

Abstract
We incorporated a previously identified mutation that reduces the fidelity of the DNA polymerase into a human adenovirus vector. Using this mutator vector, we demonstrate rapid selection of resistance to a neutralizing anti-hexon monoclonal antibody due to a G434D mutation in hexon that precludes antibody binding. Since mutator adenoviruses can accumulate compound mutations that are unattainable using traditional random mutagenesis techniques, this approach will be valuable to the study of antivirals and host factor interactions.
TEXT
Selection for viruses resistant to antivirals, either biological or chemical, can be a powerful method to elucidate antiviral mechanisms; moreover, inhibitor studies often provide insight into the biology of virus infection. Adenoviruses (AdVs) are nonenveloped, icosahedral viruses that package a single copy of a linear, nonsegmented, 36-kb double-stranded DNA (dsDNA) genome. Unlike RNA viruses with low-fidelity RNA-dependent RNA polymerases, DNA-dependent DNA polymerases like that of AdV have high fidelity (μg = mutation rate per genome per generation of ≈0.003) (1). Thus, the ability of AdVs to evade antivirals through mutation is limited, and there are few reports of AdVs selected in vitro for antiviral resistance.
Recently, mutants of the human AdV (HAdV) polymerase were described with reduced replication fidelity using pol-deleted viruses that were trans-complemented in pol-expressing cell lines (2). Based on this study, we generated a mutator vector using homologous recombination in bacteria (recombineering), as previously described (3), to introduce one of the polymerase mutations, F421Y, into an E1/E3-deleted HAdV-5 genome encoding enhanced green fluorescent protein (eGFP) in place of E1. This residue is predicted to be involved in proofreading and increases the error rate of the adenovirus polymerase to a μg value of ≥0.4 (2). Upon characterization, we found that concentrated stocks of purified HAdV-5.polF421Y from two independent preparations (7.2 × 1012 and 1.0 × 1013 particles/ml) were equivalent in titer to that of a stock of the parent vector (1.4 × 1013 particles/ml). In addition, the mutation did not adversely affect the particle/infectious unit (p/iu) ratios of these preparations of HAdV-5.polF421Y (p/iu = 76 and 40, respectively) compared to that of the parent vector (p/iu = 241). The HAdV-5.polF421Y vector grew with wild-type kinetics in a one-step growth curve in 293β5 cells, although more progeny were produced at later time points (Fig. 1). Thus, the F421Y mutation in the HAdV-5 polymerase is compatible with viral replication and permits high-titer virus production with no deleterious effect on infectivity.
Fig 1.
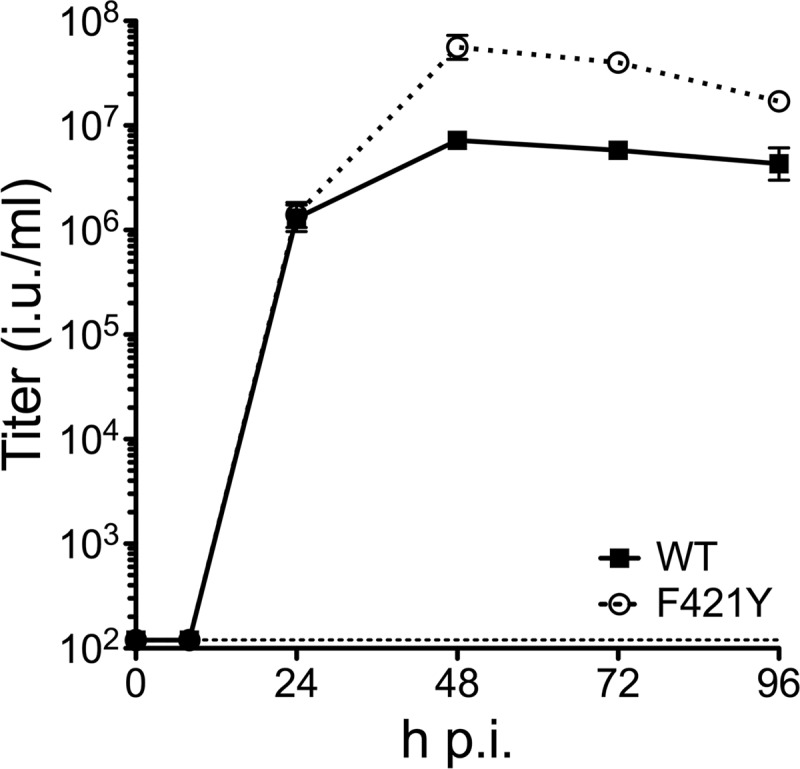
One-step growth kinetics of HAdV-5.polF421Y is equivalent to that of the wild type. 293β5 cells were infected with wild-type Ad5.eGFP or HAdV-5.polF421Y. Infectious titers of combined cell lysate and supernatant were determined at the indicated times postinfection by flow cytometry for eGFP expression of serially infected 293β5 cells. Data are the average of two titer determinations from one time course experiment ± standard deviations (SD). The limit of detection is indicated by the horizontal dotted line.
