JFH-1 strain. Interestingly, downregulation of S1R expression does not affect steady-state HCV RNA replication in persistently infected cells (Fig. 5) or in a subgenomic replicon model (Fig. 6). These results suggest that downregulation of S1R expression does not affect HCV RNA replication per se in persistent infections, but rather, plays a role in establishing HCV RNA replication early after infection. This hypothesis is similar to that postulated for autophagy-related ATG4B and Beclin genes, whose expression was shown to be rate limiting for primary translation but not for translation of viral genomes derived from viral replication (48). Alternatively, these results could indicate that the downregulation achieved by shRNA expression is not sufficient to reveal a role for these genes in persistent infections. Taken together, these results suggest that the virus takes advantage of infection-induced cellular stress-related genes, e.g., S1R- and autophagy-related genes, to deploy a cellular program that favors its early colonization of the target cell. Since early steps in the formation of replicase complexes in infected cells occur in an environment where viral protein expression levels are low, it is conceivable that expression of cellular genes that initiate the formation of HCV replicase complexes are limiting in this situation, but not once the infection has taken over the cell metabolism to set in a proviral state. Interestingly, we have observed a clear relocalization of S1R to perinuclear areas of the infected cells, where viral NS proteins start accumulating during HCV infection (Fig. 12A). It is noteworthy that such transient S1R relocalization has been described during ER stress and has been proposed to be a mechanism by which S1R contributes to the cellular response to stress by chaperoning ER membrane proteins (32, 59). One of the key cellular substrates for S1R chaperone activity is the calcium channel formed by the IP3R, which modulate Ca2+ transition from the ER to the mitochondria at the MAMs, promoting cell survival under stress conditions (32). HCV proteins have been shown to induce Ca2+ transition to the mitochondria, leading to mitochondrial dysfunction and ROS production (reviewed in reference 61). It is conceivable, although we have no evidence for S1R binding to HCV proteins during HCV infection, that accumulation of HCV proteins in the ER triggers S1R relocalization, as shown in Fig. 12, to mitigate ER stress. Interestingly, it has been proposed that HCV protein-induced changes in mitochondrial metabolism could lead to an increase in the production of certain lipid species during HCV infection (28), as well as to increased local ATP production required for efficient HCV replication (28, 61, 62, 63). Thus, it is possible that S1R downregulation leads to a different initial cellular response to infection-induced stress caused by a differential regulation of ER-mitochondria Ca2+ transition, and this situation might not satisfy the bioenergetic and metabolic needs for efficient initial HCV RNA replication.
Many studies have shown a strong dependence of HCV infection on cellular lipid metabolism, at the levels of viral entry, HCV RNA replication, and virion assembly (reviewed in reference 10). It is noteworthy that S1R-deficient primary hippocampal cultures display significant changes in the levels of expression of cellular genes related to cholesterol and fatty acid metabolism (64). Moreover, a recent study shows that steroid biosynthesis, which depends on the immediate availability of cholesterol at the outer mitochondrial membrane, is defective in S1R-deficient MA-10 cells (65). These data, together with the recognized ability of S1R to modulate cholesterol homeostasis in different cellular compartments (66) and the ability of S1R to bind cholesterol and sphingolipids (33), raise the possibility that S1R provides these and other lipids to the nascent replication complexes through nonvesicular transport to specific areas of the ER where viral proteins accumulate, a notion that is supported by our confocal microscopy data (Fig. 12A). In fact, it has been recently shown that efficient viral RNA replication requires the recruitment of specific subsets of phospho- and sphingolipids to HCV replication complexes (20, 23). During HCV infection, S1R and NS proteins appear to colocalize in specialized cholesterol- and sphingolipid-rich membranes that are resistant to Triton-X114 treatment (Fig. 12 and 13). Moreover, it has been shown that a fraction of HCV NS proteins are copurified with S1R in MAMs from Huh-7 cells (67) and that the viral replicase colocalizes with both ER and mitochondrial markers (22), supporting the notion that a fraction of the viral replication complexes is associated with MAMs.
