

Abstract
Inflammasome activation is important for the development of an effective host defense against many pathogens, including RNA viruses. However, the mechanism by which the inflammasome recognizes RNA viruses and its role in rabies virus (RABV) pathogenicity and immunogenicity remain poorly defined. To determine the function of the inflammasome in response to RABV infection, we infected murine bone marrow-derived dendritic cells (BMDCs) with RABV. Our results indicate that the infection of BMDCs with RABV induces both the production of pro-interleukin-1β (pro-IL-1β) and its processing, resulting in the secretion of active IL-1β through activation of the NLRP3-, ASC-, and caspase-1-dependent inflammasome. As previously shown for the induction of type I interferon by RABV, the induction of pro-IL-1β also depends upon IPS-1. We demonstrate that both the production of pro-IL-1β and activation of the inflammasome require viral replication. We also demonstrate that increased viral replication in BMDCs derived from IFNAR-deficient mice resulted in significantly more IL-1β release. Additionally, IL-1 receptor-deficient mice show an increase in RABV pathogenicity. Taken together, these results indicate an important role of the inflammasome in innate immune recognition of RABV.
INTRODUCTION
Early recognition of viral infection by the innate immune system is essential for an effective host defense. The innate immune response has evolved to recognize common motifs of pathogens by a variety of pattern recognition receptors (PRRs) (for a review, see reference 1). Viral motifs recognized by these sensors include distinct nucleic acids such as double-stranded RNA (dsRNA), cytosolic DNA, or uncapped single-stranded RNA. Recognition of these motifs results in the release of type I interferons (IFNs) and inflammatory cytokines, which leads to the control of the pathogen and supports the development of an adaptive immune response (1). Four main families of PRRs have been described: Toll-like receptors (TLRs), retinoic acid inducible gene I (RIG-I)-like receptors (e.g., RIG-I and MDA5), PYHIN proteins (e.g., AIM2), and nucleotide-binding oligomerization domain (NOD)-like receptors (e.g., NOD1, NOD2, NLRP1, NLRP3, and NLRC4) (1–3).
The NOD-like receptor family consists of cytoplasmic receptors that function in the regulation of inflammatory responses and contain PRRs that are involved in the assembly of large protein complexes referred to as inflammasomes (for a review, see reference 2). The inflammasome is typically composed of a NOD-like receptor, the adaptor protein ASC, and pro-caspase-1. Depending on the danger signal present, distinct inflammasome complexes are formed. This multiprotein complex regulates caspase-1 activation and serves as a scaffold for the proteolytic activation of interleukin-1β (IL-1β) and IL-18. Processed forms of IL-1β and IL-18 are potent mediators of the inflammatory response and are involved in a range of activities, including cell proliferation, activation, differentiation, apoptosis, and antibody production (4, 5).
One of the best-characterized inflammasomes consists of NLRP3/NALP3/cryopyrin (6). NLRP3 recognizes a wide range of pathogen- and danger-associated molecular patterns (2, 3). Specifically, this particular inflammasome can recognize both positive- and negative-sense RNA viruses, including influenza virus, Sendai virus, encephalomyocarditis virus (EMCV), vesicular stomatitis virus (VSV), and West Nile virus (7–15). However, the exact mechanism by which these pathogens activate the inflammasome is presently unknown.
In contrast to the immediate activation of type I interferons, the activation of IL-1β is a two-step process combining two signaling pathways found mainly within antigen-presenting cells, such as dendritic cells (DCs) and macrophages (16). The first pathway leads to the nuclear factor κB (NF-κB)-dependent transcription of pro-IL-1β and components of the inflammasome, including NLRP3 (2). The second pathway is activated through the assembly of the inflammasome, which results in the activation of caspase-1 and subsequent cleavage of pro-IL-1β to active, secreted IL-1β.
Rabies virus (RABV) is a negative-sense single-stranded RNA virus that replicates exclusively in the cytosol (17). Previous reports have shown that RABV RNA is recognized by RIG-I and MDA5, which exclusively signal via IPS-1/MAVS/VISA/Cardif to activate both NF-κB- and type I IFN-regulated responses (18–20). Like many viruses, RABV has evolved a variety of mechanisms to counteract these host defenses. For example, RABV phosphoprotein (P) is able to suppress both the induction of type I IFN and the ensuing signaling by IFN-α/β in certain cell types, including fibroblasts and neuronal cells (21–23). Despite this, RABV infection triggers type I IFN and proinflammatory responses, which contribute to an efficient immune response (20, 24, 25). RIG-I-like receptors have also been shown to activate NF-κB pathways, which leads to the production of IL-1β and IL-6 (26), but the mechanism by which RABV activates the inflammatory cytokine IL-1β has not yet been elucidated.
Here, we examine the role of the inflammasome-dependent IL-1β activation pathway in RABV infection of myeloid dendritic cells. We demonstrate that RABV functionally activates the signaling pathway leading to the release of IL-1β by primary murine bone marrow-derived dendritic cells (BMDCs). IPS-1, an adaptor for RIG-I and MDA5, is essential for the production of pro-IL-1β. An inflammasome composed of NLRP3, ASC, and caspase-1 processes pro-IL-1β into active IL-1β, which is then secreted by the cell. The increased pathogenicity of RABV in IL-1 receptor-knockout mice highlights the role of the inflammasome as a regulator of RABV infection.
MATERIALS AND METHODS
Viruses.
SPBN, a RABV vaccine strain-based vector, has been described previously (27). Vesicular stomatitis virus expressing green fluorescent protein (VSV-GFP) was obtained from John Hiscott (McGill University) (17). For pathogenicity studies, recombinant BNSP-Cre was used (28).
Mice.
Mice used were as follows: BALB/c (NIH), C57BL/6 (NIH), IPS-1 (29), IFNAR−/− (30), and IL-1R−/− (Il1r1tm1Imx/J; Jackson Laboratories; stock number 003245). Mice deficient in NLRP3 (31), ASC (Pycard/CARD5) (31), and caspase-1/11 (32, 33) were provided by Millennium Pharmaceuticals, Lexicon Genetics, and Yale University. The Institutional Animal Care and Use Committee at Thomas Jefferson University approved all animal protocols.
For the pathogenicity study, 6- to 8-week-old C57BL/6 and IL-1R−/− mice were infected intranasally with 1 × 105 focus-forming units (FFU) of BNSP-Cre (28). The mice were monitored daily for weight loss and signs of RABV pathology. Health scoring used the following conventions: 0, no sign of disease; 1, hunched back; 2, motor impairment; 3, unilateral hind limb paralysis; 4, bilateral hind limb paralysis; 5, moribund. Mice with a weight loss of 25% (an indicator of severe RABV infection) were humanely euthanized.
