

Abstract
Schmallenberg virus (SBV), a newly emerged orthobunyavirus (family Bunyaviridae), has spread rapidly across Europe and has caused congenital abnormalities in the offspring of cattle, sheep, and goats. Like other orthobunyaviruses, SBV contains a tripartite negative-sense RNA genome that encodes four structural and two nonstructural proteins. The nucleoprotein (N) encapsidates the three viral genomic RNA segments and plays a crucial role in viral RNA transcription and replication. Here we report the crystal structure of the bacterially expressed SBV nucleoprotein to a 3.06-Å resolution. The protomer is composed of two domains (N-terminal and C-terminal domains) with flexible N-terminal and C-terminal arms. The N protein has a novel fold and forms a central positively charged cleft for genomic RNA binding. The nucleoprotein purified under native conditions forms a tetramer, while the nucleoprotein obtained following denaturation and refolding forms a hexamer. Our structural and functional analyses demonstrate that both N-terminal and C-terminal arms are involved in N-N interaction and oligomerization and play an essential role in viral RNA synthesis, suggesting a novel mechanism for viral RNA encapsidation and transcription.
INTRODUCTION
Schmallenberg virus (SBV) was first identified in November 2011 in Germany and has since spread to more than 5,000 farms across Europe (1, 2). SBV infects cattle, sheep, and goats, causing transient fever, diarrhea, reduction in milk yield, and, more significantly, abortion in pregnant animals or malformations in offspring, resulting in considerable economic losses (3, 4). There is strong evidence to suggest that Culicoides midges play an essential role in transmission (5, 6). Although diagnostic methods have been established, there are no vaccines available to control the disease.
Phylogenetic analyses suggest that SBV belongs to the species Sathuperi virus (7), in the Simbu serogroup of the genus Orthobunyavirus in the family Bunyaviridae. There are more than 350 recognized bunyaviruses that are divided into five genera, Orthobunyavirus, Hantavirus, Nairovirus, Phlebovirus, and Tospovirus, based largely on biochemical and serological criteria (8). Some bunyaviruses, such as Rift Valley fever virus (RVFV), Hantaan virus, Crimean-Congo hemorrhagic fever virus (CCHFV), and sandfly fever viruses, are significant human or animal pathogens, while new bunyaviruses, such as severe fever with thrombocytopenia syndrome virus in China (9) and SBV in Europe, have emerged recently (1), causing acute human and livestock diseases. Like all bunyaviruses, SBV is an enveloped negative-stranded RNA virus with a tripartite genome that encodes two surface glycoproteins (Gc and Gn), an RNA-dependent RNA polymerase (L protein), a nucleoprotein (N), and two nonstructural proteins (NSs and NSm) (10). The nucleoprotein is the most abundant protein in the virus and in infected cells and interacts with viral genomic and antigenomic (replicative intermediate) RNA species to form ribonucleoprotein complexes (RNPs) that are the functional templates for RNA replication and transcription (10). Among bunyaviruses, structures of the N proteins from RVFV and CCHFV, belonging to the Phlebovirus and Nairovirus genera, respectively, have been reported (11–15). Those studies showed that the two structures, and their ways to bind genomic RNAs, are significantly different. Here we report the structure of the SBV nucleoprotein as a representative of a third bunyavirus genus. The SBV N protein shares a high level of amino acid identity with other orthobunyavirus N proteins (Fig. 1), indicating that the SBV N structure can serve as a model for all members of the Orthobunyavirus genus. In addition, the structure may help in designing therapies for infectious diseases caused by the emerging SBV and other clinically significant orthobunyaviruses such as Oropouche or California encephalitis viruses.
Fig 1.

Amino acid sequence alignment of orthobunyavirus N proteins. The amino acid sequence of N proteins from selected orthobunyaviruses is highly conserved. SBV, SHAV, AKBV, OROV, LACV and BUNV represent Schmallenberg virus (GenBank accession number CCF55031), Shamonda virus (accession number YP_006590077), Akabane virus (accession number YP_001497161), Oropouche virus (accession number NP_982305), La Crosse virus L78 (accession number Q8JPR0), and Bunyamwera virus (accession number NP_047213), respectively. The predicted secondary structures above the sequences were based on the SBV N structure. Conserved residues are shown in red.
MATERIALS AND METHODS
Plasmids and mutagenesis.
The bacterial expression plasmid p14SBVN was constructed by cloning the coding region of the SBV N protein into the modified pDEST14 vector (Invitrogen) downstream of the hexahistidine (6-His) tag and tobacco etch virus (TEV) protease cleavage site. Plasmids TVT7R-SBVL(+) and TVT7R-SBVS(+), for expressing SBV L and N proteins in T7 polymerase-expressing BSR-T7-5 cells (16), were described previously (17). Plasmid TVT7R-SBVM-Ren(−) was constructed by cloning the coding region of Renilla luciferase between the SBV M segment 3′ and 5′ untranslated regions (UTR) under the control of the T7 promoter in the negative-sense orientation, such that T7 transcription generates a genome-like minireplicon RNA. Plasmid pTM1-FF-Luc, which contains the firefly luciferase gene in the pTM1 vector (18), was used as an internal control (19) in minireplicon experiments.