It has been shown that NS3 exerts an important proviral function in MAMs, where it specifically cleaves mitochondrial antiviral signaling proteins (MAVS) in order to blunt the innate host response at very early stages of HCV infection (67). However, S1R dependence appears to be independent of the RIG-I-mediated innate immune response, since early HCV replication is also deficient in S1R-deficient Huh-7.5.1 cells (Fig. 9C and D), which lack a functional RIG-I (68). These precedents and our data suggest that MAMs could constitute an ideal platform for HCV to initiate active replication while inhibiting innate immune responses at the onset of the infection. Initiating HCV RNA replication at MAMs would be advantageous for the virus, since viral RNA and protein would initially accumulate in an environment rich in enzymes involved in cholesterol, triglyceride, fatty acid, and phospholipid metabolism (69), all of which are intimately involved in HCV infection (10). As viral RNA replication commences, small amounts of NS3/4A could blunt innate immune responses locally soon after replication is initiated by targeting MAVS at the MAM, where it is functionally relevant for HCV (67). This local inactivation of MAVS could pave the road for efficient viral replication throughout the ER.
The finding that S1R plays a role in HCV infection is of particular interest because, in addition to endogenous ligands, S1R activity can be modulated in vivo by a large number of drug-like ligands (70), some of which have been proven to be useful in the treatment of pain (71) and in experimental models for pathologies of the central nervous system (55). Although we have previously shown that putative S1R ligands inhibit HCV infection, most of these compounds selectively and efficiently inhibit viral entry (35), a process that is not altered by S1R downregulation (Fig. 7), probably due to their lysosomotropic properties as weak amines, a structural feature that appears to be important for synthetic ligands to bind S1R (72). Thus, in view of the evidence presented in this study and the reported ability of putative S1R ligands to modulate cellular processes in an S1R-independent manner (73), we suspect that the antiviral activity of the putative S1R ligands that prompted this study is not mediated by S1R. Nevertheless, our findings support the notion that modulation of S1R by synthetic S1R ligands capable of specifically inhibiting S1R activity in cell culture and in the liver may be therapeutically effective.
ACKNOWLEDGMENTS
We are indebted to Takaji Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for providing JFH-1 cDNA, Francois Loic Cosset for providing the HCVpp system, Ralf Bartenschlager (University of Heidelberg) for providing JFH-1 and Con1 replicons and the rabbit sera against NS proteins, Didier Trono (University of Lausanne) and Inder Verma (Salk Institute) for lentiviral plasmids, Mansun Law (Scripps Research Institute) for providing antibodies against E2, and Sylvia Gutierrez from CNB Core Microscopy for technical assistance and advice. We are thankful to Juan Ortin, Amelia Nieto, Urtzi Garaigorta, and Stefan Wieland for critically reading the manuscript.
L.M. is funded by a JAE-Pre fellowship from Consejo Superior de Investigaciones Científicas. This work was supported by a Plan Nacional De Investigación Científica, Desarrollo e Innovación Tecnológica grant from the Spanish Ministry of Science and Innovation (SAF2010-19270), a Marie Curie Career Integration grant (PCIG-9-GA-2011-293664) from the European Union 7th Framework Programme for Research (P.G.), and NIH grant R01-AI079043 (F.V.C.).
Footnotes
Published ahead of print 27 March 2013
REFERENCES
- 1. Guidotti LG, Chisari FV. 2006. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 1:23–61 [DOI] [PubMed] [Google Scholar]
- 2. Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, DiNubile MJ, Sniukiene V, Brass CA, Albrecht JK, Bronowicki JP. 2011. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364:1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci P, Flisiak R, George J, Rizzetto M, Shouval D, Sola R, Terg RA, Yoshida EM, Adda N, Bengtsson L, Sankoh AJ, Kieffer TL, George S, Kauffman RS, Zeuzem S. 2011. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364:2405–2416 [DOI] [PubMed] [Google Scholar]
- 4. Otto GA, Puglisi JD. 2004. The pathway of HCV IRES-mediated translation initiation. Cell 119:369–380 [DOI] [PubMed] [Google Scholar]
- 5. Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5:453–463 [DOI] [PubMed] [Google Scholar]
- 6. Ploss A, Dubuisson J. 2012. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut 61(Suppl. 1):i25–i35 [DOI] [PubMed] [Google Scholar]
