

Abstract
Bovine herpesvirus 1 (BHV-1), like other members of the Alphaherpesvirinae subfamily, establishes latency in sensory neurons. The virally encoded latency-related RNA (LR-RNA) is expressed abundantly in latently infected sensory neurons and encodes several proteins, including ORF2. An LR mutant virus with stop codons at the amino terminus of ORF2 does not reactivate from latency after treatment with the synthetic corticosteroid dexamethasone, in part because it induces higher levels of apoptosis during the establishment of latency. ORF2 inhibits apoptosis, interacts with three cellular transcription factors (Notch1, Notch3, and C/EBP-α), and interferes with Notch-mediated signaling. Consequently, we predict that ORF2 expression is crucial for the latency reactivation cycle in cattle. In this study, we tested whether ORF2 interacts with nucleic acids, because it contains 18% basic amino acids and localizes to the nucleus. A subset of ORF2 proteins was associated with chromatin and preferentially associated with single-stranded DNA in transfected neuroblastoma cells (Neuro-2A). Alanine substitution of serine, threonine, and tyrosine residues in ORF2 increased the steady-state protein levels in Neuro-2A cells, and this protein preferentially interacted with double-stranded DNA. Certain in-frame transposon insertion mutants did not interact with DNA as efficiently as wild-type (wt) ORF2 did. ORF2 purified from bacteria under denaturing conditions preferentially interacted with double-stranded DNA, suggesting that the interaction between ORF2 and DNA was direct. In contrast, ORF2 purified under native conditions preferentially interacted with single-stranded DNA. We suggest that interactions between ORF2 and DNA mediate certain aspects of the latency reactivation cycle.
INTRODUCTION
Bovine herpesvirus 1 (BHV-1), a member of the Alphaherpesvirinae subfamily, causes significant economic losses to the cattle industry (1). For example, the ability of BHV-1 to suppress the immune system can result in life-threatening bacterial pneumonia. This polymicrobial disease is known as the bovine respiratory disease complex (reviewed in references 2 and 3). When acute infection occurs on mucosal linings within the ocular, nasal, or oral cavity, sensory neurons within trigeminal ganglia (TG) become the primary site for BHV-1 latency. Abundant viral gene expression (4) and infectious virus (5) are detected during acute infection, but viral gene expression is subsequently extinguished and latency is established (3, 6). Stress (due to confinement, transportation of cattle, restriction of food and water, or weaning) increases corticosteroid levels and can initiate reactivation from latency (7). Administration of the synthetic corticosteroid dexamethasone to calves latently infected with BHV-1 reproducibly induces reactivation from latency (5, 6, 8–11). Induction of lytic cycle viral gene transcription is also consistently detected in TG neurons of calves latently infected with BHV-1 following dexamethasone treatment.
Abundant expression of the BHV-1-encoded latency-related RNA (LR-RNA) occurs in latently infected neurons; however, infectious virus is not detected by standard assays (because of maintenance of latency) (6, 8, 9, 11–13). LR-RNA is antisense relative to the bICP0 gene and has a unique start site in TG (14, 15). The LR gene has two open reading frames (ORF1 and ORF2) and two reading frames that lack an initiating ATG (RF-B and RF-C). An LR mutant virus strain with 3 stop codons at the N terminus of ORF2 results in diminished clinical symptoms and reduced virus shedding from the eye, TG, or tonsils of infected calves (5, 16, 17). ORF1, ORF2, and RF-C are expressed when bovine cells are infected with wild-type (wt) or LR-rescued virus but have reduced or no expression following infection with the LR mutant virus (18, 19). Wild-type LR gene expression is necessary for dexamethasone-induced reactivation from latency (5), in part because the antiapoptosis activity of ORF2 appears to be crucial for the latency reactivation cycle (20–23). Additional studies demonstrated that ORF2 interacts with the cellular transcription factors Notch1, Notch3, and C/EBP-α (19, 24, 25). ORF2 reduces Notch-mediated trans-activation of the bICP0 early promoter and the glycoprotein C promoter. ORF2 also promotes neurite differentiation in the presence of Notch (26), which correlates with maintaining a mature neuronal phenotype. Transposon insertion mutants and site-directed mutants of ORF2 were prepared and analyzed (23) (see Fig. 1 for a schematic of the mutants used for this study). These studies revealed that ORF2 contains sequences necessary for inhibiting apoptosis; conversely, nonoverlapping sequences were identified that interfere with Notch-mediated trans-activation of the bICP0 early promoter (23). In addition to ORF2, LR-specific micro-RNAs inhibit bICP0 expression and apoptosis (27, 28). Collectively, these studies indicate that the LR gene promotes the establishment and maintenance of latency by enhancing the survival of infected neurons.
Fig 1.

Schematic of LR gene and expression of ORF2 in transfected Neuro-2A cells. (A) Schematic of the LR gene. A partial restriction map of the LR gene is shown. The numbering system for the LR gene and the position of ORF2 was derived from a previous study (12). The locations of LR-RNA start sites were previously published (14, 15). The positions of the 3′ terminus of bICP0 and the bICP0 stop codon were previously described (53–55). (B) Locations of ORF2 transposon mutants used in this study. Construction and cloning of ORF2 transposon insertion mutants and an NLS deletion mutant were described previously (23). Vertical lines with numbers indicate the nucleotide positions of the respective transposon insertions. The relative position of the NLS, which spans nucleotides 192 to 210 (amino acids 64 to 70), is denoted by the checkered rectangle. (C) Amino acid sequence of ORF2. The black-shaded amino acids are basic amino acids, the underlined amino acids contain the NLS, and the gray-shaded amino acids are the threonine or serine residues that are within consensus PKA or PKC phosphorylation sites. (D) Neuro-2A cells were transfected with the wt ORF2 construct, the ORF2-P construct, or the ORF2-AP construct. Forty-eight hours after transfection, cell lysate was prepared and wt or mutant ORF2 detected by Western blotting using a Flag-specific monoclonal antibody.