Two sets of mutant N constructs were generated by using PCR-based mutagenesis based on either plasmid p14SBVN for bacterial expression or plasmid TVT7R-SBVS(+) for mammalian cell expression. SBV N truncations with a deletion of the N-terminal arm (residues 1 to 19) or C-terminal arm (residues 217 to 233) and single (R41G, K48E, and K51Q), double (R41G/K51Q), and triple (R41G/K51Q/W95Q) substitution mutants were created, and the entire coding region of each mutant was confirmed by DNA sequencing.
Minireplicon assay.
The SBV minireplicon assay was based on a method described previously by Weber et al. (19) and was developed to examine the effect of mutation of N on viral RNA replication. Briefly, subconfluent BSR/T7-5 cells were transfected with 35 ng TVT7R-SBVM-Ren(−), 50 ng TVT7R-SBVL(+), and 300 ng TVT7R-SBVS(+), or one of SBV N mutants, together with 2.5 ng pTM1-FF-Luc by using Lipofectamine 2000 transfection reagent (Invitrogen). At 24 h posttransfection, Renilla and firefly luciferase activities were measured by using a commercial kit (Promega) according to the manufacturer's instructions.
Protein expression and purification.
Escherichia coli Rosetta 2 cells (Merck) were transformed with p14SBVN, cultures were incubated overnight at 20°C in LB broth, and N protein expression was induced by the addition of 0.2 mM isopropyl β-d-1-thiogalactopyranoside. The cells were harvested by centrifugation at 6,000 × g for 20 min at 4°C and resuspended in cold lysis buffer (20 mM Tris [pH 7.5], 300 mM NaCl, 10 mM imidazole, and 10% glycerol containing cOmplete EDTA-free protease inhibitor cocktail [Roche]). Cells were lysed by sonication on ice, and debris was removed by centrifugation (30 min at 11,000 rpm in a Beckman JA20 rotor). The supernatant was loaded onto a Ni-nitrilotriacetic acid (NTA) column, and the column was washed with 20 mM Tris (pH 7.5)–300 mM NaCl–30 mM imidazole–10% glycerol. The proteins were eluted with 20 mM Tris (pH 7.5)–300 mM NaCl–300 mM imidazole–10% glycerol. The His tag was removed by TEV protease digestion, and the protein was further purified by passage over another Ni-NTA column followed by a S200 size-exclusion column. The protein was concentrated to 10 mg/ml for crystallization screening and biochemical analysis; where required, host RNA was removed by RNase A digestion. For denaturation and refolding, His-tagged SBV N bound to the Ni-NTA column was washed by using 10 column volumes of 8 M urea in 20 mM Tris (pH 7.5)–0.5 M NaCl buffer and then refolded by using a gradient of 8 M to 0 M urea in 20 mM Tris (pH 7.5)–0.5 M NaCl. The protein was then eluted and purified as described above.
Structure determination.
SBV N purified under native conditions was crystallized in a solution containing 0.2 M sodium formate and 20% polyethylene glycol 3350 (PEG 3350), while the denatured and refolded N was crystallized in a solution containing 0.075 M Tris (pH 8.5), 1.5 M ammonium sulfate, and 25% glycerol. The SBV N structure was determined by multiwavelength anomalous diffraction (MAD) using selenomethionine (SeMet)-labeled crystals. The hexamer structure was determined by molecular replacement using Phaser (20). Models were built by using Coot (21), and the structure was refined by using REFMAC5 (22). The structure was validated by MolProbity (23). The statistics of data collection and structure refinement are listed in Table 1.
Table 1.
Statistics of data collection and structure refinement
| Data collection parameterb | Value (value for highest-resolution shell)a |
|||
|---|---|---|---|---|
| Multiwavelength expt |
Refolded SBV N | |||
| Peak | Inflection | Remote | ||
| Wavelength (Å) | 0.9797 | 0.9799 | 0.9218 | 0.9919 |
| Resolution range (Å) | 29.82–3.11 (3.19–3.11) | 29.61–3.22 (3.30–3.22) | 29.71–3.08 (3.16–3.08) | 49.94–3.21 (3.28–3.21) |
| Space group | P21 | P21 | P21 | I422 |
| Cell constants (Å) | a = 76.57 b = 86.73 | a = 76.40 b = 86.46 | a = 76.32 b = 86.44 | a = b = 159.20 |
| c = 77.72 | c = 77.51 | c = 77.40 | c = 157.80 | |
| Cell constants (°) | α = γ = 90, β = 101.28 | α = γ = 90, β = 101.23 | α = γ = 90, β = 101.26 | α = β = γ = 90 |
| No. of unique reflections | 17,901 (1,070) | 16,103 (1,067) | 18,405 (1,260) | 16,505 (1,080) |
| Avg redundancy | 20.50 (13.10) | 7.50 (6.60) | 7.50 (6.90) | 5.60 (5.50) |
| I/σ | 24.00 (2.70) | 17.00 (2.90) | 18.60 (2.60) | 16.40 (2.20) |
| Completeness (%) | 98.40 (80.30) | 99.10 (89.70) | 99.40 (93.80) | 99.70 (99.90) |
| Anomalous completeness (%) | 97.30 (67.80) | 98.70 (84.40) | 99.00 (89.00) | |
| Rmerge (%) | 11.20 (86.20) | 8.80 (65.20) | 9.30 (77.30) | 7.30 (68.30) |
| Refinement | ||||
| Rfactor | 0.3219 | 0.3071 | ||
| Rfree | 0.3653 | 0.3723 | ||
| RMSD bonds (Å)/angles (°) | 0.007/1.257 | 0.006/1.162 | ||
| PDB accession no. | 4IDU | 4IDX | ||
Values in parentheses are for the highest-resolution shell.