BMDC differentiation.
BMDCs were differentiated as described previously (20, 34, 35). Briefly, bone marrow was obtained from the mouse femur and tibia. Red blood cells were lysed using ACK lysing buffer (Lonza). The remaining cells were seeded into 24-well plates at a density of 1 × 106 cells per ml in differentiation medium supplemented with 10 ng/ml mouse granulocyte-macrophage colony-stimulating factor (GM-CSF; Shenandoah Biotechnology Inc.). On day 7, cells were washed twice with differentiation medium supplemented with GM-CSF and then immature BMDCs were harvested. Differentiation was confirmed by flow cytometry with surface staining using hamster anti-mouse CD11c (Becton Dickinson [BD]) as described below.
BMDC stimulation.
BMDCs were plated in 12-well plates at a density of 1 × 106 cells per ml of differentiation medium. At 6 h after plating, BMDCs were left uninfected, infected with RABV at a multiplicity of infection (MOI) of 10, or stimulated with UV-inactivated RABV, followed by incubation at 34°C and 5% CO2. RABV was UV inactivated with a 254-nm UV light source for 15 min. When indicated in the figures, BMDCs were primed by stimulation with 50 ng per ml lipopolysaccharide (LPS) from Escherichia coli serotype O111:B4 (Invivogen) for 3 h before stimulation (36). As a positive control for NLRP3 inflammasome activation, 5 mM ATP (Sigma-Aldrich) was added to LPS-primed cells for 20 min (37). For caspase inhibitor treatment, cells were preincubated for 30 min with 25 μM Z-VAD-fmk (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone; pancaspase inhibitor; Calbiochem) before stimulation. At the indicated time after stimulation, both detached and adherent cells were collected and spun down at 510 × g for 10 min to separate the cells from supernatant. Cells were evenly divided into two harvests: one for flow cytometry (fluorescence-activated cell sorting [FACS]) as described below and one to prepare cell lysate. Lysate harvests were washed with phosphate-buffered saline (PBS) and lysed in RIPA buffer (10 mM Tris [pH 7.6], 1% NP-40, 0.4% deoxycholate, 66 mM EDTA) supplemented with HALT protease inhibitor cocktail (Thermo Scientific). Lysates were stored at −20°C until analysis.
FACS analysis.
BMDCs were collected as described above and blocked at 4°C for 30 min with 2 μl Fc block (anti-CD16/32; Biolegend) in 100 μl FACS buffer (2% bovine serum albumin [BSA] in PBS) per 1 × 106 cells. Cells were then stained with antibodies against cell surface markers: allophycocyanin (APC)-CD11c (clone HL3; BD) and brilliant violet 605-CD86 (clone GL-1; Biolegend) for 30 min at room temperature. After staining, cells were washed with FACS buffer and fixed with 4% paraformaldehyde for 20 min at 4°C. Cells were incubated in Perm/Wash buffer (BD) overnight at 4°C. Cells were stained with fluorescein isothiocyanate (FITC)-conjugated rabbit anti-RABV-N (FITC-RVN; Centacor) in Perm/Wash buffer for 1 h at room temperature. After staining, cells were washed and resuspended in FACS buffer and analyzed on an LSRII (BD) cytometer; 50,000 events were collected. When analyzing flow cytometry data from BMDCs, the cells were first gated on viability (using forward and side scatter) and then gated for CD11c. Infectivity was analyzed by looking at FITC-conjugated RABV nucleoprotein (FITC-RVN), and activation was analyzed by looking at the upregulation of CD86. All FACS data were analyzed using FlowJo (version 9.5.3) software.
Intracellular staining.
BMDCs were plated and collected as described above. BMDCs were left uninfected or infected with RABV at an MOI of 10 in the presence of GolgiPlug (BD) and GolgiStop (BD) followed by incubation at 34°C and 5% CO2 for 6 h. Cells were fixed with 4% paraformaldehyde for 20 min at 4°C, after which cells were incubated in Perm/Wash buffer (BD) overnight at 4°C. Cells were stained for tumor necrosis factor alpha (TNF-α) (BD) in Perm/Wash buffer for 1 h at room temperature. FACS analysis was performed as described above.
IFN sensitivity assay.
Supernatants were assessed for the ability to inhibit vesicular stomatitis virus (VSV) replication. Cell culture supernatants were analyzed for type I IFN as described previously (17). Briefly, supernatant collected at 48 h postinfection as described above was UV inactivated. Dilutions of the supernatant in RPMI 1640 were then added to the reporter cell line (3T3 cells). After a 24-h incubation, the reporter cells were then infected with VSV-GFP at an MOI of 5 for 6 h. VSV replication was determined by the presence of GFP as measured by FACS analysis.
IL-1β ELISA.
Cell-free supernatants and cell lysates collected as described above were analyzed for the amount of secreted IL-1β and intracellular pro-IL-1β, respectively. Detection was performed by sandwich enzyme-linked immunosorbent assay (ELISA) using a mouse IL-1β ELISA kit (BD OptEIA) according to the manufacturer's recommendations. O-Phenylenediamine dihydrochloride (OPD) peroxidase substrate (SigmaFast OPD tablets; Sigma-Aldrich) was used for color development. Light absorbance at 450 nm was read on a BioTek EL800 microplate reader (BioTek Instruments, Inc.).
Statistical analysis.
All data were analyzed by Prism software (GraphPad, version 5.0d). Statistical analysis was performed using Student's t test to compare two groups and represented as two-tailed P value with a confidence interval of 95%. Presented results show the mean ± standard deviation (SD) of measurements within a group. Statistical significance of mouse survival between groups was calculated by log-rank Mantel-Cox test. For all statistics, the following notations are used to indicate significance between two groups: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
RESULTS
IL-1β is secreted after RABV infection.
Whereas the interaction of RABV with the innate immune system is well studied, the role of the inflammasome during RABV infection is unknown. Previous results indicate that both murine and human dendritic cells can be infected and activated by RABV (19, 20, 34), and such cells are widely used to study inflammasome activation. Therefore, we first determined whether IL-1β was activated and secreted in BMDCs from mice after infection with RABV. For this approach, BMDCs were either infected with RABV at a multiplicity of infection (MOI) of 10 or mock infected for various periods of time. After 48 h, BMDCs infected with RABV produced a significantly larger amount of pro-IL-1β than did the uninfected control as determined by t test analysis (Fig. 1A, left panel; ***, P < 0.001). Moreover, significantly greater amounts of IL-1β were released from infected BMDCs than from uninfected cells as early as 12 h postinfection (hpi) and continued to increase over time (Fig. 1A, right panel; **, P < 0.01). The production of pro-IL-1β as well as the secretion of IL-1β was dependent upon infection with replication-competent RABV, secreting between 200 and 400 pg/ml IL-1β, and was not observed when cells were treated with UV-inactivated RABV (UV-RABV) (Fig. 1B and C).