- 7. Ploss A, Evans MJ. 2012. Hepatitis C virus host cell entry. Curr. Opin. Virol. 2:14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dubuisson J, Helle F, Cocquerel L. 2008. Early steps of the hepatitis C virus life cycle. Cell Microbiol. 10:821–827 [DOI] [PubMed] [Google Scholar]
- 9. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097 [DOI] [PubMed] [Google Scholar]
- 10. Alvisi G, Madan V, Bartenschlager R. 2011. Hepatitis C virus and host cell lipids: an intimate connection. RNA Biol. 8:258–269 [DOI] [PubMed] [Google Scholar]
- 11. Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. 2010. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 84:10999–11009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shimizu Y, Hishiki T, Ujino S, Sugiyama K, Funami K, Shimotohno K. 2011. Lipoprotein component associated with hepatitis C virus is essential for virus infectivity. Curr. Opin. Virol. 1:19–26 [DOI] [PubMed] [Google Scholar]
- 13. Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286:3018–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. den Boon JA, Diaz A, Ahlquist P. 2010. Cytoplasmic viral replication complexes. Cell Host Microbe 8:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moradpour D, Gosert R, Egger D, Penin F, Blum HE, Bienz K. 2003. Membrane association of hepatitis C virus nonstructural proteins and identification of the membrane alteration that harbors the viral replication complex. Antiviral Res. 60:103–109 [DOI] [PubMed] [Google Scholar]
- 17. Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461 [DOI] [PubMed] [Google Scholar]
- 18. Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301–50308 [DOI] [PubMed] [Google Scholar]
- 19. El-Hage N, Luo G. 2003. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 84:2761–2769 [DOI] [PubMed] [Google Scholar]
- 20. Hirata Y, Ikeda K, Sudoh M, Tokunaga Y, Suzuki A, Weng L, Ohta M, Tobita Y, Okano K, Ozeki K, Kawasaki K, Tsukuda T, Katsume A, Aoki Y, Umehara T, Sekiguchi S, Toyoda T, Shimotohno K, Soga T, Nishijima M, Taguchi R, Kohara M. 2012. Self-enhancement of hepatitis C virus replication by promotion of specific sphingolipid biosynthesis. PLoS Pathog. 8:e1002860 doi:10.1371/journal.ppat.1002860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 85:6963–6976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056 doi:10.1371/journal.ppat.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85:8870–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shulla A, Randall G. 2012. Hepatitis C virus-host interactions, replication, and viral assembly. Curr. Opin. Virol. 2:725–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moriishi K, Matsuura Y. 2007. Host factors involved in the replication of hepatitis C virus. Rev. Med. Virol. 17:343–354 [DOI] [PubMed] [Google Scholar]
- 27. Barbaro G, Di Lorenzo G, Asti A, Ribersani M, Belloni G, Grisorio B, Filice G, Barbarini G. 1999. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: ultrastructural and biochemical findings. Am. J. Gastroenterol. 94:2198–2205 [DOI] [PubMed] [Google Scholar]
- 28. Quarato G, Scrima R, Agriesti F, Moradpour D, Capitanio N, Piccoli C. 2013. Targeting mitochondria in the infection strategy of the hepatitis C virus. Int. J. Biochem. Cell Biol. 45:156–166 [DOI] [PubMed] [Google Scholar]
- 29. Piccoli C, Scrima R, Quarato G, D'Aprile A, Ripoli M, Lecce L, Boffoli D, Moradpour D, Capitanio N. 2007. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 46:58–65 [DOI] [PubMed] [Google Scholar]
- 30. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766 [DOI] [PubMed] [Google Scholar]
- 31. de Brito OM, Scorrano L. 2010. An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 29:2715–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayashi T, Su TP. 2007. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610 [DOI] [PubMed] [Google Scholar]
- 33. Hayashi T, Su TP. 2003. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 306:718–725 [DOI] [PubMed] [Google Scholar]
- 34. Hayashi T, Su TP. 2004. Sigma-1 receptors at galactosylceramide-enriched lipid microdomains regulate oligodendrocyte differentiation. Proc. Natl. Acad. Sci. U. S. A. 101:14949–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gastaminza P, Whitten-Bauer C, Chisari FV. 2010. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. U. S. A. 107:291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhong J, Gastaminza P, Chung J, Stamataki Z, Isogawa M, Cheng G, McKeating JA, Chisari FV. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J. Virol. 80:11082–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858–3863 [PubMed] [Google Scholar]
- 39. Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–74 [DOI] [PubMed] [Google Scholar]