RMSD, root mean square deviation.
Chemical cross-linking of SBV N protein.
Cross-linking of purified N protein was performed by using dithiobis(succinimidyl propionate) (DSP), a thiol-cleavable cross-linking reagent, according to the manufacturer's instructions (Thermo Scientific Pierce). Briefly, 10 to 20 μg of purified N protein was treated with 1 mM DSP for 30 min at room temperature in phosphate-buffered saline (PBS). The cross-linked N protein was analyzed by SDS-PAGE under nonreducing conditions and visualized by Coomassie blue staining.
RNA binding ability of SBV N protein.
Natively purified wild-type (wt) and mutant N proteins were examined for their RNA binding ability. Briefly, equal amounts of each protein (20 μg) were dissolved in 2× RNA gel loading buffer (Invitrogen) and electrophoresed on a 2% agarose gel. RNA gel loading buffer contains 95% formamide, which can effectively denature and dissociate RNA from the protein. RNA was visualized under UV light after staining with GelRed (Cambridge Bioscience).
Protein structure accession numbers.
The atomic coordinates and the structure factors have been deposited in the Protein Data Bank (PDB) under accession numbers 4IDU for the native SBV N structure (tetramer) and 4IDX for the refolded N structure (hexamer).
RESULTS
Purified native SBV N forms oligomers and binds RNA.
The SBV N protein was expressed in E. coli Rosetta 2 cells and purified by using standard protocols (see Materials and Methods). Gel filtration chromatography showed that the N protein appeared in oligomeric forms in solution (Fig. 2A). The oligomeric state of the N protein was further investigated by chemically cross-linking purified N with DSP and analysis by SDS-PAGE, which showed that N existed predominantly as a tetramer (Fig. 2B and C). It has been reported that nucleoproteins of other negative-stranded viruses, when expressed in bacteria, can bind host RNA. The absorption ratio of the natively purified SBV N at 260 nm/280 nm was 1.3, indicating that SBV N binds bacterial RNA. To confirm the RNA binding ability of recombinant N, an aliquot of the natively purified protein was electrophoresed on a 2% agarose gel, and bound RNA, about 30 to 40 nucleotides (nt) in length, was visualized by using GelRed staining (Fig. 2D).
Fig 2.
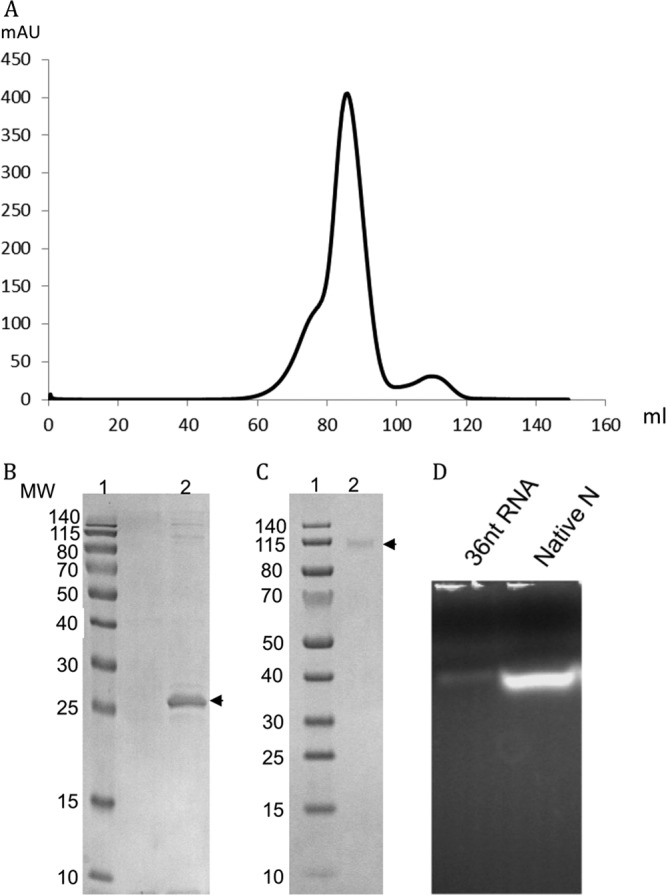
Purified SBV N exists as an oligomer in solution and contains RNA. (A) Gel filtration chromatography of native SBV N protein. After TEV cleavage, the His tag and the TEV protease were removed by binding to a nickel column, and SBV N was applied onto a Hiload 16/600 Superdex 200 column. The main peak represents the tetrameric form of SBV N. mAU, milli-absorbance unit at 280 nm. (B and C) Oligomerization studies. Bacterially expressed wild-type SBV N protein was cross-linked with 1 mM dithiobis(succinimidyl propionate), resolved by SDS-PAGE under reducing (B) or nonreducing (C) conditions, and visualized by Coomassie blue staining (lanes 2). N protein is indicated by arrows. Protein molecular weight (MW) markers (in thousands) are shown in lanes 1. (D) Purified N contains RNA. The purified N protein was loaded onto a 2% agarose gel, and the RNA was visualized by staining with GelRed. The first lane is a 36-nt synthetic RNA marker, and the second lane shows RNA from the native protein.