To determine whether the increased mutation rate of the HAdV-5.polF421Y vector accelerated viral evolution, we imposed selective pressure with the neutralizing anti-hexon monoclonal antibody 9C12 (4–6). We serially passaged HAdV-5.polF421Y for 10 rounds in 293β5 cells to generate diversity in the starting population, purified the virus, incubated it with or without 4.3 μg/ml 9C12 in Dulbecco's modified Eagle's medium (DMEM)-10% fetal bovine serum (FBS) for 45 min at room temperature (RT), and then added the resulting mixture to 293β5 cells. Under these conditions, the multiplicity of infection (MOI) was ∼2 in the absence of 9C12. The cultures were monitored for the development of complete cytopathic effect, which typically developed 3 to 4 days postinfection (p.i.) in the antibody-treated well. For subsequent rounds of selection, we infected at a similar MOI using cell lysates rather than purifying the virus at each step.
We then compared the 9C12 50% inhibitory concentrations (IC50s) for seven rounds of selection (Fig. 2A and data not shown). For this assay, cell lysate from each passage was used at a dilution that produced ∼80% infection in the absence of 9C12. Lysates were incubated with increasing concentrations of 9C12 for 45 min at RT and then added to 293β5 cells in black-wall, clear-bottom 96-well plates. Total eGFP expression in each well was quantified 24 h p.i. using a Typhoon scanner (GE Healthcare). The IC50 for the first round of selection was 168 ng/ml (95% confidence interval [CI] = 135 to 210 ng/ml). By the fourth round, this had increased slightly to 256 ng/ml (95% CI = 115 to 568 ng/ml). We observed no neutralization of virus in the lysates from the fifth and subsequent rounds of selection. In contrast, HAdV-5.polF421Y cultured in parallel in the absence of 9C12 selection remained sensitive to 9C12 after seven rounds of passage (data not shown). We then repeated this process to select 9C12-resistant viruses using the wild-type vector, which had also been multiply passaged on 293β5 cells prior to selection. Through 10 rounds of selection, we have not observed resistance. Instead, the IC50 was 159 ng/ml (95% CI = 113 to 224 ng/ml) at round 6 and remained at 158 ng/ml (95% CI = 80 to 310 ng/ml) at round 8 (Fig. 2B). Thus, the F421Y mutation accelerated HAdV evolution under neutralizing antibody (NAb) selection.
Fig 2.

HAdV-5.polF421Y evolved resistance to 9C12 neutralizing antibody. The 9C12 IC50s were determined on 293β5 cells of lysates from cells infected with HAdV-5.polF421Y after the indicated rounds of 9C12 selection (A); purified HAdV-5.polF421Y, a plaque-purified HAdV-5.polF421Y bearing the G434D hexon mutation (9R-01), and wild-type vector engineered with the G434D mutation (HAdV-5.hexonG434D) (B); or lysates from cells infected with wild-type Ad5.eGFP after the indicated rounds of 9C12 selection (C). Data are the mean percentages of control infection from three independent experiments ± SD.