Fig 1.
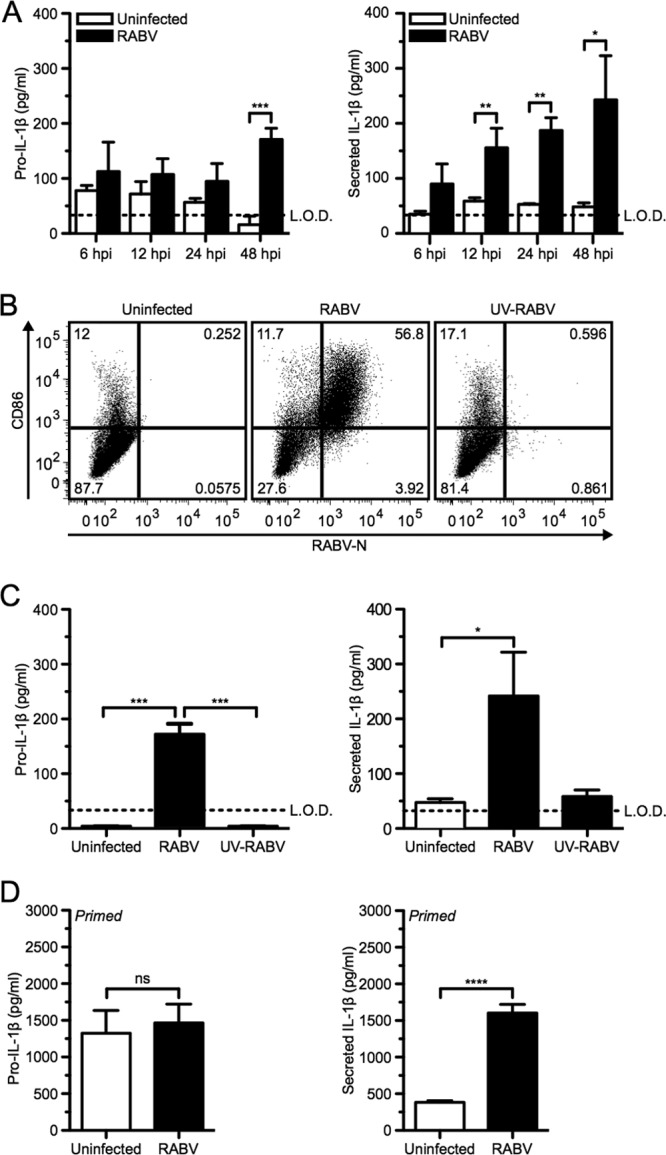
RABV infects murine dendritic cells and induces pro-IL-1β and the secretion of IL-1β in a time- and replication-dependent manner. (A) Bone marrow-derived DCs (BMDCs) from C57BL/6 mice were left uninfected (open bars) or infected at a multiplicity of infection (MOI) of 10 with RABV (closed bars) for 6, 12, 24, and 48 h postinfection (hpi). ELISA measuring pro-IL-1β in cell lysates (left) and secreted IL-1β in BMDC supernatants (right). (B and C) BMDCs left uninfected, infected for 48 h with RABV (MOI of 10), or treated for 48 h with UV-inactivated RABV (MOI of 10). (B) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (C) ELISA measuring pro-IL-1β in cell lysates (left) and secreted IL-1β in supernatants (right). (D) BMDCs primed with 50 ng LPS per ml for 3 h before being left uninfected or infected with RABV (MOI of 10) for 6 hpi. Results shown are representative of four independent experiments (n = 12 mice per group). L.O.D., limit of detection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. All error bars show standard deviations (SD).
Even though RABV is not a cytolytic virus, some cell death of BMDCs has been described at 48 hpi (34). Necrotic cell death can result in the release of intracellular pro-IL-1β and IL-1β. To determine if the IL-1β secretion after RABV infection was solely dependent upon the infection or cell death induced over time, pro-IL-1β was induced using LPS priming. Priming alone supplies a reservoir of immature pro-IL-1β but does not result in the secretion of mature IL-1β (36). As expected, infected and uninfected cells showed similar amounts of pro-IL-1β within the BMDCs (Fig. 1D, left panel). However, a significant amount of IL-1β was detected in the supernatant of the RABV-infected cells compared to that for uninfected control BMDCs within 6 hpi (Fig. 1D, right panel; ****, P < 0.0001). Taken together, these results indicate that RABV replication activates the inflammasome and the subsequent secretion of IL-1β during infection of murine BMDCs in a time-dependent manner.
Increased viral infection results in increased IL-1β secretion.
It has previously been shown that although antigen-presenting cells are susceptible to RABV infection, there is limited viral replication (20). To further investigate the impact of RABV replication on IL-1β secretion in BMDCs, we infected cells with a multiplicity of infection (MOI) between 0.01 and 10 (Fig. 2A). We found that increased IL-1β release was proportional to the MOI.
Fig 2.