Novel structure of SBV N.
Crystals of the SBV N protein purified under native conditions were obtained, but diffractions were isotropic to only a 6-Å resolution. To improve the crystals, the N protein was treated with the RNase A before gel filtration, and new crystallization conditions were screened. Crystals were obtained that belonged to space group P21 with the following unit cell dimensions: a = 76.11 Å, b = 86.33 Å, c = 76.90 Å, and β = 100.98°. The SBV N structure was determined by multiwavelength anomalous dispersion to 3.06 Å (Table 1). The protomer of N consists of two domains (N-terminal domain and C-terminal domain) and two flexible arms (N-terminal arm and C-terminal arm) with a novel fold (Fig. 3A). The N-terminal domain spans residues 19 to 127 and is formed by five α helices (α1 to α5) and two antiparallel β strands (β1 and β2), while the C-terminal domain consists of residues 128 to 213, which form six helices (α6 to α11). The flexible N-terminal arm (residues 1 to 18) and C-terminal arm (residues 214 to 230) extend outward from the body of the protein. Like the nucleoproteins of other negative-stranded viruses, there is a highly positively charged cleft between the N- and C-terminal domains (Fig. 3B), which we speculate is the genomic RNA binding site.
Fig 3.

Crystal structures of protomeric and tetrameric forms of SBV N. (A) Protomer structure showing that SBV N consists of an N-terminal arm (NTA), an N-terminal domain (NTD), a C-terminal domain (CTD), and a C-terminal arm (CTA). The N-terminal arm is in blue, and the C-terminal arm is in red. (B) Electrostatic surface potential map of the N protomer. Positively charged residues are in blue, and negatively charged residues are in red. (C) Cartoon of the tetrameric structure of SBV N showing that the left dimer is formed by a C-terminal arm interaction and that the right dimer is formed by an N-terminal arm interaction. (D) Surface representation of the tetrameric structure reveals that each dimer has a free C-terminal arm (red) stretched out. The four protomers are shown in yellow, green, cyan, and orange. (E) The structures of the protomers in the tetramer are similar. The four protomers, represented in yellow, green, orange, and cyan, are superimposed. The conformations of the N-terminal arms are quite different, with three binding at the potential RNA binding cleft (green, orange, and yellow) and the fourth pointing away (cyan). (F) The N-terminal arm holds a hydrophobic patch formed by residues at the middle region of another protomer. The N-terminal arm is in cyan, and the protomer is in green. (G) Dimer-dimer interactions of the tetramer. The side chain L113 locks into a hydrophobic region on the opposing dimer. The dark dotted lines indicate hydrogen bonds.
Tetrameric structure of SBV N.
There are four N molecules in the asymmetric unit, and the four molecules have identical N-terminal and C-terminal domains but differ in the conformation and position of the two arms. In particular, three of the N-terminal arms are located at the RNA binding cleft at different positions, whereas the fourth points away from the cleft (Fig. 3E). The tetramer is composed of two dimers at a 2-fold rotational axis (Fig. 3C and D). One dimer is formed by the N-terminal arm of one protomer stretching out to hold the C-terminal edge of the RNA binding cleft of the second protomer in a head-to-tail manner, while the second dimer is generated by the C-terminal arm of a protomer binding to a hydrophobic area formed by another N protein molecule, which is close to the outside of the C-terminal cleft (discussed below). Residues S3, Q4, F5, I6, and F7 of the N-terminal arm of the first N protomer are located in a hydrophobic area formed by residues M124, L126, V129, F44, L45, and F144 of the second protomer (Fig. 3F). It is worth noting that the side chain of K48 forms a hydrogen bond with the side chain of S3, which may play an important role in anchoring the N-terminal arm. There are two interaction faces between the dimers, and both comprise helix α5 from one dimer and helices α1 and α4 from the other dimer (Fig. 3G). The side chain of L113 from helix α5 is located in a hydrophobic area lined by residues F27, W103, and I28 and the side chain of residue R102 from helices α1 and α4, while the side chain of Y24 forms hydrogen bonds with the side chains of E117 and Q121.