In this study, we found that ORF2 extracted from mouse neuroblastoma cells (Neuro-2A cells) preferentially interacted with single-stranded DNA (ssDNA)-cellulose compared to double-stranded DNA (dsDNA)-cellulose. In contrast, a mutant ORF2 protein with all potential phosphorylation sites replaced with alanine residues preferentially interacted with dsDNA. A similar result was observed when the five consensus protein kinase A (PKA) and/or PKC phosphorylation sites were mutated to alanine. Two transposon insertion mutants in the amino-terminal half of ORF2 did not interact with DNA as efficiently as wt ORF2 did, indicating that certain domains are necessary for DNA binding. ORF2 partially purified under denaturing conditions from Escherichia coli preferentially interacted with dsDNA-cellulose, while ORF2 partially purified under nondenaturing conditions preferentially interacted with ssDNA-cellulose. When ORF2 was purified from bacteria under denaturing conditions, dsDNA, but not RNA, inhibited binding to dsDNA-cellulose. Collectively, these studies provide evidence that ORF2 interacts with DNA, suggesting that this function plays a role in the latency reactivation cycle.
MATERIALS AND METHODS
Cells.
Murine neuroblastoma cells (Neuro-2A) were grown in Earle's modified Eagle's medium (EMEM) supplemented with 5% fetal bovine serum (FCS), penicillin (10 U/ml), and streptomycin (100 μg/ml).
ORF2 expression constructs and mutants used in this study.
The mammalian ORF2 expression construct was generated in the vector pCMV-Tag-2B (Stratagene). From this vector, a Flag epitope is expressed at the N terminus of ORF2, and the human immediate-early (IE) cytomegalovirus (CMV) promoter drives expression of ORF2. Further details of this plasmid were described previously (21–23). Two ORF2 phosphorylation mutants were synthesized. ORF2-AP contains alanine mutations at all potential phosphorylation sites (Ser8, Ser31, Tyr39, Thr40, Ser71, Ser79, Ser 98, Ser116, Ser117, Ser120, Ser121, Ser134, Ser 138, Thr174, and Ser176). ORF2-P contains alanine substitutions at consensus cyclic AMP (cAMP)-dependent PKA and/or serine/threonine-specific PKC (Ser31, Thr40, Ser71, Ser121, and Thr174) sites. ORF2-P also contains alanine substitutions of Tyr39 and Ser120, which are adjacent to the consensus PKA/PKC sites. Both mutants were synthesized by IDT (Coralville, IA) and ligated into the ORF2 expression vector (pCMV-Tag-2B; Stratagene) at the BamHI-HindIII restriction enzyme sites.
Transposon mutants of ORF2 were described previously (23) (see Fig. 1 for a schematic of the locations of the transposon insertion constructs used in this study). In brief, a 57-bp linker (Epicentre) was randomly inserted into the ORF2 DNA sequences according to the manufacturer's instructions. The site of transposon insertion was determined by DNA sequencing, and the respective inserts were recloned into the Flag-tagged pCMV-Tag-2B vector at the BamHI-Sal site. The Flag-tagged mutant constructs were confirmed by sequencing. Each transposon mutant expressed a full-length ORF2 with a 19-amino-acid transposon insertion in frame with ORF2. The transposon mutants used for this study were previously shown to be expressed in Neuro-2A cells, at levels similar to those of wt ORF2 (23).
ORF2 was also synthesized by IDT as a codon-optimized protein for bacterial expression. Briefly, codon-optimized ORF2 was synthesized with XhoI and HindIII sites introduced at the 5′ and 3′ ends of the gene, respectively. ORF2 sequences were then cloned downstream of six histidine residues and an Xpress epitope in the vector (pRSET-A; Invitrogen), at unique XhoI and HindIII sites.
Western blot analysis.
Neuro-2A cells in 60-mm dishes were transfected with the designated plasmids. Forty-eight hours after transfection, cells were collected, washed once with phosphate-buffered saline (PBS), and suspended in lysis buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% Triton X-100) with protease inhibitors (Roche). Cell lysate was subsequently incubated at 4°C for 30 min and centrifuged at 16,000 × g for 10 min at 4°C, and the supernatant was collected. The pellet was suspended in lysis buffer with Benzonase (Sigma), incubated at 4°C for 30 min, and then centrifuged for 10 min at 16,000 × g, and the supernatant was collected. The pellet was suspended in RIPA buffer (50 mM Tris-HCl, pH 8, 500 mM NaCl, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100) and incubated at 4°C for 30 min with rotation. The lysate was centrifuged at 16,000 × g for 10 min, and the supernatant was collected. The pellet or the respective samples from each fraction were boiled in Laemmli sample buffer for 5 min, and all samples were separated in a 12% SDS-polyacrylamide gel. Immunodetection of ORF2 and its mutants was performed using a mouse anti-Flag antibody (F1804; Sigma) (1:500). Endogenous cyclin-dependent kinase 2 (cdk2) was used as a nuclear control and was detected using a mouse anti-cdk2 antibody (SC-6248; Santa Cruz).
ORF2 protein extraction and DNA binding assays by affinity chromatography.
The pBad/His expression vector with a six-histidine N-terminal tag (Invitrogen) was used to overexpress ORF2 in Escherichia coli BL21. A bacterial codon-optimized ORF2 was synthesized by IDT and then transformed into Escherichia coli BL21. The recombinant protein was expressed by induction with isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h at mid-log phase. The histidine-tagged protein was then purified through Ni2+-agarose beads under nondenaturing or denaturing conditions, according to the manufacturer's instructions (Clontech). Briefly, cells were lysed by sonication in binding buffer (50 mM phosphate buffer [pH 7.4], 300 mM NaCl, and 10 mM imidazole), and cell debris was removed by centrifugation at 46,500 × g for 30 min. The supernatant was mixed with a 50% slurry of Ni2+-agarose beads for 2 h at 4°C. After the beads were washed 5 times with wash buffer (50 mM phosphate buffer [pH 7.4], 500 mM NaCl, and 20 mM imidazole), bound protein was eluted with 0.5 M imidazole in binding buffer.
For partial purification of bacterially expressed ORF2 by denaturing purification, 8 M urea was added during binding and the respective washing steps. ORF2 in 8 M urea was then subjected to dialysis in a buffer containing 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, 10% glycerol, and 100 μg/ml bovine serum albumin (BSA) for 16 h. This protein was operationally defined as renatured and was then used for DNA chromatography studies. The purity of the protein was examined by silver staining, and Western analysis of ORF2 was performed using an anti-Xpress antibody to confirm which protein was ORF2 (Invitrogen).