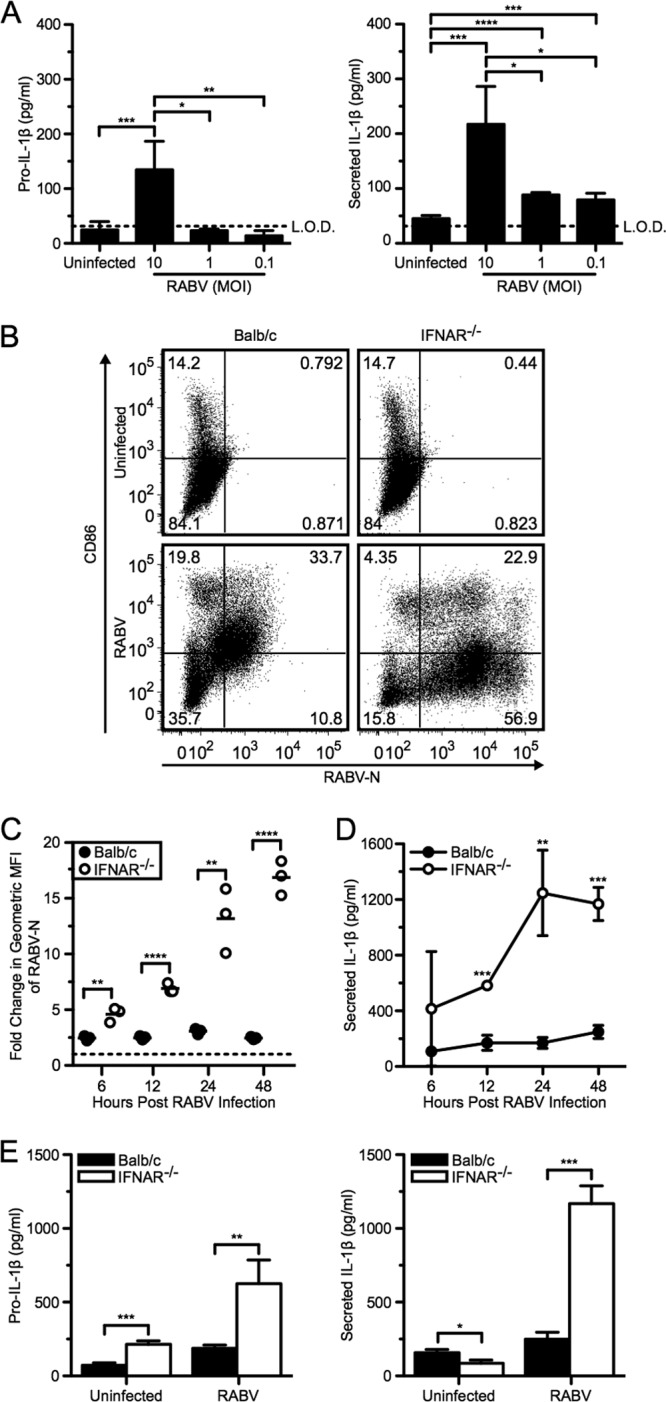
Increased RABV replication amplifies IL-1β secretion. (A) BMDCs from C57BL/6 mice were left uninfected or infected at an MOI of 10, 1, and 0.1 with RABV for 48 hpi. ELISA measuring pro-IL-1β in cell lysates (left) and secreted IL-1β in supernatants (right). (B to E) BMDCs from BALB/c (wild type; closed symbols and bars) and IFNAR−/− (open symbols and bars) mice were left uninfected or infected with RABV (MOI of 10) for 6, 12, 24, and 48 hpi. (B) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (C) For each sample, the fold increase in RABV-N was determined by dividing the geometric mean fluorescence intensity (MFI) of the infected BMDCs by the MFI of the uninfected BMDCs. Each point is representative of BMDCs from one mouse (n = 3 mice per group). (D) ELISA measuring IL-1β in supernatants over time. (E) ELISA measuring pro-IL-1β in cell lysates (left) and secreted IL-1β in supernatants (right) at 48 hpi. Results shown are representative of two independent experiments (n = 6 mice per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars show SDs.
Based on these results, we next analyzed IL-1β secretion in BMDCs derived from IFNAR-deficient mice, as previous studies have shown robust RABV replication within this model (20, 38). BMDCs from wild-type BALB/c and IFNAR−/− mice were infected at an MOI of 10 and analyzed 6, 12, 24, and 48 hpi. To assess the level of viral replication within BMDCs and DC activation, cells were stained for both RABV-N and CD86 (Fig. 2B and C). As observed previously (20), within 6 hpi we detected significantly more replication of RABV in IFNAR-deficient BMDCs than in wild-type cells, which further increased over time (Fig. 2C, open symbols; **, P < 0.01). In contrast, infection in wild-type BMDCs remained constant over time (Fig. 2C, closed symbols). In cells lacking IFNAR, increased RABV replication does not upregulate CD86, a costimulatory molecule on mature DCs, compared to wild type (Fig. 2B). Thus, although RABV infection is sufficient to induce IL-1β secretion in IFNAR-deficient BMDCs in a replication-dependent manner (Fig. 2D), RABV-induced DC maturation requires stimulation by type I IFNs, as has been previously reported (20). The level of RABV replication correlated with the level of released IL-1β (Fig. 2C and D). Additionally, we observed that increased virus replication correlated with both increased production and secretion of IL-1β at 48 hpi (Fig. 2E). Together, these results demonstrate that increased replication of RABV correlates with increased IL-1β secretion.
IPS-1 is required for the secretion of IL-1β following RABV infection.
In order to better characterize the RABV and host cell interaction, we sought to define the activated signaling pathways leading to IL-1β secretion by BMDCs in response to viral infection. RABV activates antiviral immune responses in BMDCs via the adaptor protein IPS-1 following recognition by RIG-I and MDA5 (20). It has been previously shown that IPS-1-mediated signaling is capable of activating the NF-κB signaling cascade following infection with RABV (19, 20). Furthermore, transcription of pro-IL-1β is NF-κB dependent (39). Therefore, we hypothesized that IPS-1 contributes to the production of pro-IL-1β in BMDCs infected with RABV.
BMDCs were collected from IPS-1+/+ (wild-type) and IPS-1−/− mice, infected with RABV at an MOI of 10, and then analyzed at 6, 12, 24, and 48 hpi. As previously shown (20), IPS-1+/+ BMDCs express high levels of CD86 on their surface at 48 hpi, whereas on IPS-1−/− BMDCs there is a reduction of CD86 expression (Fig. 3A). However, in IPS-1−/− BMDCs, RABV undergoes significantly more replication than it does in the wild-type cells (Fig. 3B; **, P < 0.01).
Fig 3.
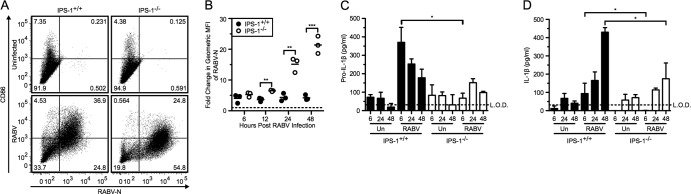
RABV-induced pro-IL-1β depends on IPS-1-mediated signaling pathways. BMDCs from IPS-1+/+ (wild type; closed bars) and IPS-1−/− (open bars) mice were left uninfected or infected with RABV (MOI of 10) for 6, 12, 24, and 48 hpi. (A) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (B) For each sample, the fold increase in RABV-N was determined by dividing the geometric MFI of the infected BMDCs by the MFI of the uninfected BMDCs. Each point is representative of BMDCs from one mouse (n = 3 mice per group). ELISA measuring pro-IL-1β in cell lysates (C) and secreted IL-1β in supernatants (D). Results shown are representative of four independent experiments (n = 12 mice per group). L.O.D., limit of detection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars show SDs.
Based on the fact that increased RABV replication in IFNAR−/− BMDCs resulted in increased IL-1β secretion, we could also expect to see increased IL-1β secretion in the IPS-1−/− BMDCs. However, we found that IPS-1−/− BMDCs infected with RABV had significantly lower pro-IL-1β production at early time points following infection (Fig. 3C; *, P < 0.05). Infection of IPS-1−/− BMDCs induced much less IL-1β release by 24 hpi, whereas infection with RABV in wild-type BMDCs induced robust IL-1β release in the supernatant (Fig. 3D). Therefore, it is likely that IPS-1 mediates synthesis of pro-IL-1β as a response to RABV infection.