It should be noted that one protomer has a free C-terminal arm in the dimer (Fig. 3C and D). The RNA binding cleft of the protomer without a free C-terminal arm is exposed to the outside, but the RNA binding cleft of the promoter with a free C-terminal arm is largely covered at the interface (Fig. 3D).
Hexameric structure of SBV N.
In order to investigate the non-RNA-binding-state structure of SBV N, a denaturation and refolding experiment was performed. Unlike the RVFV nucleoprotein (14), the refolded SBV N protein remains in an oligomeric state and forms a hexamer (Fig. 4A to C). The refolded SBV N protein was crystallized, and the structure was determined by molecular replacement by Phaser (20), using the SBV N protomer as a search model. The crystals belong to space group I422 with the following cell dimensions: a = b = 159.2 Å and c = 157.8 Å. By comparison with the previous tetrameric structure, significant changes were seen in the hexameric form: the N-terminal arm becomes disordered and the C-terminal arm becomes ordered. The hexamer is formed from three dimers at a 3-fold symmetry axis (Fig. 4B and C). The dimers are formed in a tail-to-tail manner by the C-terminal arm from one protomer forming a hand that holds a hydrophobic patch formed by helix α10 and loops from another protomer (Fig. 4E). The hydrophobic patch consists of residues M164, L168, V174, L181, W193, I201, K202, K178, and E192, while the hand of the C-terminal arm is formed by residues F218, A221, A222, R223, T224, F225, L226, and Q227. The dimer-dimer interfaces are formed in a head-to-head manner and are composed of helices α1 and α2 from each dimer. Hydrophobic interactions play essential roles in stabilizing the interaction, comprising residues E21, Y24, I28, and K30 from both dimers (Fig. 4D). It is interesting that there is one free C-terminal arm in each dimer (Fig. 4B and C). In summary, both N-terminal and C-terminal arms are involved in SBV N multimerization, as are some residues in the N-terminal and C-terminal domains of SBV N, which is consistent with results of multimerization studies of Bunyamwera orthobunyavirus N protein mutants reported previously (24). Notably, however, protomers in neither the SBV tetramer nor the SBV hexamer display the rotation required to form ring structures as reported for all other RNA negative-sense virus nucleoproteins. Instead, SBV N has a special arrangement in which half of the protomers are in one rotation and the other half are in a different rotation (Fig. 3C and D and 4B and C).
Fig 4.

Hexameric structure of SBV N. (A) Gel filtration chromatography of denatured and refolded SBV N. The N protein was denatured and refolded on a nickel column by using 8 M urea. The refolded N protein was cleaved by TEV protease, further purified by nickel column chromatography, and then applied onto a Hiload 16/600 Superdex 200 column. The main peak represents the hexameric form of SBV N. (B) Cartoon of the hexameric SBV N structure. The six protomers are in six different colors, and the C-terminal arms are in red. The C-terminal arm is essential for formation of hexamer. (C) Surface representation of the hexameric structure of SBV. Each protomer is shown in a different color. Three protomers (orange, cyan, and yellow) have their free C-terminal arm standing out. (D) Dimer-dimer interaction in the hexamer, formed by helices α1 and α2 of the protomers. One protomer is in orange, and the other is in gray. (E) The C-terminal arm is located in a hydrophobic region of the neighboring protomer. The C-terminal arm is in gray, and the protomer is in yellow.
N protein mutations impair RNA binding activity.
The N-terminal arm binds to the protomer's RNA binding cleft and is very flexible, suggesting that the N-terminal arm may play a role in binding and protecting the RNA, while the central cleft, with its highly positively charged residues, has the potential to bind the RNA. To examine these structural predictions in more detail, we generated several N protein mutants: truncations of either N-terminal (Δ1-19) or C-terminal (Δ217-233) arms and single (R41G, K48E and K51Q), double (R41G/K51Q), or triple (R41G/K51Q/W95Q) amino acid substitutions in the potential RNA binding cleft (Fig. 5A). The mutant proteins were expressed and purified by using the same protocol as that used for wild-type SBV N, and the RNA bound to all mutant proteins was examined. Briefly, equal amounts of each purified protein preparation were dissolved in RNA denaturation buffer to dissociate RNA from protein and loaded onto a 2% agarose gel, followed by visualization using GelRed staining. The N-terminal-arm-truncated protein and the triple mutant were shown to have no RNA bound, suggesting that the N terminus and the three residues in the potential RNA binding cleft are crucial for RNA binding. The amount of RNA bound to single-site N protein R41G and K51Q mutants was reduced, and the amount of RNA bound to N protein with the C-terminal arm truncated, the K48E single mutant, or the R41G/K51Q double mutant was significantly less than that bound to the wt SBV N protein (Fig. 5B).