To identify mutations that confer resistance to 9C12, the hexon genes were amplified by PCR and sequenced from 20 individual plaques from the seventh round of 9C12 selection of the HAdV-5.polF421Y virus. In each case, we also confirmed that the F421Y mutation in the polymerase was retained. We identified the same single nonsynonymous base change in hexon genes (G20142A when numbering the HAdV-5 genome using NCBI ID AC_000008) in all of the clones, and the change results in a G434D amino acid change in hypervariable region 7. To exclude the possibility that unidentified mutations in other parts of the genome were required for 9C12 resistance, we engineered the hexon G434D mutation into the wild-type vector by recombineering. We then compared the 9C12 IC50s of preparations purified through two sequential CsCl gradients of HAdV-5.polF421Y, a plaque-purified HAdV-5.polF421Y bearing the G434D hexon mutation (9R-01), and the wild-type vector engineered with the G434D mutation (HAdV-5.hexonG434D) (Fig. 2C). The IC50 for HAdV-5.polF421Y was 31 ng/ml (95% CI = 25 to 38 ng/ml). The lower IC50 for purified HAdV-5.polF421Y compared to lysates likely reflects removal of excess hexon from the lysates during purification. In contrast to HAdV-5.polF421Y, no reduction in 9R-01 or HAdV-5.hexonG434D infection was observed at 9C12 concentrations up to 4.3 μg/ml. Thus, a single mutation in hexon at position G434 is sufficient to confer resistance to 9C12 neutralization.
9C12-dependent neutralization is dependent upon binding of the NAb to the viral capsid (4, 6). To determine whether the G434D mutation alters 9C12 binding, we compared the ability of 9C12 to immunoprecipitate purified HAdV-5.polF421Y, 9R-01, and HAdV-5.hexonG434D. We incubated 1 μg of each virus with 2.6 μg 9C12 in FAK buffer (6). 9C12 was precipitated with protein G beads, and bound proteins were probed for penton base by immunoblot using rabbit antiserum (a kind gift of Glen Nemerow, The Scripps Research Institute). 9C12 efficiently immunoprecipitated HAdV-5.polF421Y but not 9R-01 or HAdV-5.hexonG434D (Fig. 3A). Thus, the G434D mutation in hexon disrupts 9C12 binding to the capsid.
Fig 3.
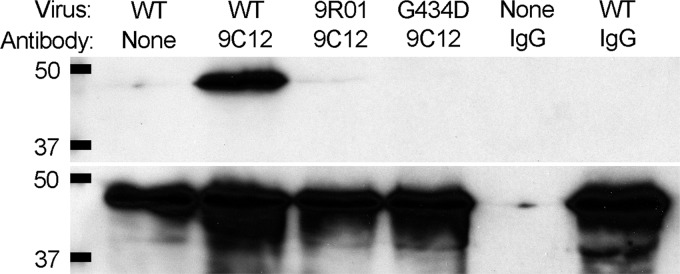
The hexon G434D mutation abrogates 9C12 binding. Wild-type, 9R-01, or HAdV-5.hexonG434D (G434D) viruses were immunoprecipitated with equal amounts of 9C12 or control IgG. Bound viruses (top) or input samples (bottom) were probed for penton base. Molecular mass markers are indicated in kDa on the left.
By engineering a mutation in the DNA polymerase of HAdV-5, we have shown that the virus is able to rapidly evolve resistance to a potent anti-hexon NAb. In contrast, under these experimental conditions, wild-type HAdV was unable to evolve resistance even after prolonged passage under selective pressure. This is particularly striking because antibody resistance was conferred by a single nonsynonymous base change in hexon, underscoring the high fidelity of the HAdV polymerase. In a similar effort to evolve HAdV-5 isolates resistant to the polymerase inhibitor cidofovir, Gordon et al. reported that 16 to 22 passages in increasing concentrations of cidofovir were required to generate resistant viruses with a 6-fold increase in IC50 (7, 8). As in our study, a single nonsynonymous base change was sufficient to confer resistance. Thus, the rapid isolation of 9C12-resistant virus after 5 passages of HAdV-5.polF421Y is contrasted by the prolonged passaging required to generate resistance to two classes of antivirals using HAdV-5 or HAdV-5-based vectors bearing a wild-type polymerase.