The mammalian ORF2 expression construct was transfected into Neuro-2A cells by using TransIT-Neural (Mirus, Madison, WI) according to the manufacturer's instructions. Forty-eight hours after transfection, monolayers were washed twice with cold phosphate-buffered saline, scraped from plates, and then lysed in cell lysis buffer (20 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 500 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, 10% glycerol, and 100 μg/ml BSA). The lysate was sonicated for 15 s prior to incubating with rotation at 4°C for 30 min. Cell debris was centrifuged at 46,500 × g for 15 min. The supernatant was subsequently desalted against dialysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1 mM β-mercaptoethanol, 10% glycerol, and 100 μg/ml BSA) and then used for DNA chromatography. ORF2 was detected by Western blotting using an anti-Flag antibody (Sigma, St. Louis, MO).
DNA-cellulose chromatography was performed by incubating 100 μg of ds- or ssDNA-cellulose (Sigma) with 100 μg of Ni2+-purified ORF2 for 2 h or 1 mg of mammalian cell lysate containing ORF2 or ORF2 mutants for 24 h at 4°C in binding buffer (20 mM Tris-HCl [pH 7.4], 50 mM NaCl, 1 mM β-mercaptoethanol, 1 mM EDTA, 100 μg/ml BSA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1× protease inhibitor [Roche]). After the protein was incubated with the designated DNA-cellulose beads, the beads were washed 5 times in binding buffer containing 100 mM NaCl, and bound proteins were eluted with Laemmli SDS-PAGE buffer. Samples were subjected to electrophoresis by SDS-PAGE and analyzed by Western blotting. To determine the preference of ORF2 for nucleic acid binding, yeast total RNA or herring dsDNA was added at different concentrations to the DNA-cellulose column with the extract prior to the washing step in competition experiments. DNA-free cellulose (Sigma) was used as a negative control.
RESULTS
ORF2 is a basic protein that localizes to the nucleus.
ORF2 is the first open reading frame downstream of the start site of LR-RNA (Fig. 1A); the coding sequences of ORF2 completely overlap the bICP0 transcript, and ORF2 is expressed in a subset of latently infected neurons (29, 30). The nuclear localization signal (NLS) within ORF2 (Fig. 1B) is identical to the NLS in the transcription factor Sp1 (31), and deletion of amino acids comprising the ORF2 NLS prevents nuclear localization (23). ORF2 contains approximately 18% basic amino acids (Fig. 1C, black-shaded amino acids). BLAST analysis of the entire ORF2 sequence did not reveal extensive similarity to other known proteins.
For the studies described below, we used a mouse neuroblastoma cell line (Neuro-2A) because these cells are readily transfected, ORF2 is readily expressed in these cells, and ORF2 promotes neurite sprouting in these cells following growth factor withdrawal (23, 25, 26). ORF2 contains five consensus PKA and PKC phosphorylation sites (Fig. 1C, gray-shaded amino acids). Activation of PKA stimulates herpes simplex virus 1 (HSV-1) reactivation from latency (32, 33), suggesting that PKA regulates ORF2 phosphorylation and certain aspects of the BHV-1 latency reactivation cycle. Consequently, two phosphorylation mutants of ORF2 were synthesized and their expression compared to that of wt ORF2 in transfected Neuro-2A cells. ORF2-AP contains alanine mutations of all serine, threonine, and tyrosine residues, and ORF2-P contains all of the PKA/PKC consensus sites mutated to alanine (23). ORF2-P also contains mutations in Tyr39 and Ser120, which are adjacent to PKA/PKC sites. In transfected Neuro-2A cells, wt ORF2 migrated as 20- and 33-kDa bands (Fig. 1D, closed circles). The ORF2-P and ORF2-AP mutant proteins were expressed at higher steady-state levels (open circle) than that of wt ORF2, which is consistent with a recent study (23). The predominant band expressed by ORF2-P and ORF2-AP migrated as a slightly larger protein than the faster-migrating wt ORF2 band and was the only band detected unless the blot was overexposed to allow detection of wt ORF2. The predicted molecular mass of ORF2 is approximately 20 kDa. For cells transfected with the blank expression vector, the Flag-specific antibody did not readily detect specific bands. When ORF2 is expressed in Neuro-2A cells, it localizes to the nuclear periphery (23, 25). When all of the potential phosphorylation sites (ORF2-AP) or the 5 consensus PKA or PKC phosphorylation sites were mutated to alanine, the mutant proteins and wt ORF2 had similar subnuclear localizations as judged by confocal microscopy (23; our unpublished data).
To further examine the localization of ORF2 in transfected Neuro-2A cells, biochemical fractionation was performed and compared to that of a protein that localizes to the nucleus, i.e., cdk2 (Fig. 2A and B). Treatment of cells with 0.15 M NaCl released most of the ORF2 and cdk2 (Fig. 2A and B). Readily detectable levels of the high-molecular-weight ORF2-specific bands were released from the nucleus after DNase I treatment followed by extraction with RIPA buffer. In contrast to the case for cdk2, ORF2-specific bands were readily detected in the nuclear pellet even after extraction with RIPA buffer.
Fig 2.

Localization of ORF2 in transfected Neuro-2A cells after biochemical fractionation. (A) Neuro-2A cells were transfected with wt ORF2 or the designated ORF2 mutant construct. Forty-eight hours after transfection, cells were collected and processed for Western blot analysis as designated in Materials and Methods. Asterisks denote wt ORF2- and mutant ORF2-specific bands. One hundred micrograms of protein from the indicated fraction was loaded in each lane. cdk2 was used as a control for a protein residing in the nucleus. (B) The ORF2 band intensity in each lane in panel A was quantified using a Bio-Rad FX molecular imager. The graph is representative of three independent experiments and presents percentages of the total ORF2 detected in transfected cells.