Fig 5.

RNA binding and RNA replication activities of SBV N mutants. (A) Location of the mutated residues in the RNA binding cleft, and positions of the C-terminal arm (CTA) and N-terminal arm (NTA). (B) In vivo RNA binding ability of wt and mutant N proteins. Equal amounts of the purified proteins were loaded onto a 2% agarose gel, and the gel was stained with GelRed. (C) Ability of mutant SBV N to support minireplicon activity. The graph shows luciferase activity as a measure of minireplicon activity normalized with respect to the wt SBV N protein. Mutant N proteins are indicated on the x axis. TVT, empty vector.
Minireplicon assays.
To further examine the effect of mutations in N on viral RNA synthesis, we performed minireplicon assays using the C- and N-terminally truncated proteins as well as the mutant N proteins containing single-, double-, and triple-point substitutions of potential RNA binding residues in the RNA binding cleft. In these assays, luciferase activity was measured as an indication of viral RNA synthesis. As expected, the N-terminal- and C-terminal-arm truncations and the R41G/K51Q/W95Q triple mutant did not support minireplicon activity. Interestingly, the K48E single mutant and R41G/K51Q double mutant lost more than 98% of their activity, while the K51Q single mutant lost 31% of its activity. On the other hand, it is intriguing that the R41G single mutant had increased activity (Fig. 5C). The data strongly suggest that both the C-terminal and the N-terminal arms are essential for viral RNA replication. Considering that residue K48 is located at the edge of the RNA binding cleft and is involved in interactions with the N-terminal arm in the tetrameric structure and genomic RNA binding, it is not surprising that K48 is critical.
DISCUSSION
To date, the structures of N proteins from several negative-strand RNA viruses with a segmented genome (influenza virus, Lassa virus, CCHFV, and RVFV) have been determined (11–15, 25–28). Although the structures of the nucleoproteins are quite different, they all share a feature in having a highly positively charged cleft for binding genomic RNA. We have determined the crystal structure of the SBV N protein, which showed that the structure is overall dissimilar to the known viral nucleoprotein structures. However, it does share the property of having a highly positively charged cleft between the N-terminal and C-terminal domains. Biochemical studies and functional assays strongly indicate that this cleft contains the genomic RNA binding site (Fig. 6A and B). Unlike the situation with RVFV N, the N-terminal and C-terminal arms of SBV N play an important role in forming dimers within the tetramer, while the C-terminal arm is essential for forming dimers in the hexamer. Furthermore, the dimer-dimer interface in the tetrameric form is different from the dimer-dimer interfaces of the hexamer, indicating that the SBV N protein is relatively flexible in its ability to multimerize. This suggests that residues in the N-terminal, C-terminal, and middle regions of the SBV N protein are involved in oligomerization, which is consistent with previous biochemical studies on mutant N proteins of Bunyamwera virus (24), the prototypic orthobunyavirus, suggesting that all N proteins of orthobunyaviruses may share this feature.
Fig 6.

Proposed rotation of N for RNA encapsidation and RNA replication. The six protomers are shown in a surface representation in six different colors, with the RNA in blue. (A) Modeling showing that the RNA binds at the cleft of a protomer of SBV N. (B) Hexameric structure of SBV N with RNA modeled at the RNA binding cleft. The six protomers are in different colors. The RNAs in blue are modeled at the RNA binding cleft to show the RNA binding sites. The C-terminal arms are in red. (C) Protomers that have the C-terminal arm free hide the RNA inside the circle of the hexamer, indicating that the genomic RNA is located inside the circle during encapsidation. (D) The structure in panel C becomes the structure in panel D by rotation 180° along the y axis. Protomers without the C-terminal arm free expose the RNA to the outside of the hexamer, suggesting that the genomic RNA is located outside during RNA replication transcription. (E) Proposed rotation mechanism for encapsidation. The protomers without a free C-terminal arm (C) rotate about 180° to hide the RNAs inside the inner circle, resulting in all protomers having free C-terminal arms. (F) Proposed rotation mechanism for replication and transcription. The protomers with a free C-terminal arm (D) rotate about 180° to expose the RNA for replication and transcription. The L protein may be involved in this process.