RABV engages an NLRP3-, ASC-, and caspase-1-dependent inflammasome.
The processing of pro-IL-1β to mature IL-1β requires the cleavage of pro-caspase-1 to caspase-1, which is the common step for all inflammasomes (2). One of the best-characterized inflammasomes is activated by NLRP3, which recognizes a variety of pathogens, including RNA viruses (11); therefore, we investigated whether NLRP3 activation is required for IL-1β secretion after RABV infection.
For this approach, we infected BMDCs derived from NLRP3-deficient and C57BL/6 wild-type mice with RABV at an MOI of 10 and followed viral replication, BMDC activation, and production of pro-IL-1β as well as IL-1β secretion over time. There was no difference in viral replication or activation in NLRP3−/− or wild-type BMDCs (Fig. 4A and B). However, NLRP3 was required for RABV-triggered release of IL-1β in BMDCs, while RABV induced similar levels of pro-IL-1β production in NLRP3-deficient and wild-type BMDCs (Fig. 4C and D). ATP can trigger the activation of the NLRP3 inflammasome and served as a control in these experiments (37). Compared to wild-type BMDCs, NLRP3−/− BMDCs treated with ATP plus LPS priming secreted significantly less active IL-1β (Fig. 4D, right panel; **, P < 0.01). Taken together, this experiment indicates an essential role of NLRP3 in RABV-induced inflammasome activation.
Fig 4.

RABV-induced IL-1β secretion is dependent on the NLRP3 inflammasome. (A to C) BMDCs from C57BL/6 (wild type; closed symbols and bars) and NLRP3−/− (open symbols and bars) mice were left uninfected or infected with RABV (MOI of 10) for 6, 12, 24, and 48 hpi. (A) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (B) For each sample, the fold increase in RABV-N was determined by dividing the geometric MFI of the infected BMDCs by the MFI of the uninfected BMDCs. Each point is representative of BMDCs from one mouse (n = 3 mice per group). (C) ELISA measuring secreted IL-1β in supernatants over time. (D) C57BL/6 and NLRP3−/− BMDCs were left uninfected, infected with RABV for 48 hpi, or stimulated with 5 mM ATP for 30 min after priming with LPS. ELISA measuring pro-IL-1β in cell lysates (left) and measuring secreted IL-1β in supernatants (right). Results shown are representative of two independent experiments (n = 6 mice per group). L.O.D., limit of detection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars show SDs.
Many inflammasomes require the adaptor protein ASC to bridge the receptor and pro-caspase-1 (2). We therefore assessed ASC-induced secretion of IL-1β using ASC-deficient BMDCs. Compared to the wild-type BMDCs, the ASC−/− BMDCs showed no observable difference in viral replication or activation (Fig. 5A and B). C57BL/6 BMDCs showed robust pro-IL-1β production and IL-1β release in response to infection with RABV and ATP plus LPS (Fig. 5C and D). However, removing ASC significantly reduced RABV-induced secretion of IL-1β by 48 hpi (Fig. 5D, right panel; *, P < 0.05). Thus, ASC is essential for coupling recognition of RABV infection by NLRP3 to caspase-1-dependent activation of IL-1β production.
Fig 5.

RABV-induced IL-1β secretion requires the adaptor protein ASC. (A to C) BMDCs from C57BL/6 (wild type; closed symbols and bars) and ASC−/− (open symbols and bars) mice were left uninfected or infected with RABV (MOI of 10) for 6, 12, 24, and 48 hpi. (A) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (B) For each sample, the fold increase in RABV-N was determined by dividing the geometric MFI of the infected BMDCs by the MFI of the uninfected BMDCs. Each point is representative of BMDCs from one mouse (n = 3 mice per group). (C) ELISA measuring secreted IL-1β in supernatants over time. (D) C57BL/6 and ASC−/− BMDCs were left uninfected, infected with RABV for 48 hpi, or stimulated with 5 mM ATP for 30 min after priming with LPS. ELISA measuring pro-IL-1β in cell lysates (left) and measuring secreted IL-1β in supernatants (right). Results shown are representative of two independent experiments (n = 6 mice per group). L.O.D., limit of detection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars show SDs.
The primary function of the inflammasome complex is to mediate the conversion of pro-caspase-1 to active caspase-1. Initially, to examine the involvement of caspase activity in secretion of IL-1β after RABV infection, we pretreated BMDCs with a pancaspase inhibitor, Z-VAD-fmk. Adding a caspase inhibitor prior to stimulation with RABV dramatically reduced the level of IL-1β detected in the supernatants of BMDCs (data not shown). Therefore, together with the above knockouts, we studied IL-1β release in BMDCs from caspase-1-deficient mice. As seen for NLRP3−/− and ASC−/− cells, replication of RABV and activation of BMDC were also unaffected in caspase-1−/− cells compared to the wild-type BMDCs (Fig. 6A and B). However, the lack of caspase-1 resulted in significantly less secretion of IL-1β after RABV infection compared to wild-type BMDC controls, while pro-IL-1β production remained unaffected (Fig. 6C and D; *, P < 0.05).
Fig 6.

RABV induced IL-1β secretion in a caspase-1-dependent manner. (A to C) BMDCs from C57BL/6 (wild type; closed symbols and bars) and caspase-1−/− (open symbols and bars) mice were left uninfected or infected with RABV (MOI of 10) for 6, 12, 24, and 48 hpi. (A) BMDCs were stained with an antibody against RABV-N and CD86. Results shown from one representative mouse at 48 hpi. (B) For each sample, the fold increase in RABV-N was determined by dividing the geometric MFI of the infected BMDCs by the MFI of the uninfected BMDCs. Each point is representative of BMDCs from one mouse (n = 3 mice per group). (C) ELISA measuring secreted IL-1β in supernatants over time. (D) C57BL/6 and caspase-1−/− BMDCs were left uninfected, infected with RABV for 48 hpi, or stimulated with 5 mM ATP for 30 min after priming with LPS. ELISA measuring pro-IL-1β in cell lysates (left) and measuring secreted IL-1β in supernatants (right). Results shown are representative of two independent experiments (n = 6 mice per group). L.O.D., limit of detection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. All error bars show SDs.