Subsequent studies were performed to compare the subnuclear localization of certain mutants to that of wt ORF2. Three transposon mutants, ORF2-134, ORF2-240, and ORF2-529, were chosen for this study because ORF2-134 and ORF2-240 do not stimulate viral promoters or productive infection and do not promote neurite formation in the presence of Notch (23, 26). Conversely, ORF2-529 behaves like wt ORF2. Most of the ORF2-134 and ORF2-240 protein was released after treatment with 0.15 M NaCl and DNase I (Fig. 2A and B). ORF2-134 was not readily detectable following extraction with RIPA buffer and was not associated with the final pellet. ORF2-529 was released following digestion of nuclei with DNase I and extraction with RIPA and was found in the pellet, indicating that this protein was associated with chromatin and/or nuclear structures. Relative to wt ORF2, the alanine substitution mutants (ORF2-P and ORF2-AP) contained high levels of protein that were tightly associated with the nucleus (Fig. 2A and B). The relative levels of the 33-kDa ORF2-specific band varied from experiment to experiment, and the fact that this higher-molecular-mass band was not detected in cells transfected with ORF2-P or ORF2-AP further suggested that ORF-2-specific high-molecular-mass bands might be due to phosphorylation. In summary, biochemical fractionation demonstrated that there were differences in the subnuclear localization of specific ORF2 mutants relative to that of wt ORF2. These subtle differences in nuclear localization were not readily observed by confocal microscopy (23).
ORF2 expressed in Neuro-2A cells interacts with DNA.
Release of ORF2 from transfected cells by DNase I treatment added support to our prediction that ORF2 interacts with DNA or nuclear structures that contain DNA. To test whether ORF2 interacted with DNA, we performed DNA binding assays with DNA-cellulose. For the DNA binding assays, a Flag-tagged ORF2- or mutant ORF2-expressing plasmid was released from transfected cells with a buffer that contained 500 mM NaCl, and this lysate was then dialyzed. Under these conditions, we believe that ORF2 and the designated mutant ORF2 proteins were not denatured. We compared the ability of ORF2 to interact with ssDNA versus dsDNA coupled to cellulose, because certain DNA binding proteins preferentially bind ssDNA versus dsDNA (reviewed in reference 34). Cell lysate was incubated with DNA-cellulose beads, and binding of ORF2 to DNA-cellulose beads was detected by Western blotting using a Flag-specific monoclonal antibody. In several independent studies, ORF2 prepared from transfected Neuro-2A cells preferentially interacted with ssDNA beads relative to dsDNA beads or blank cellulose beads (Fig. 3A). As expected, ORF2-specific bands were not detected by the Flag monoclonal antibody in mock-transfected cells (Fig. 3A, “mock” lanes).
Fig 3.

ORF2 interacts with DNA following extraction from transfected Neuro-2A cells. Neuro-2A cells (5 × 106) were transfected with plasmids expressing wt ORF2 (A) or ORF2 mutants (B and C). DNA chromatography was performed as described in Materials and Methods, with ssDNA-cellulose, dsDNA-cellulose, or blank cellulose beads. As a negative control, Neuro-2A cells were transfected with just transfection reagent. One milligram of cell extract was used for cells that expressed wt and mutant ORF2, except for the ORF2-P and ORF2-AP mutants, for which 300 μg of cell extract was incubated with DNA-cellulose. A lower concentration of cell extract was used after transfection with ORF2-AP or ORF2-P because the respective plasmids expressed higher levels of ORF2. Input (I) equivalent to 10% cell extract used in the experiments was loaded in the respective panels to verify that ORF2 was expressed. Expression of wt or mutant ORF2 in Neuro-2A cells was analyzed by Western blotting using an anti-Flag antibody. Asterisks denote ORF2-specific bands. Molecular size standards denote the positions of ORF2-specific bands.
To identify domains that were important for interacting with DNA, we utilized previously described ORF2 mutants (23, 26) and tested them for the ability to bind DNA. Although ORF2ΔNLS does not localize to the nucleus (23), it interacted with both ssDNA and dsDNA, but with reduced affinity (Fig. 3B). ORF2ΔNLS has preferentially interacted with ssDNA-cellulose beads in several independent experiments, similar to wt ORF2. Previous studies demonstrated that the ORF2ΔNLS protein migrated as the larger protein species (23). Three in-frame transposon insertion mutants were examined: ORF2-134, ORF2-240, and ORF2-529 (Fig. 3C). ORF2-134 and ORF2-240 have insertions in the N-terminal region (Fig. 1B), which is adjacent to the NLS, and are defective in known ORF2 functions (23, 26). ORF2-529 contains an in-frame transposon insertion in the C-terminal part of ORF2 and functions similarly to wt ORF2. ORF2-134 and -240 were unable to bind dsDNA-cellulose, and only a weak interaction with ssDNA-cellulose was detected (Fig. 3C). In contrast, ORF2-529 interacted with both ssDNA- and dsDNA-cellulose, but preferentially with ssDNA-cellulose (Fig. 3C), similar to the case with wt ORF2. In contrast to the other mutants and wt ORF2, the ORF2-AP and ORF2-P mutants preferentially interacted with dsDNA-cellulose. In general, this study provided evidence that wt ORF2 preferentially interacted with ssDNA-cellulose compared to dsDNA-cellulose. The N-terminal domain of ORF2 was important for interacting with DNA, whereas putative PKA/PKC consensus phosphorylation sites in ORF2 influenced the interaction with dsDNA- versus ssDNA-cellulose.
ORF2 purified from bacteria interacts with DNA.
Although the studies in Fig. 3 indicated that ORF2 interacted with DNA, the fact that ORF2 interacts with three cellular transcription factors, i.e., Notch1, Notch3, and c/EBP-α (25, 35), suggested that the interaction between ORF2 and DNA occurred by an indirect mechanism. To address this concern, we overexpressed His-tagged ORF2 in E. coli, purified this protein, and performed DNA chromatography. Following nickel-affinity chromatography of proteins extracted from E. coli under denaturing conditions, ORF2 was the predominant protein eluted from the Ni2+ column (Fig. 4A). The lower-molecular-weight proteins migrating below intact ORF2 are likely degraded ORF2 products, because an antibody directed against a region that lies between a polyhistidine tract and an Xpress-specific antibody epitope specifically recognized these bands (Fig. 4B and C, lane Ni2+). As expected, ORF2 was not readily detected in the unbound extract (Fig. 4C, lane unbound).
Fig 4.