Although some segmented negative-stranded RNA viral nucleoproteins have been reported to have other functions (13, 27), their primary role is to form a ribonucleoprotein complex to protect the genomic RNA, and the RNP is the functional template for both genome transcription and replication. Structures of nonsegmented negative-stranded RNA virus N proteins show that for human respiratory syncytial virus, the nucleoprotein forms a helical ribonucleoprotein complex with the RNA located on the outside (29), while for rabies and vesicular stomatitis viruses, the helical ribonucleoprotein complex has the RNA located inside (30, 31). Only two nucleoprotein structures from the entire bunyavirus family have been reported to date: one is the nucleoprotein from Rift Valley fever phlebovirus (12), and the other is the nucleoprotein of Crimean-Congo hemorrhagic fever nairovirus (11, 13, 15). The nucleoprotein of RVFV forms a hexameric ring structure, and the genomic RNA segments are indicated to bind inside the ring. More recently, the crystal structures of RVFV N in complex with different lengths of single-stranded RNA or DNA were reported, which revealed that 4 nucleotides sit inside a hydrophobic slot, and the nucleic acid is bound inside circles of tetrameric, pentameric, and hexameric forms (32). In contrast, the CCHFV N protein was suggested to form a superhelix with the genomic RNA bound in clefts on the outer side of the helix (15). It is easy to explain how the viral genomic RNAs are protected by these N proteins, as the RNA is wrapped inside the nucleoprotein ring structures, but it is more difficult to explain how the RNA, in the form of the RNP, can be accessed by the RNA-dependent RNA polymerase (L protein). To our knowledge, this is the first nucleoprotein structure that shows a third arrangement, in which half of the protomers keep the potential RNA binding clefts inside the oligomers and half of the protomers expose the RNA binding clefts outside the oligomers. This arrangement may be an intermediate that indicates how the genomic RNAs are protected and how the RNA can be accessed in the RNP template for replication and transcription.
In both tetrameric and hexameric structures, one protomer in each dimer has a free C-terminal arm stretched out, and the RNA binding cleft faces toward the inside of the hexamer (Fig. 6B to D) or tetramer (Fig. 3D), while another protomer rotates about 180° so that its C-terminal arm can hold the neighboring protomer at the hydrophobic patch, resulting in exposure of the RNA binding cleft to the outside of the hexamer (Fig. 6D) or tetramer (Fig. 3D). We have modeled RNA in the cleft of the hexameric structure, which clearly shows that the protomers that have free C-terminal arms hide the RNA inside the ring of the hexamer (Fig. 6B and C), while the protomers that have their C-terminal arms attached to neighboring protomers expose the RNA on the outside of the ring (Fig. 6D). Based on this observation, we therefore propose that the SBV N protomers without the free C-terminal arm will rotates about 180° to hide the genomic RNA inside the ring, resulting in all protomers having free C-terminal arms (Fig. 6E), while N protomers with the free C-terminal arms will rotate about 180° to expose the RNA for replication and transcription (Fig. 6F). We further propose that the N-terminal and C-terminal arms may play an important role in rotation of the N protein, perhaps aided by the viral RNA-dependent RNA polymerase (L protein). The detailed mechanism by which SBV N rotates for encapsidation and RNA replication/transcription is under investigation. In addition, the binding sites for the N-terminal and C-terminal arms, as well as the RNA binding cleft, are potential targets for rational drug discovery.
ACKNOWLEDGMENTS
We thank the staff of the IO4 and I24 beam stations at Diamond Light Resources UK and Xiaohong Shi for critical reading of the manuscript.
This work was supported by the Medical Research Council (grant no. G1100110/1 to C.D.) and the Wellcome Trust (career development fellowship WT083501MA to C.D. and program grant 079810 and project grant 091783 to R.M.E.). R.M.E. is a Wellcome Trust senior investigator.
Footnotes
Published ahead of print 6 March 2013
REFERENCES
- 1. Hoffmann B, Scheuch M, Höper D, Jungblut R, Holsteg M, Schirrmeier H, Eschbaumer M, Goller KV, Wernike K, Fischer M, Breithaupt A, Mettenleiter TC, Beer M. 2012. Novel orthobunyavirus in cattle, Europe, 2011. Emerg. Infect. Dis. 18:469–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beer M, Conraths FJ, van der Poel WH. 2013. ‘Schmallenberg virus’—a novel orthobunyavirus emerging in Europe. Epidemiol. Infect. 141:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garigliany MM, Bayrou C, Kleijnen D, Cassart D, Jolly S, Linden A, Desmecht D. 2012. Schmallenberg virus: a new Shamonda/Sathuperi-like virus on the rise in Europe. Antiviral Res. 95:82–87 [DOI] [PubMed] [Google Scholar]
- 4. Tarlinton R, Daly J, Dunham S, Kydd J. 2012. The challenge of Schmallenberg virus emergence in Europe. Vet. J. 194:10–18 [DOI] [PubMed] [Google Scholar]