We also examined whether the effect of these knockouts was limited to the release of IL-1β. We thus measured TNF-α and type I IFN secretion as markers for NF-κB and IRF3/7 activation, respectively (Fig. 7A and B). NLRP3, ASC, and caspase-1 were completely dispensable for TNF-α and type I IFN secretion. We did not detect any significant differences in either TNF-α or type I IFN in the absence of NLRP3, ASC, or caspase-1 compared to wild-type BMDCs following RABV infection. Taken together, these results indicate that release of IL-1β from RABV-infected BMDCs is NLRP3, ASC, and caspase-1 dependent, whereas the production of pro-IL-1β is not dependent on the inflammasome components.
Fig 7.
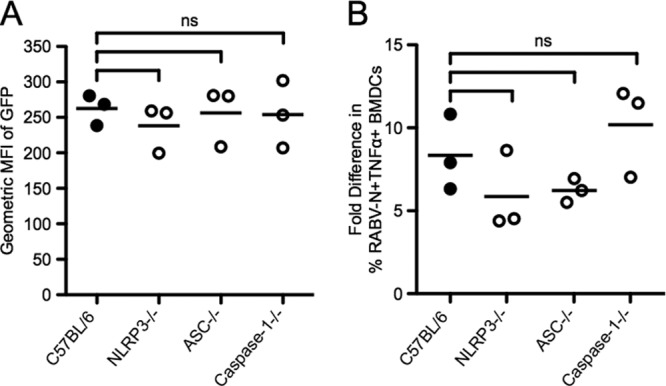
IFN and TNF-α secretion are independent of NLRP3, ASC, and caspase-1. (A) BMDCs from C57BL/6 (wild type; closed symbols) and NLRP3−/−, ASC−/−, and caspase-1−/− (open symbols) mice were infected with RABV (MOI of 10) for 48 hpi, and supernatants were collected. Type I IFN levels were detected in the supernatants by the sensitivity of reporter cells to VSV-GFP infection. After incubation with the UV-inactivated supernatant for 24 h, reporter cells were infected with VSV-GFP for 6 hpi. GFP fluorescence indicates VSV replication and, therefore, lack of type I IFN. (B) BMDCs from C57BL/6 (wild type; closed symbols) and NLRP3−/−, ASC−/−, and caspase-1−/− (open symbols) mice were left uninfected or infected with RABV (MOI of 10) for 6 hpi. Expression of TNF-α by BMDCs was measured by intracellular cytokine staining. For each sample, the fold difference in TNF-α was determined by subtracting the percent RABV-N+ TNF-α+ infected BMDCs from uninfected BMDCs. Results shown are representative of two independent experiments (n = 6 mice per group). ns, not significant.
IL-1 receptor signaling affects RABV pathogenicity.
The above-described experiments suggested that IL-1 receptor signaling would be important in the mediation of the innate immune response to RABV in vivo. To study the importance of this pathway in vivo, we infected wild-type and IL-1R-deficient mice with a strain of RABV that is moderately pathogenic after intranasal inoculation (28). As shown in Fig. 8A, 13 out of 23 IL-1R−/− mice (56.5%) succumbed to the infection whereas only 3 of 24 mice (12.5%) died within the control group (**, P < 0.01). As shown in a previous study (28), the pathogenicity of the virus correlated with the weight loss occurring at around 14 days postinfection (Fig. 8B). Previously published work indicates that paralysis induced by RABV infection is an indicator of disease; therefore, the morbidity score was also recorded, with a score of 0 indicating no signs of rabies and a score of 5 indicating a moribund state (40). As shown in Fig. 8C, the IL-1R-deficient mice experienced greater levels of paralysis starting at 11 days postinfection and either succumbed to disease or were euthanized by day 16. Those IL-1R−/− mice that did survive took longer to recover from the weight loss, as indicated by the majority of mice scoring a 1 or 2 on days 17 through 21 compared to the wild-type mice, which quickly recovered to a score of 0. The result of the infection of IL-1R−/− mice compared to that of wild-type mice in vivo indicates that IL-1 receptor signaling plays a critical role in the control of RABV infection.
Fig 8.

Lack of IL-1 receptor signaling in a mouse model of RABV infection increases viral pathogenicity. C57BL/6 (n = 24; closed symbols) and IL-1R−/− (n = 23; open symbols) mice were infected intranasally with 1 × 105 focus-forming units of RABV. Mice were monitored daily over time for their survival (A), weight change (B), and morbidity score (C). Results shown are cumulative from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
Recently, the inflammasome has gained considerable attention for its ability to control viral infection. The inflammasome is an immunological protein scaffold that recognizes danger signals within the cytoplasm of infected host cells and is essential for the production and secretion of the inflammatory cytokines IL-1β and IL-18. However, the activation of the inflammasome by RNA viruses has been largely unexplored.
Previous work has demonstrated that RABV is detected by RIG-I and MDA5 and signals through the adaptor IPS-1 for activation of NF-κB and IRF signaling pathways (20). Elimination of either RIG-I or MDA5 resulted in a decrease of BMDC activation as indicated by the upregulation of CD86 and IFN production. The genetic experiments that we show here demonstrated a signaling cascade downstream of IPS-1 that controlled the production of IL-1β in response to innate immune recognition of RABV. This makes it likely that RIG-I and/or MDA5 also plays a role in the production of IL-1β by signaling through the adaptor IPS-1.
As active IL-1β secretion in response to RABV infection was defective in BMDCs from NLRP3-, ASC-, and caspase-1-knockout mice, we conclude that RABV infection activated the inflammasome by forming a signaling complex comprised of NLRP3, ASC, and pro-caspase-1. We propose the following IL-1β activation pathway triggered by RABV infection: after IPS-1 signaling induces the production of pro-IL-1β, RABV activates an NLRP3-, ASC-, and caspase-1-dependent inflammasome, which then activates secretion of IL-1β. We found that NLRP3-, ASC-, and caspase-1-deficient DCs had pronounced defects in IL-1β secretion after RABV infection but maintained typical TNF-α and interferon responses. This indicates that the NLRP3/ASC/caspase-1 inflammasome controls the proinflammatory cytokine IL-1β after RABV recognition and suggests that the inflammasome functions in the innate immune recognition of RABV infection.
Poeck et al. demonstrated that RNA viruses, namely, VSV, were recognized by a RIG-I-, ASC-, caspase-1-dependent inflammasome, whereas double-stranded RNA (dsRNA) that lacked the 5′-triphosphate did not induce IL-1β or caspase-1 activation (12). As RABV is a related rhabdovirus, we expect that RIG-I is involved in sensing infection. However, we do not suspect that RIG-I is involved in the formation of the NLRP3 inflammasome, but rather, we think that RIG-I and MDA5 signal through IPS-1 for the synthesis of pro-IL-1β via the NF-κB pathway. Therefore, our findings support the conclusion that secretion of IL-1β, which is dependent upon cleavage of pro-IL-1β, is dependent upon NLRP3 activity following RABV infection. Additionally, this work suggests that NLRP3 activation by itself is sufficient for inflammasome activation.