Expression and purity of histidine-tagged ORF2 during Ni2+ agarose purification. ORF2 was expressed in E. coli as described in Materials and Methods. A cell lysate from bacteria was prepared and ORF2 partially purified using a Ni2+ column under denaturing conditions as described in Materials and Methods. Two micrograms of protein from the indicated sample was separated by 12% SDS-PAGE and visualized by silver staining (A) or analyzed by Western blotting (B). Standard molecular size markers were used to estimate the sizes of the respective proteins. Lanes: extract, cell extract from induced BL21 cells; unbound, fraction after the extract was incubated with the Ni2+ column; Ni2+, proteins bound to the Ni2+ column prior to elution. Panel C is a longer exposure of panel B and is shown to visualize ORF2 in the total extract.
We purified ORF2 under denaturing conditions to rule out the possibility that it interacted with a bacterial DNA binding protein and then consequently interacted with DNA. Following purification of ORF2 under denaturing conditions, dialysis was performed to promote renaturing of ORF2. ORF2 prepared under these conditions preferentially interacted with dsDNA (Fig. 5A). As expected, ORF2 purified under denaturing conditions was not readily detected when it was incubated with blank cellulose beads. When ORF2 was purified by Ni2+ chromatography under nondenaturing conditions, the ORF2 prepared under these conditions preferentially interacted with ssDNA-cellulose and, to a lesser extent, dsDNA-cellulose (Fig. 5B). As expected, ORF2 purified from E. coli under nondenaturing conditions did not interact with blank cellulose beads. We suggest that ORF2 partially purified from E. coli under denaturing conditions and then dialyzed had a different conformation from that of ORF2 partially purified under nondenaturing conditions, because it preferentially interacted with dsDNA-cellulose. It is also conceivable that ORF2 prepared under nondenaturing conditions copurified with a bacterial protein that enhanced the association with ssDNA.
Fig 5.
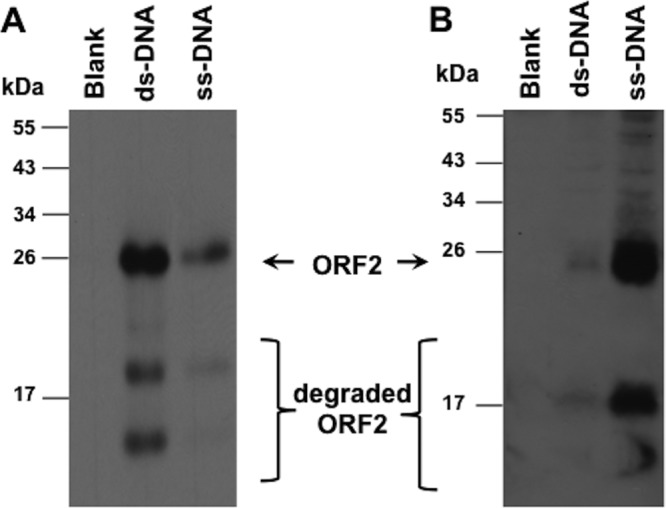
ORF2 expressed in bacteria interacts with DNA. (A) ORF2 was partially purified by Ni2+ column chromatography under denaturing conditions. The partially purified ORF2 protein (approximately 1 μg total protein) was incubated with dsDNA-cellulose, ssDNA-cellulose, or blank cellulose as described in Materials and Methods. After extensive washing, bound proteins were eluted using SDS-PAGE buffer. Samples were separated by 12% SDS-PAGE, proteins were transferred to nylon membranes, and ORF2 was detected using an anti-Xpress antibody. Molecular size markers are shown on the left. (B) ORF2 was partially purified by Ni2+ column chromatography under native conditions. The partially purified ORF2 protein (approximately 2 μg protein) was incubated with the designated cellulose beads and analyzed as described for panel A. The results in panels A and B are representative of at least 3 independent experiments.
To examine the specificity of the DNA binding activity of ORF2, competition studies were performed using ORF2 prepared from E. coli under denaturing conditions. In these experiments, double-stranded DNA from herring or total yeast RNA was added in increasing amounts (12.5- to 250-fold excess) to dsDNA-cellulose columns, along with partially purified ORF2. Binding of ORF2 to dsDNA-cellulose beads was competed for by dsDNA (Fig. 6). In contrast, yeast RNA did not have an effect on the levels of ORF2 that interacted with dsDNA-cellulose beads, even when the RNA was present in 250-fold excess. Similar results were obtained when ORF2 was purified under nondenaturing conditions (data not shown). In summary, these studies suggested that ORF2 interacts directly with DNA.
Fig 6.
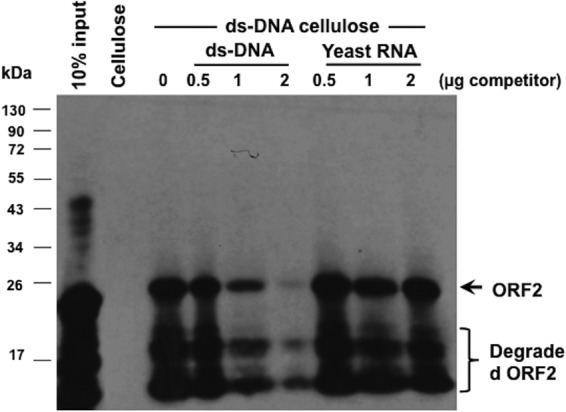
dsDNA, but not RNA, competes for binding of ORF2 to dsDNA-cellulose. DNA binding reactions were performed with dsDNA-cellulose and ORF2 partially purified by a Ni2+ column under denaturing conditions (approximately 1 μg protein). Where indicated, increasing concentrations (μg) of herring DNA (ds-DNA) or yeast RNA were added to the binding reaction mix. Blank cellulose beads (cellulose lane) were used as a negative control to verify that ORF2 did not stick to the beads. The results are representative of four independent experiments.
DISCUSSION
In this study, we have provided evidence that ORF2, in the absence of other viral proteins, stably associates with DNA. Native ORF2 (purified from bacteria or expressed in transiently transfected Neuro-2A cells) interacted preferentially with ssDNA (see Fig. 7A for a schematic summarizing these studies). Conformational changes, as a result of partial purification of ORF2 under denaturing conditions and then dialysis to allow refolding of the protein, yielded a protein that preferentially interacted with dsDNA. Mutating serine or threonine residues in the consensus PKA/PKC phosphorylation sites in ORF2 to alanine (ORF2-P) also appeared to alter the conformation of ORF2, as the mutant protein preferentially bound dsDNA versus ssDNA. Phosphorylation-dependent degradation of the oncogene c-Myc (36), the Notch1 intercellular domain (37), and the cyclin-dependent kinase inhibitor p27 (38) occurs, suggesting that cellular protein kinases phosphorylate PKA/PKC consensus sites in ORF2 and promote its degradation via the ubiquitin pathway. In spite of the alanine substitutions in ORF2-P, the resulting protein still inhibits apoptosis and Notch-mediated signaling properties with similar efficiencies to those of wt ORF2 (23, 26). However, ORF2-P interfered with activation of a Notch-dependent promoter, hairy enhancer of split 5, less efficiently than wt ORF2 did (23, 26).