- 40. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kronke J, Kittler R, Buchholz F, Windisch MP, Pietschmann T, Bartenschlager R, Frese M. 2004. Alternative approaches for efficient inhibition of hepatitis C virus RNA replication by small interfering RNAs. J. Virol. 78:3436–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T. 2008. Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J. Virol. 82:7034–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. 2003. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J. Virol. 77:3007–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82:2120–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159 [DOI] [PubMed] [Google Scholar]
- 50. Garaigorta U, Chisari FV. 2009. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 6:513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232 [DOI] [PubMed] [Google Scholar]
- 52. Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. 2004. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 86:3993–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Steensel B, van Binnendijk EP, Hornsby CD, van der Voort HT, Krozowski ZS, de Kloet ER, van Driel R. 1996. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J. Cell Sci. 109:787–792 [DOI] [PubMed] [Google Scholar]
- 54. Hayashi T, Fujimoto M. 2010. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 77:517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hayashi T, Su TP. 2004. Sigma-1 receptor ligands: potential in the treatment of neuropsychiatric disorders. CNS Drugs 18:269–284 [DOI] [PubMed] [Google Scholar]
- 56. Pietschmann T, Lohmann V, Rutter G, Kurpanek K, Bartenschlager R. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gastaminza P, Pitram SM, Dreux M, Krasnova LB, Whitten-Bauer C, Dong J, Chung J, Fokin VV, Sharpless KB, Chisari FV. 2011. Antiviral stilbene 1,2-diamines prevent initiation of hepatitis C viral RNA replication at the outset of infection. J. Virol. 85:5513–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hayashi T, Su TP. 2003. Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J. Pharmacol. Exp. Ther. 306:726–733 [DOI] [PubMed] [Google Scholar]
- 60. Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77:4160–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Piccoli C, Quarato G, Ripoli M, D'Aprile A, Scrima R, Cela O, Boffoli D, Moradpour D, Capitanio N. 2009. HCV infection induces mitochondrial bioenergetic unbalance: causes and effects. Biochim. Biophys. Acta 1787:539–546 [DOI] [PubMed] [Google Scholar]
- 62. Ando T, Imamura H, Suzuki R, Aizaki H, Watanabe T, Wakita T, Suzuki T. 2012. Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 8:e1002561 doi:10.1371/journal.ppat.1002561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KA, Proll SC, McDermott JE, Gritsenko MA, Zhang Q, Zhao R, Metz TO, Camp DG, II, Waters KM, Smith RD, Rice CM, Katze MG. 2010. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 6:e1000719 doi:10.1371/journal.ppat.1000719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tsai SY, Rothman RK, Su TP. 2012. Insights into the Sigma-1 receptor chaperone's cellular functions: a microarray report. Synapse 66:42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marriott KS, Prasad M, Thapliyal V, Bose HS. 2012. Sigma-1 receptor at the mitochondrial associated ER-membrane is responsible for mitochondrial metabolic regulation. J. Pharmacol. Exp. Ther. 343:578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hayashi T, Su TP. 2010. Cholesterol at the endoplasmic reticulum: roles of the sigma-1 receptor chaperone and implications thereof in human diseases. Subcell. Biochem. 51:381–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr 2011. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 108:14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schon EA, Area-Gomez E. 2012. Mitochondria-associated ER membranes in Alzheimer disease. Mol. Cell Neurosci. 24:S1044–7431(12)00127-3. doi:10.1016/j.mcn.2012.07.011 [DOI] [PubMed] [Google Scholar]
- 70. Hayashi T, Su T. 2005. The sigma receptor: evolution of the concept in neuropsychopharmacology. Curr. Neuropharmacol. 3:267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Diaz JL, Zamanillo D, Corbera J, Baeyens JM, Maldonado R, Pericas MA, Vela JM, Torrens A. 2009. Selective sigma-1 (sigma1) receptor antagonists: emerging target for the treatment of neuropathic pain. Cent. Nerv. Syst. Agents Med. Chem. 9:172–183 [DOI] [PubMed] [Google Scholar]
- 72. Ablordeppey SY, Fischer JB, Glennon RA. 2000. Is a nitrogen atom an important pharmacophoric element in sigma ligand binding? Bioorg. Med. Chem. 8:2105–2111 [DOI] [PubMed] [Google Scholar]
- 73. Amer MS, McKeown L, Tumova S, Liu R, Seymour VA, Wilson LA, Naylor J, Reenhalgh K, Hou B, Majeed Y, Turner P, Sedo A, O'Regan DJ, Li J, Bon RS, Porter KE, Beech DJ. 20132 Inhibition of endothelial cell Ca(2+) entry and transient receptor potential channels by Sigma-1 receptor ligands. Br. J. Pharmacol. 168:1445–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]