RABV activates the inflammasome and triggers the subsequent release of active IL-1β through a mechanism requiring viral replication as shown above, even though viral replication in DCs is limited and ultimately results in a nonproductive infection (20). However, the exact viral stimulants that trigger the NLRP3 inflammasome remain to be elucidated. In IFNAR−/− BMDCs, we saw significantly more RABV-N expression over time, together with increased induction of pro-IL-1β and secretion of IL-1β. This indicates that with viral replication occurring at a higher rate there is also an increased induction and processing of pro-IL-1β. Another RNA virus, influenza virus, activates the NLRP3 inflammasome by lysosomal damage and reactive oxygen species (ROS) production during entry and virus replication (8, 10), whereas recent work demonstrates that encephalomyocarditis virus (EMCV), a positive-stranded RNA virus, activates the NLRP3 inflammasome through the elevation of intracellular Ca2+ levels (41). Additional studies on the role of viral entry and Ca2+ levels may further identify the mechanism by which RABV activates the inflammasome.
Alternatively, noncanonical inflammasomes utilizing caspase-8 are capable of cleaving pro-IL-1β (42, 43). However, we would expect to see a caspase-1-independent IL-1β secretion in RABV-infected BMDCs. Additionally, while we are aware that there are alternative mechanisms for IL-1β activation that are independent of the caspase-1 inflammasome, they are limited to other cell types such as neutrophils via cleavage by serine proteases, including proteinase-3, elastase, or cathepsin G (44, 45).
As NLRP3 inflammasome-dependent IL-1β secretion occurs in RABV-infected BMDCs, RABV also induces type I interferons (20). IFN-α/β signaling can also inhibit the inflammasome from being activated by NLRP3 and therefore results in decreased IL-1β secretion (46). However, we believe that the differences observed in this study may result from the fact that RABV induces a marginal amount of IFN-β compared to the amount needed to see inhibition of IL-1β production.
The aim of this study was to investigate which inflammasomes in dendritic cells regulate the response to RABV. However, the inflammasome is found in numerous antigen-presenting cells, including macrophages, monocytes, neutrophils, and microglial cells. Bone marrow-derived macrophages were found to be nonpermissive for viral infection and resulted in lack of IL-1β secretion (T. M. Lawrence and M. J. Schnell, unpublished data). During in vivo studies, IL-1β was found in the brains of mice infected with RABV (Lawrence and Schnell, unpublished data). Mice were immunized intranasally with RABV similarly to this study. ELISA of brain lysates collected at 15 days postinfection showed significantly more IL-1β in RABV-infected mice than in mock-immunized mice, confirming previously published results (47–49). Marquette et al. found that resident microglial cells and infiltrating macrophages were positive for IL-1β (47); Laothamatas et al. showed that IL-1β appeared during the early stages of infection with RABV (48). In view of these results, our data strongly suggest that in cells found at the site of RABV infection in the brain, the increased IL-1β production is the result of NLRP3 inflammasome activation.
The production of IL-1β in the brain may have a beneficial role in the recruitment of inflammatory cells as suggested by Baloul and Lafon (50) and by our observations that without the IL-1 receptor there is increased viral pathogenicity. Previous studies in mice deficient in IL-1R did not show increased susceptibility to infection (51). It is important to note that these experiments utilized a different strain of RABV as well as another route of infection. Of note, results from several groups in IL-1R−/− mice demonstrate that the IL-1 receptor is critical for infiltration of neutrophils and development of local and systemic inflammation (4). The presence of inflammatory cells recruited into the central nervous system (CNS) by inflammasome activation may lead to the recognition of RABV, at least in the case of attenuated RABV strains. Whereas some viral studies, including those on Japanese encephalitis virus, suggest that cytokines produced by infiltrating inflammatory cells, such as TNF-α and IL-1β, are associated with the death of patients (52), in RABV infections inflammation has been clearly shown to be beneficial for clearances of the virus (53, 54).
In this study, we examined the IL-1β activation pathway triggered by RABV infection. We found that NLRP3-, ASC-, and caspase-1-deficient DCs had pronounced defects in IL-1β secretion after RABV infection but had typical TNF-α and interferon responses. This indicates that NLRP3, ASC, and caspase-1 act together to form an inflammasome complex that controls the proinflammatory cytokine IL-1β release after RABV recognition. These data suggest that the inflammasome functions in the innate immune recognition of RABV infection. Further in vitro work would clarify the mechanism by which the NLRP3 inflammasome recognizes RABV and how this recognition contributes to the control of viral infection in vivo.
ACKNOWLEDGMENTS
We thank John Hiscott (McGill University) for providing the VSV-expressing GFP, Joan Durbin (Ohio State University) for providing the IFNAR−/− mice, and Shizuo Akira (Osaka University) for providing the IPS-1 mice.