Fig 7.
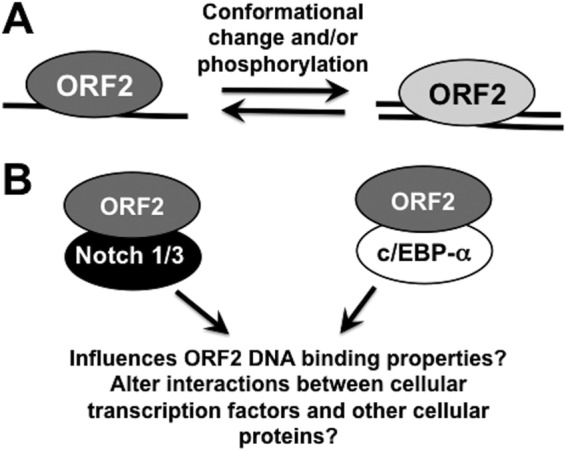
Schematic of ORF2 interactions with DNA and cellular transcription factors. See the text for further details.
Previous studies have demonstrated that ORF2 directly interacts with Notch1, Notch3, and c/EBP-α (19, 25) (Fig. 7B). This raises the possibility that interactions between ORF2 and Notch1, Notch3, or c/EBP-α may influence the DNA binding properties of ORF2. It is also possible that interactions between ORF2 and these cellular transcription factors interfere with the known functions of these transcription factors. For example, Notch family members must interact with Mastermind and a CSL family member in the nucleus to activate transcription (37, 39–41). Since ORF2, in general, interferes with Notch-dependent signaling pathways (25, 26), ORF2 may recruit Notch1 or Notch3 to novel DNA sequences or interfere with interactions between Notch, Mastermind, and CSL family members. Furthermore, c/EBP-α cooperates with the BHV-1 VP16 homologue to trans-activate viral immediate-early promoters (24), suggesting that an interaction between ORF2 and c/EBP-α reduces immediate-early gene expression.
To date, we have no evidence indicating that ORF2 binds DNA in a sequence-specific fashion. However, we cannot rule out the possibility that ORF2 can bind specific DNA sequences in the presence of a neuronal factor or following specific posttranslational modifications. It is also possible that ORF2 preferentially binds a broad-based consensus sequence that is frequently present in the viral genome. Our studies suggest that the first 240 amino acids of ORF2 contain a DNA interaction domain, because two transposon insertion mutants (ORF2-134 and ORF2-240) were unable to efficiently interact with DNA. These two transposon mutants also do not (i) interfere with stimulation of productive infection or trans-activation of certain viral promoters by Notch1 or Notch 3 (23), (ii) promote neurite growth in Neuro-2A cells when Notch is present (26), or (iii) inhibit Notch-mediated activation of the hairy enhancer of split 5 promoter (23). These observations indicate that there is a correlation between the ability of ORF2 to interact with DNA and its ability to perform the known functions of ORF2. Attempts to generate ORF2 deletion mutants to further localize the DNA binding domain of ORF2 failed because proteins encoded by the deletion mutants were not stable in bacteria or in transiently transfected Neuro-2A cells. These negative results also provide evidence that the half-life of ORF2 is tightly regulated and has functional significance.
Previous studies concluded that ORF2 is a crucial product encoded by the LR gene that promotes the latency reactivation cycle (reviewed in reference 30). For example, an LR mutant virus containing stop codons at the N terminus of ORF2 induces higher levels of apoptosis in sensory neurons during the establishment of latency (21), but the LR mutant virus does not reactivate from latency following dexamethasone treatment (5). Consequently, the ability of ORF2 (20, 22, 23) to inhibit apoptosis is proposed to promote the survival of infected sensory neurons, in particular during the establishment of latency (21). The ability of ORF2 to interact with cellular transcription factors, for example, Notch1, Notch3, and c/EBP-α (25, 35), also appears to be important during the latency reactivation cycle. These interactions reduced the ability of Notch1 to stimulate productive infection, the ability of Notch1 or Notch3 to trans-activate certain viral promoters, and the ability of Notch1 or Notch3 to inhibit neuronal differentiation (23, 25, 26). Finally, support for ORF2 playing a role in the latency reactivation cycle comes from studies demonstrating that the wt LR gene, but not the ORF2-mutated LR gene, restores wt reactivation levels to an HSV-1 LAT null mutant (42, 43). Collectively, these studies suggest that ORF2 represses viral transcription and productive infection during the establishment or maintenance of latency. In the context of productive infection, ORF2 may not be important, because the splicing pattern of the LR transcript is quite different from that in sensory neurons, and thus, intact ORF2 may not be expressed (44). Additional support for this prediction comes from the finding that expression of ORF2 is not readily detected in productively infected cells until 24 h after infection (18); by then, productive infection is completed and most cells are dead or dying. The LR gene also encodes two micro-RNAs that interfere with bICP0 expression and promote cell survival (27, 28), indicating that ORF2 is not the only viral factor that regulates the latency reactivation cycle.