AdV elicits a potent humoral immune response, primarily directed against hexon, although antibodies against fiber and penton base can also be neutralizing (9). Reported efforts to identify epitopes rely primarily on the construction of chimeric viruses (10–12), the interrogation of overlapping peptide libraries or capsid protein fragments (13–15), phage display (16, 17), or structural studies of HAdV/antibody complexes (6, 18). These approaches are complex and, in the case of peptide libraries, limited to linear epitopes. Our approach using directed evolution of resistant viruses is a technically less demanding alternative method to identify neutralizing antibody epitopes and could be similarly applied to the identification of binding sites for other antivirals.
In other viral systems, biological functions of viral gene products and targets of antivirals have been identified or verified by studies of naturally arising or intentionally evolved resistant viruses. Our study supports a straightforward mechanism by which the G434D mutation abrogates 9C12 binding to the capsid, which is required for neutralization. In the original publication describing 9C12, the epitope was believed to involve the hexon DE1 and FG1 loops and to be centered on residues 178 and 179 in the DE1 loop (6). In a more recent high-resolution structure of HAdV-5 hexon (Protein Data Bank [PDB] 1VSZ [19]), G434 is located in the FG1 loop and protrudes into the space occupied by the 9C12 density in the cryoelectron microscopy (cryoEM) structure of the 9C12/HAdV-5 complex (Fig. 4) (6). Thus, G434 and additional residues in the DE1 loop likely comprise the 9C12 epitope.
Fig 4.
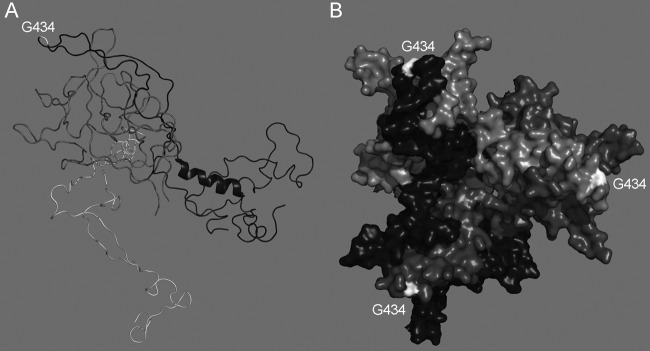
G434 is located on a surface-exposed hexon loop. (A) Ribbon diagram of the DE1 (dark gray), FG1 (black), and FG2 (light gray) loops of HAdV-5 hexon (PDB 1VSZ), with G434 indicated in the FG1 loop. The hexon is orientated for direct comparison with the cryoEM structure of 9C12 in complex with hexon (6). (B) Top view of a surface-rendered model of the HAdV-5 hexon trimer, illustrating the surface exposure of G434. Created with PyMOL.
As HAdV infection causes significant morbidity affecting multiple organ systems and can be fatal in immunocompromised patients, specific antivirals are needed. pol is highly conserved among AdVs from diverse species, and the sequences surrounding F421Y are almost completely conserved (data not shown). Therefore, creating mutator AdVs may be a general approach to elucidate mechanisms of novel antivirals by studying the phenotypes of resistant viruses. As previously discussed, this approach may also be used to develop gene therapy vectors with altered tropisms or to select mutants with expanded host ranges (2).
ACKNOWLEDGMENT
This work was supported by Royalty Research Fund grant A70250 from the University of Washington.