This work was supported by NIH grants P01AI082325, R21NS074006, and P40OD010996 to M.J.S. and a Rubicon Fellowship from the Netherlands Organization of Scientific Research (NWO) to M.R.D.Z. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 13 March 2013
REFERENCES
- 1. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801 [DOI] [PubMed] [Google Scholar]
- 2. Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832 [DOI] [PubMed] [Google Scholar]
- 3. Kanneganti TD. 2010. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10:688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dinarello CA. 2009. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27:519–550 [DOI] [PubMed] [Google Scholar]
- 5. Sims JE, Smith DE. 2010. The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10:89–102 [DOI] [PubMed] [Google Scholar]
- 6. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA. 2008. The NLR gene family: a standard nomenclature. Immunity 28:285–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. 2009. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30:556–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. 2009. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ichinohe T, Pang IK, Iwasaki A. 2010. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11:404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N, Nunez G. 2006. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281:36560–36568 [DOI] [PubMed] [Google Scholar]
- 12. Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschlager N, Schlee M, Rothenfusser S, Barchet W, Kato H, Akira S, Inoue S, Endres S, Peschel C, Hartmann G, Hornung V, Ruland J. 2010. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 11:63–69 [DOI] [PubMed] [Google Scholar]
- 13. Rajan JV, Rodriguez D, Miao EA, Aderem A. 2011. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 85:4167–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M., Jr 2012. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 8:e1003039 doi:10.1371/journal.ppat.1003039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kumar M, Roe K, Orillo B, Muruve DA, Nerurkar VR, Gale M, Jr, Verma S. 2013. Inflammasome adaptor protein apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in West Nile virus encephalitis. J. Virol. 87:3655–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, Tschopp J. 2011. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 186:2529–2534 [DOI] [PubMed] [Google Scholar]
- 17. Faul EJ, Wanjalla CN, McGettigan JP, Schnell MJ. 2008. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology 382:226–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997 [DOI] [PubMed] [Google Scholar]
- 19. Li J, McGettigan JP, Faber M, Schnell MJ, Dietzschold B. 2008. Infection of monocytes or immature dendritic cells (DCs) with an attenuated rabies virus results in DC maturation and a strong activation of the NFkappaB signaling pathway. Vaccine 26:419–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. 2010. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016 doi:10.1371/journal.ppat.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vidy A, Chelbi-Alix M, Blondel D. 2005. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J. Virol. 79:14411–14420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brzozka K, Finke S, Conzelmann KK. 2006. Inhibition of interferon signaling by rabies virus phosphoprotein P: activation-dependent binding of STAT1 and STAT2. J. Virol. 80:2675–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rieder M, Conzelmann KK. 2009. Rhabdovirus evasion of the interferon system. J. Interferon Cytokine Res. 29:499–509 [DOI] [PubMed] [Google Scholar]
- 24. Wang ZW, Sarmento L, Wang Y, Li XQ, Dhingra V, Tseggai T, Jiang B, Fu ZF. 2005. Attenuated rabies virus activates, while pathogenic rabies virus evades, the host innate immune responses in the central nervous system. J. Virol. 79:12554–12565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Prehaud C, Megret F, Lafage M, Lafon M. 2005. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 79:12893–12904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kawai T, Akira S. 2008. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 1143:1–20 [DOI] [PubMed] [Google Scholar]
- 27. McGettigan JP, Pomerantz RJ, Siler CA, McKenna PM, Foley HD, Dietzschold B, Schnell MJ. 2003. Second-generation rabies virus-based vaccine vectors expressing human immunodeficiency virus type 1 gag have greatly reduced pathogenicity but are highly immunogenic. J. Virol. 77:237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gomme EA, Wirblich C, Addya S, Rall GF, Schnell MJ. 2012. Immune clearance of attenuated rabies virus results in neuronal survival with altered gene expression. PLoS Pathog. 8:e1002971 doi:10.1371/journal.ppat.1002971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, Yamamoto M, Uematsu S, Ishii KJ, Takeuchi O, Akira S. 2006. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203:1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Martinez-Sobrido L, Gitiban N, Fernandez-Sesma A, Cros J, Mertz SE, Jewell NA, Hammond S, Flano E, Durbin RK, Garcia-Sastre A, Durbin JE. 2006. Protection against respiratory syncytial virus by a recombinant Newcastle disease virus vector. J. Virol. 80:1130–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. 2006. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24:317–327 [DOI] [PubMed] [Google Scholar]
- 32. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. 1995. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267:2000–2003 [DOI] [PubMed] [Google Scholar]
- 33. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121 [DOI] [PubMed] [Google Scholar]
- 34. Wanjalla CN, Faul EJ, Gomme EA, Schnell MJ. 2010. Dendritic cells infected by recombinant rabies virus vaccine vector expressing HIV-1 Gag are immunogenic even in the presence of vector-specific immunity. Vaccine 29:130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wanjalla CN, Goldstein EF, Wirblich C, Schnell MJ. 2012. A role for granulocyte-macrophage colony-stimulating factor in the regulation of CD8(+) T cell responses to rabies virus. Virology 426:120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. 2009. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183:787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232 [DOI] [PubMed] [Google Scholar]
- 38. Mendonca RZ, Pereira CA. 1994. Relationship of interferon synthesis and the resistance of mice infected with street rabies virus. Braz. J. Med. Biol. Res. 27:691–695 [PubMed] [Google Scholar]
- 39. Weber A, Wasiliew P, Kracht M. 2010. Interleukin-1 (IL-1) pathway. Sci. Signal. 3:cm1 doi:10.1126/scisignal.3105cm1 [DOI] [PubMed] [Google Scholar]
- 40. Hemachudha T, Wacharapluesadee S, Mitrabhakdi E, Wilde H, Morimoto K, Lewis RA. 2005. Pathophysiology of human paralytic rabies. J. Neurovirol. 11:93–100 [DOI] [PubMed] [Google Scholar]
- 41. Ito M, Yanagi Y, Ichinohe T. 2012. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 8:e1002857 doi:10.1371/journal.ppat.1002857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. 2008. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 205:1967–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB. 2012. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13:246–254 [DOI] [PubMed] [Google Scholar]
- 44. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, Leimer AH, Cheronis J. 1999. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. U. S. A. 96:6261–6266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA. 2010. IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog. 6:e1000661 doi:10.1371/journal.ppat.1000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. 2011. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223 [DOI] [PubMed] [Google Scholar]
- 47. Marquette C, Van Dam AM, Ceccaldi PE, Weber P, Haour F, Tsiang H. 1996. Induction of immunoreactive interleukin-1β and tumor necrosis factor-α in the brains of rabies virus infected rats. J. Neuroimmunol. 68:45–51 [DOI] [PubMed] [Google Scholar]
- 48. Laothamatas J, Wacharapluesadee S, Lumlertdacha B, Ampawong S, Tepsumethanon V, Shuangshoti S, Phumesin P, Asavaphatiboon S, Worapruekjaru L, Avihingsanon Y, Israsena N, Lafon M, Wilde H, Hemachudha T. 2008. Furious and paralytic rabies of canine origin: neuroimaging with virological and cytokine studies. J. Neurovirol. 14:119–129 [DOI] [PubMed] [Google Scholar]
- 49. Solanki A, Radotra BD, Vasishta RK. 2009. Correlation of cytokine expression with rabies virus distribution in rabies encephalitis. J. Neuroimmunol. 217:85–89 [DOI] [PubMed] [Google Scholar]
- 50. Baloul L, Lafon M. 2003. Apoptosis and rabies virus neuroinvasion. Biochimie 85:777–788 [DOI] [PubMed] [Google Scholar]
- 51. Li J, Faber M, Dietzschold B, Hooper DC. 2011. The role of Toll-like receptors in the induction of immune responses during rabies virus infection. Adv. Virus Res. 79:115–126 [DOI] [PubMed] [Google Scholar]
- 52. Kaushik DK, Gupta M, Kumawat KL, Basu A. 2012. NLRP3 inflammasome: key mediator of neuroinflammation in murine Japanese encephalitis. PLoS One 7:e32270 doi:10.1371/journal.pone.0032270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hooper DC, Morimoto K, Bette M, Weihe E, Koprowski H, Dietzschold B. 1998. Collaboration of antibody and inflammation in clearance of rabies virus from the central nervous system. J. Virol. 72:3711–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Niu X, Wang H, Fu ZF. 2011. Role of chemokines in rabies pathogenesis and protection. Adv. Virus Res. 79:73–89 [DOI] [PMC free article] [PubMed] [Google Scholar]