The findings that ORF2 is tightly associated with the nuclear periphery in transiently transfected cells (23, 25) and that ORF2 interacts with ss- and dsDNA imply that ORF2 recruits the viral genome to certain subnuclear structures in latently infected neurons. Several studies have concluded that if genes are localized to the nuclear periphery or nuclear envelope, gene expression is reduced or silenced (45–48). The organization of genes at the nuclear periphery or nuclear envelope can also lead to activation depending on factors located at these respective specific sites (reviewed in reference 49). A yeast protein, enhancer of silent chromatin 1, localizes to nuclear peripheral areas much like ORF2 does, and the enhancer of silent chromatin 1 protein recruits heterochromatin to the nuclear periphery (50–52). If ORF2 recruits viral genomes to the nuclear periphery, this may interfere with viral genomes entering replication compartments in infected sensory neurons and consequently promote the establishment and/or maintenance of latency. ORF2 does not regulate basal levels of the bICP0 early promoter, the immediate-early promoter that activates bICP4 and bICP0 expression, or the glycoprotein C promoter in transient-transfection assays (23, 25), suggesting that ORF2 is not a sequence-specific repressor of transcription. Conversely, our studies suggest that ORF2 regulates gene expression by sequestering specific cellular transcription factors, and this function may not require sequence-specific binding of ORF2 to DNA. Studies designed to understand the precise role that the DNA binding properties of ORF2 play in the latency reactivation cycle of BHV-1 are under way.
ACKNOWLEDGMENTS
This research was supported by a grant from the USDA Agriculture and Food Research Initiative Competitive Grants Program (09-01653). A grant to the Nebraska Center for Virology (1P20RR15635) also supported certain aspects of these studies. Devis Sinani was partially supported by the Ruth L. Kirschstein National Research Service Award (grant 1 T32 AIO60547) (through the National Institute of Allergy and Infectious Diseases).
Footnotes
Published ahead of print 6 March 2013
REFERENCES
- 1. Turin L, Russo S, Poli G. 1999. BHV-1: new molecular approaches to control a common and widespread infection. Mol. Med. 5:261–284 [PMC free article] [PubMed] [Google Scholar]
- 2. Jones C. 2009. Regulation of innate immune responses by bovine herpesvirus 1 and infected cell protein 0. Viruses 1:255–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones C, Chowdhury S. 2007. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex, and development of improved vaccines. Adv. Anim. Health 8:187–205 [DOI] [PubMed] [Google Scholar]
- 4. Schang L, Jones C. 1997. Analysis of bovine herpesvirus 1 transcripts during a primary infection of trigeminal ganglia of cattle. J. Virol. 71:6786–6795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inman M, Lovato L, Doster A, Jones C. 2002. A mutation in the latency-related gene of bovine herpesvirus 1 interferes with the latency-reactivation cycle of latency in calves. J. Virol. 76:6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones C, Geiser V, Henderson G, Jiang Y, Meyer F, Perez S, Zhang Y. 2006. Functional analysis of bovine herpesvirus 1 (BHV-1) genes expressed during latency. Vet. Microbiol. 113:199–210 [DOI] [PubMed] [Google Scholar]
- 7. Jones C, Chowdhury S. 2010. Bovine herpesvirus type 1 is an important cofactor in the bovine respiratory disease complex, p 303–321 In Cooper VL, Broderson BW. (ed), Veterinary clinics of North America, food animal practice, bovine respiratory disease. W B Saunders Company, Philadelphia, PA: [DOI] [PubMed] [Google Scholar]
- 8. Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv. Virus Res. 51:81–133 [DOI] [PubMed] [Google Scholar]
- 9. Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin. Microbiol. Rev. 16:79–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones C, Newby TJ, Holt T, Doster A, Stone M, Ciacci-Zanella J, Webster CJ, Jackwood MW. 2000. Analysis of latency in cattle after inoculation with a temperature sensitive mutant of bovine herpesvirus 1 (RLB106). Vaccine 18:3185–3195 [DOI] [PubMed] [Google Scholar]
- 11. Rock D, Lokensgard J, Lewis T, Kutish G. 1992. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J. Virol. 66:2484–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kutish G, Mainprize T, Rock D. 1990. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J. Virol. 64:5730–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rock DL, Beam SL, Mayfield JE. 1987. Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits. J. Virol. 61:3827–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bratanich AC, Hanson ND, Jones C. 1992. The latency-related gene of bovine herpesvirus 1 inhibits the activity of immediate-early transcription unit 1. Virology 191:988–991 [DOI] [PubMed] [Google Scholar]
- 15. Hossain A, Schang LM, Jones C. 1995. Identification of gene products encoded by the latency-related gene of bovine herpesvirus 1. J. Virol. 69:5345–5352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inman M, Lovato L, Doster A, Jones C. 2001. A mutation in the latency-related gene of bovine herpesvirus 1 leads to impaired ocular shedding in acutely infected calves. J. Virol. 75:8507–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perez S, Inman M, Doster A, Jones C. 2005. Latency-related gene encoded by bovine herpesvirus 1 promotes virus growth and reactivation from latency in tonsils of infected calves. J. Clin. Microbiol. 43:393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiang Y, Inman M, Zhang Y, Posadas NA, Jones C. 2004. A mutation in the latency related gene of bovine herpesvirus 1 (BHV-1) inhibits protein expression of a protein from open reading frame 2 (ORF-2) and an adjacent reading frame during productive infection. J. Virol. 78:3184–3189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meyer F, Perez S, Jiang Y, Zhou Y, Henderson G, Jones C. 2007. Identification of a novel protein encoded by the latency-related gene of bovine herpesvirus 1. J. Neurovirol. 13:569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ciacci-Zanella J, Stone M, Henderson G, Jones C. 1999. The latency-related gene of bovine herpesvirus 1 inhibits programmed cell death. J. Virol. 73:9734–9740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lovato L, Inman M, Henderson G, Doster A, Jones C. 2003. Infection of cattle with a bovine herpesvirus 1 (BHV-1) strain that contains a mutation in the latency related gene leads to increased apoptosis in trigeminal ganglia during the transition from acute infection to latency. J. Virol. 77:4848–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen W, Jones C. 2008. Open reading frame 2, encoded by the latency-related gene of bovine herpesvirus 1, has antiapoptotic activity in transiently transfected neuroblastoma cells. J. Virol. 82:10940–10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sinani D, Jones C. 2011. Localization of sequences in a protein encoded by the latency related gene of bovine herpesvirus 1 (ORF2) that inhibits apoptosis and interferes with Notch1-mediated trans-activation of the bICP0 promoter. J. Virol. 85:12124–12133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meyer F, Jones C. 2008. C/EBP-alpha cooperates with bTIF to activate the bovine herpesvirus 1 immediate early transcription unit 1 promoter. J. Neurovirol. 2:1–8 [DOI] [PubMed] [Google Scholar]