Footnotes
Published ahead of print 13 March 2013
REFERENCES
- 1. Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uil TG, Vellinga J, de Vrij J, van den Hengel SK, Rabelink MJ, Cramer SJ, Eekels JJ, Ariyurek Y, van Galen M, Hoeben RC. 2011. Directed adenovirus evolution using engineered mutator viral polymerases. Nucleic Acids Res. 39:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith JG, Silvestry M, Lindert S, Lu W, Nemerow GR, Stewart PL. 2010. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog. 6:e1000959 doi:10.1371/journal.ppat.1000959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McEwan WA, Hauler F, Williams CR, Bidgood SR, Mallery DL, Crowther RA, James LC. 2012. Regulation of virus neutralization and the persistent fraction by TRIM21. J. Virol. 86:8482–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith JG, Cassany A, Gerace L, Ralston R, Nemerow GR. 2008. Neutralizing antibody blocks adenovirus infection by arresting microtubule-dependent cytoplasmic transport. J. Virol. 82:6492–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Varghese R, Mikyas Y, Stewart PL, Ralston R. 2004. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J. Virol. 78:12320–12332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gordon YJ, Araullo-Cruz TP, Johnson YF, Romanowski EG, Kinchington PR. 1996. Isolation of human adenovirus type 5 variants resistant to the antiviral cidofovir. Invest. Ophthalmol. Vis. Sci. 37:2774–2778 [PubMed] [Google Scholar]
- 8. Kinchington PR, Araullo-Cruz T, Vergnes JP, Yates K, Gordon YJ. 2002. Sequence changes in the human adenovirus type 5 DNA polymerase associated with resistance to the broad spectrum antiviral cidofovir. Antiviral Res. 56:73–84 [DOI] [PubMed] [Google Scholar]
- 9. Sumida SM, Truitt DM, Lemckert AA, Vogels R, Custers JH, Addo MM, Lockman S, Peter T, Peyerl FW, Kishko MG, Jackson SS, Gorgone DA, Lifton MA, Essex M, Walker BD, Goudsmit J, Havenga MJ, Barouch DH. 2005. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 174:7179–7185 [DOI] [PubMed] [Google Scholar]
- 10. Gall J, Kass-Eisler A, Leinwand L, Falck-Pedersen E. 1996. Adenovirus type 5 and 7 capsid chimera: fiber replacement alters receptor tropism without affecting primary immune neutralization epitopes. J. Virol. 70:2116–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gall JG, Crystal RG, Falck-Pedersen E. 1998. Construction and characterization of hexon-chimeric adenoviruses: specification of adenovirus serotype. J. Virol. 72:10260–10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roy S, Clawson DS, Calcedo R, Lebherz C, Sanmiguel J, Wu D, Wilson JM. 2005. Use of chimeric adenoviral vectors to assess capsid neutralization determinants. Virology 333:207–214 [DOI] [PubMed] [Google Scholar]
- 13. Pichla-Gollon SL, Drinker M, Zhou X, Xue F, Rux JJ, Gao GP, Wilson JM, Ertl HC, Burnett RM, Bergelson JM. 2007. Structure-based identification of a major neutralizing site in an adenovirus hexon. J. Virol. 81:1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toogood CI, Crompton J, Hay RT. 1992. Antipeptide antisera define neutralizing epitopes on the adenovirus hexon. J. Gen. Virol. 73(Part 6):1429–1435 [DOI] [PubMed] [Google Scholar]
- 15. Watson G, Burdon MG, Russell WC. 1988. An antigenic analysis of the adenovirus type 2 fibre polypeptide. J. Gen. Virol. 69(Part 3):525–535 [DOI] [PubMed] [Google Scholar]
- 16. Hong SS, Bardy M, Monteil M, Gay B, Denesvre C, Tournier J, Martin G, Eloit M, Boulanger P. 2000. Immunoreactive domains and integrin-binding motifs in adenovirus penton base capsomer. Viral Immunol. 13:353–371 [DOI] [PubMed] [Google Scholar]
- 17. Hong SS, Habib NA, Franqueville L, Jensen S, Boulanger PA. 2003. Identification of adenovirus (ad) penton base neutralizing epitopes by use of sera from patients who had received conditionally replicative ad (addl1520) for treatment of liver tumors. J. Virol. 77:10366–10375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart PL, Chiu CY, Huang S, Muir T, Zhao Y, Chait B, Mathias P, Nemerow GR. 1997. Cryo-EM visualization of an exposed RGD epitope on adenovirus that escapes antibody neutralization. EMBO J. 16:1189–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reddy VS, Natchiar SK, Stewart PL, Nemerow GR. 2010. Crystal structure of human adenovirus at 3.5 A resolution. Science 329:1071–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]