- 5. De Regge N, Deblauwe I, De Deken R, Vantieghem P, Madder M, Geysen D, Smeets F, Losson B, van den Berg T, Cay AB. 2012. Detection of Schmallenberg virus in different Culicoides spp. by real-time RT-PCR. Transbound. Emerg. Dis. 59:471–475 [DOI] [PubMed] [Google Scholar]
- 6. Rasmussen LD, Kristensen B, Kirkeby C, Rasmussen TB, Belsham GJ, Bodker R, Botner A. 2012. Culicoids as vectors of Schmallenberg virus. Emerg. Infect. Dis. 18:1204–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goller KV, Hoper D, Schirrmeier H, Mettenleiter TC, Beer M. 2012. Schmallenberg virus as possible ancestor of Shamonda virus. Emerg. Infect. Dis. 18:1644–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plyusnin A, Beaty BJ, Elliott RM, Goldbach R, Kormelink R, Lundkvist Å, Schmaljohn CS, Tesh RB. 2012. Bunyaviridae, p 725–741 In King AMQ, Adams MJ, Carstens EB, Lefkowits EJ. (ed), Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 9. Yu XJ, Liang MF, Zhang SY, Liu Y, Li JD, Sun YL, M DL, Zhang QF, Popov VL, Li C, Qu J, Li Q, Zhang YP, Hai R, Wu W, Wang Q, Zhan FX, Wang XJ, Kan B, Wang SW, Wan KL, Jing HQ, Lu JX, Yin WW, Zhou H, Guan XH, Liu JF, Bi ZQ, Liu GH, Ren J, Wang H, Zhao Z, Song JD, He JR, Wan T, Zhang JS, Fu XP, Sun LN, Dong XP, Feng ZJ, Yang WZ, Hong T, Zhang Y, Walker DH, Wang Y, Li DX. 2011. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 364:1523–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elliott RM, Blakqori G. 2011. Molecular biology of orthobunyaviruses, p 1–39 In Plyusnin A, Elliott RM. (ed), Bunyaviridae. Molecular and cellular biology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 11. Carter SD, Surtees R, Walter CT, Ariza A, Bergeron E, Nichol ST, Hiscox JA, Edwards TA, Barr JN. 2012. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J. Virol. 86:10914–10923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferron F, Li Z, Danek EI, Luo D, Wong Y, Coutard B, Lantez V, Charrel R, Canard B, Walz T, Lescar J. 2011. The hexamer structure of Rift Valley fever virus nucleoprotein suggests a mechanism for its assembly into ribonucleoprotein complexes. PLoS Pathog. 7:e1002030 doi:10.1371/journal.ppat.1002030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo Y, Wang W, Ji W, Deng M, Sun Y, Zhou H, Yang C, Deng F, Wang H, Hu Z, Lou Z, Rao Z. 2012. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. U. S. A. 109:5046–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raymond DD, Piper ME, Gerrard SR, Smith JL. 2010. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. U. S. A. 107:11769–11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y, Dutta S, Karlberg H, Devignot S, Weber F, Hao Q, Tan YJ, Mirazimi A, Kotaka M. 2012. Structure of Crimean-Congo hemorrhagic fever virus nucleoprotein: superhelical homo-oligomers and the role of caspase-3 cleavage. J. Virol. 86:12294–12303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elliott RM, Blakqori G, van Knippenberg I, Koudriakova E, Li P, McLees A, Shi X, Szemiel AM. 2013. Establishment of a reverse genetic system for Schmallenberg virus, a newly emerged orthobunyavirus in Europe. J. Gen. Virol. 94:851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moss B, Elroy-Stein O, Mizukami T, Alexander WA, Fuerst TR. 1990. New mammalian expression vectors. Nature 348:91–92 [DOI] [PubMed] [Google Scholar]
- 19. Weber F, Dunn EF, Bridgen A, Elliott RM. 2001. The Bunyamwera virus nonstructural protein NSs inhibits viral RNA synthesis in a minireplicon system. Virology 281:67–74 [DOI] [PubMed] [Google Scholar]
- 20. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67:355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eifan SA, Elliott RM. 2009. Mutational analysis of the Bunyamwera orthobunyavirus nucleocapsid protein gene. J. Virol. 83:11307–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brunotte L, Kerber R, Shang W, Hauer F, Hass M, Gabriel M, Lelke M, Busch C, Stark H, Svergun DI, Betzel C, Perbandt M, Gunther S. 2011. Structure of the Lassa virus nucleoprotein revealed by X-ray crystallography, small-angle X-ray scattering, and electron microscopy. J. Biol. Chem. 286:38748–38756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hastie KM, Liu T, Li S, King LB, Ngo N, Zandonatti MA, Woods VL, Jr, de la Torre JC, Saphire EO. 2011. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. U. S. A. 108:19365–19370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qi X, Lan S, Wang W, Schelde LM, Dong H, Wallat GD, Ly H, Liang Y, Dong C. 2010. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 468:779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ye Q, Krug RM, Tao YJ. 2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082 [DOI] [PubMed] [Google Scholar]
- 29. Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283 [DOI] [PubMed] [Google Scholar]
- 30. Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363 [DOI] [PubMed] [Google Scholar]
- 31. Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360 [DOI] [PubMed] [Google Scholar]
- 32. Raymond DD, Piper ME, Gerrard SR, Skiniotis G, Smith JL. 2012. Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc. Natl. Acad. Sci. U. S. A. 109:19208–19213 [DOI] [PMC free article] [PubMed] [Google Scholar]