- 25. Workman A, Sinani D, Pittayakhajonwut D, Jones C. 2011. A protein (ORF2) encoded by the latency-related gene of bovine herpesvirus 1 interacts with Notch1 and Notch3. J. Virol. 85:2536–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sinani D, Frizzo da Silva L, Jones C. 2013. A bovine herpesvirus 1 protein expressed in latently infected neurons (ORF2) promotes neurite sprouting in the presence of activated Notch1 or Notch3. J. Virol. 87:1183–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. da Silva LF, Jones C. 2012. Two micro-RNAs encoded within the BHV-1 latency related (LR) gene promote cell survival by interacting with RIG-I and stimulating nuclear factor-kappa B (NF-kB) dependent transcription and beta-interferon signaling pathways. J. Virol. 86:1670–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jaber T, Workman A, Jones C. 2010. Small noncoding RNAs encoded within the bovine herpesvirus 1 latency-related gene can reduce steady-state levels of infected cell protein 0 (bICP0). J. Virol. 84:6297–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jiang Y, Hossain A, Winkler MT, Holt T, Doster A, Jones C. 1998. A protein encoded by the latency-related gene of bovine herpesvirus 1 is expressed in trigeminal ganglionic neurons of latently infected cattle and interacts with cyclin-dependent kinase 2 during productive infection. J. Virol. 72:8133–8142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones C, da Silva LF, Sinani D. 2011. Regulation of the latency-reactivation cycle by products encoded by the bovine herpesvirus 1 (BHV-1) latency-related gene. J. Neurovirol. 17:535–545 [DOI] [PubMed] [Google Scholar]
- 31. Devireddy L, Zhang Y, Jones C. 2003. Cloning and initial characterization of an alternatively spliced transcript encoded by the bovine herpes virus 1 latency related (LR) gene. J. Neurovirol. 9:612–622 [DOI] [PubMed] [Google Scholar]
- 32. Leib DA, Nadeau KC, Rundle SA, Schaffer PA. 1991. The promoter of the latency-associated transcripts of herpes simplex virus type 1 contains a functional cAMP-response element: role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc. Natl. Acad. Sci. U. S. A. 88:48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smith RL, Pizer LI, Johnson EMJR, Wilcox CL. 1992. Activation of second-messenger pathways reactivates latent herpes simplex virus in neuronal cultures. Virology 188:311–318 [DOI] [PubMed] [Google Scholar]
- 34. Marceau AH. 2012. Functions of single-stranded DNA binding proteins in DNA replication, recombination, and repair. Methods Mol. Biol. 922:1–21 [DOI] [PubMed] [Google Scholar]
- 35. Meyer F, Perez S, Geiser V, Sintek M, Inman M, Jones C. 2007. A protein encoded by the bovine herpes virus 1 (BHV-1) latency related gene interacts with specific cellular regulatory proteins, including the CCAAT enhancer binding protein alpha (C/EBP-a). J. Virol. 81:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K. 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fryer CJ, White JB, Jones KA. 2004. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16:509–520 [DOI] [PubMed] [Google Scholar]
- 38. Vlach J, Hennecke S, Amati B. 1997. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J. 16:5334–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brakenhoff RH. 2011. Another NOTCH for cancer. Science 333:1102–1103 [DOI] [PubMed] [Google Scholar]
- 40. Bray SJ. 2006. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 7:678–689 [DOI] [PubMed] [Google Scholar]
- 41. Ehebauer M, Penelope P, Arias AM. 2006. Notch, a universal arbiter of cell fate decisions. Science 314:1414–1415 [DOI] [PubMed] [Google Scholar]
- 42. Mott K, Osorio N, Jin L, Brick D, Naito J, Cooper J, Henderson G, Inman M, Jones C, Wechsler SL, Perng G-C. 2003. The bovine herpesvirus 1 LR ORF2 is crucial for this gene's ability to restore the high reactivation phenotype to a herpes simplex virus-1 LAT null mutant. J. Gen. Virol. 84:2975–2985 [DOI] [PubMed] [Google Scholar]
- 43. Perng G-C, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. 2002. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J. Virol. 76:1224–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Devireddy LR, Jones C. 1998. Alternative splicing of the latency-related transcript of bovine herpesvirus 1 yields RNAs containing unique open reading frames. J. Virol. 72:7294–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finlan LE, Sproul D, Thompson I, Boyle S, Kerr E, Perry P, Ylstra B, Chubb JR, Bickmore WA. 2008. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 4:e1000039 doi:10.1371/journal.pgen.1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Green EM, Jiang Y, Joyner R, Weis K. 2012. A negative feedback loop at the nuclear periphery regulates GAL gene expression. Mol. Biol. Cell 23:1367–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shaklai S, Amariglio N, Rechavi G, Simon AJ. 2007. Gene silencing at the nuclear periphery. FEBS J. 274:1383–1392 [DOI] [PubMed] [Google Scholar]
- 48. Vande Vosse D, Wan Y, Wozniak RW, Aitchison JD. 2011. Role of the nuclear envelope in genome organization and gene expression. Syst. Biol. Med. 3:147–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akhtar A, Gasser SM. 2007. The nuclear envelope and transcriptional control. Nat. Rev. Genet. 8:507–517 [DOI] [PubMed] [Google Scholar]
- 50. Andrulis ED, Zapulla DC, Ansari A, Perrod S, Laiosa CV, Gartenberg MR, Sternglanz R. 2001. Esc1, a nuclear periphery protein required for Sir4-based plasmid anchoring and partitioning. Mol. Biol. Cell 22:8292–8301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gartenberg MR. 2000. The Sir proteins of Saccharomyces cerevisiae: mediators of transcriptional silencing and much more. Curr. Opin. Microbiol. 3:132–137 [DOI] [PubMed] [Google Scholar]
- 52. Taddei A, Hediger F, Neumann FR, Bauer C, Gasser SM. 2004. Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4, and Esc1 proteins. EMBO J. 23:1301–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, Schwyzer M. 1992. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′ coterminal and encode a putative zinc finger transactivator protein. J. Virol. 66:2763–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wirth UV, Gunkel K, Engels M, Schwyzer M. 1989. Spatial and temporal distribution of bovine herpesvirus 1 transcripts. J. Virol. 63:4882–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wirth UV, Vogt B, Schwyzer M. 1991. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J. Virol. 65:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]