

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the cause of Kaposi's sarcoma and body cavity lymphoma. In cell culture, KSHV results in a latent infection, and lytic reactivation is usually induced with the expression of K-Rta or by treatment with phorbol 12-myristate 13-acetate (TPA) and/or n-butyrate. Lytic infection is marked by the activation of the entire viral genomic transcription cascade and the production of infectious virus. KSHV-infected cells express a highly abundant, long, noncoding transcript referred to as polyadenylated nuclear RNA (PAN RNA). PAN RNA interacts with specific demethylases and physically binds to the KSHV genome to mediate activation of viral gene expression. A recombinant BACmid lacking the PAN RNA locus fails to express K-Rta and does not produce virus. We now show that the lack of PAN RNA expression results in the failure of the initiation of the entire KSHV transcription program. In addition to previous findings of an interaction with demethylases, we show that PAN RNA binds to protein components of Polycomb repression complex 2 (PRC2). RNA-Seq analysis using cell lines that express PAN RNA shows that transcription involving the expression of proteins involved in cell cycle, immune response, and inflammation is dysregulated. Expression of PAN RNA in various cell types results in an enhanced growth phenotype, higher cell densities, and increased survival compared to control cells. Also, PAN RNA expression mediates a decrease in the production of inflammatory cytokines. These data support a role for PAN RNA as a major global regulator of viral and cellular gene expression.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is an oncogenic gamma herpesvirus that is the cause of Kaposi's sarcoma (1, 2). KSHV infection in cell culture manifests mainly as a latent infection; however, there is a percentage of spontaneous lytic reactivation. Lytic infection is marked by the production of infectious virus, whereas latency is characterized by the lack of infectious virus production and the expression of a limited number of viral transcripts and proteins. One of the most abundant transcripts present in KSHV-infected cells is a long noncoding RNA, referred to as polyadenylated nuclear RNA (PAN RNA) (3, 4).
We previously showed that PAN RNA interacts with cell- and virus-encoded factors and mediates the regulation of immune response gene expression (5, 6). Data from our laboratory and others indicated that PAN RNA is a major regulator of viral gene expression through a mechanism that involves a direct interaction with the viral chromosome (5, 7). PAN RNA interacts with the demethylases UTX and JMJD3 to remove the suppressive H3K23me3 mark within the KSHV viral genome. In a recent study describing the transcriptome of KSHV-infected cell lines, PAN RNA was detected in the absence of lytic-phase induction, suggesting that PAN RNA is expressed during chronic latent infection in cell culture (8). This observation suggests that PAN RNA has the potential to influence viral and cellular gene expression during all phases of the viral life cycle.
One of the main regulatory mechanisms used by PAN RNA is through the physical interaction with the ORF50 promoter (5). However, we speculate that PAN RNA interacts with and regulates gene expression from other loci within the KSHV genome. Also, since PAN RNA fits the criteria of a regulatory long noncoding RNA (lncRNA), it is also likely that this transcript regulates cellular gene expression. Although we reported that PAN RNA interacts with specific demethylases, lncRNAs also mediate gene-repressive properties through an association with chromatin-modifying complexes such as components of Polycomb repressive complex 2 (PRC2) (9). PRC2 is composed of EZH2, SUZ12, and EED-1. EZH2 is a protein that adds three methyl groups to lysine 27 of histone 3 (10). SUZ12 contains a zinc finger domain that is the point of contact with RNA (11), and EED-1 interacts with HDAC1, histone deacetylase, and various other proteins to mediate gene repression (12). These PRC2 proteins can mediate changes in histone modifications (methylation) and subsequent repression of gene expression from various genetic loci.
As part of an effort to determine the wider role of PAN RNA in the regulation of virus and cellular gene expression, we used chromatin isolation by RNA purification followed by next-generation (deep) sequencing (ChIRP-Seq) to show that PAN RNA broadly interacts with the KSHV genome. RNA-Seq analyses show that the lack of PAN RNA leads to a complete abrogation of the initiation of the viral lytic-phase transcription program. We also show that PAN RNA is the most abundant transcript found within infectious virions and at early times postinfection. With respect to the influence of PAN RNA on cellular gene expression, we demonstrate that PAN RNA interacts with components of PRC2 and can globally influence cellular gene expression. PAN RNA expression primarily mediates changes in cellular gene expression pathways that regulate cell cycle, immune response, and production of inflammatory cytokines. These data strongly suggest that PAN RNA is a multifunctional transcript and can globally regulate viral and cellular gene expression.
MATERIALS AND METHODS
Cells and plasmids.
HEK293L and RPE cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Atlanta Biological). BCBL-1, BJABs, Jurkat, and THP-1 cells were maintained in RPMI supplemented with 10% fetal bovine serum. BAC36CR and BAC36CRΔPAN were described previously (5). 293L cells containing the bacterial artificial chromosome (BAC) constructs were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 125 μg/ml hygromycin B. TREx BCBL-1 Rta cells, provided by J. Jung (University of Southern California), were maintained in RPMI supplemented with 10% FBS and 20 μg/ml hygromycin B. Peripheral blood mononuclear cells (PBMCs) were purchased from Lonza, and donors were deidentified and hence are not considered human subjects.
PAN RNA-expressing lentivirus.
To generate the pLVX-PAN lentivirus, the PAN locus was cloned into the pLVX-Puro vector (Clontech) using a CloneEZ seamless PCR cloning system (Genscript) according to the manufacturer's instructions. First, primers were generated to amplify the PAN locus and add ends homologous to the pLVX vector that was linearized by digestion with EcoRI (New England BioLabs). The forward primer was 5′-cggactcagatctcgagctcaagcttcgaattctTTTAGCACTGGGACTGCCCAGTCA-3′, and the reverse primer was 5′-atcccgggcccgcggtaccgtcgactggaattccTGGATTAAACATTGACCTTTATTT-3′ (lowercase indicates pLVX vector sequences; uppercase indicates the PAN locus). Then, 5 μl of cloning reaction mixture was added to 50 μl chemically competent DH5α Escherichia coli, heat shocked for 30 s at 42°C, and recovered for 1 h in 1 ml SOC medium with shaking in a 37°C incubator. After 1 h, the bacteria were pelleted at 4,000 rpm for 5 min, the supernatant was poured off, and the bacteria were resuspended in the residual liquid and plated on LB agar containing ampicillin and incubated overnight at 37°C. The resulting colonies were grown, and plasmid DNA was isolated and subjected to restriction digestion with EcoRI. Correct colonies were further analyzed for the correct insert by sequencing. Lentivirus was then generated by transfecting the pLVX-PAN plasmid along with packaging plasmids into 293FT cells according to the manufacturer's instructions. At 48 h posttransfection, PAN lentivirus was harvested by collecting cells and supernatant and subjecting the mixture to one round of freeze-thawing to release lentivirus into the supernatant.
PAN RNA-expressing cell lines.
Cell lines were created in BJABs, THP-1, Jurkat, and RPE that stably express PAN RNA by either electroporating pcPAN plasmid (provided by N. Conrad, University of Texas) or transducing with pLVX-PAN lentivirus. To electroporate cells with the pcPAN plasmid, 10 × 106 cells were pelleted at 400 × g for 10 min, the supernatant was removed, and cells were resuspended in 0.5 ml of Dulbecco's modified Eagle medium (DMEM). Then, 100 μg of sheared salmon sperm DNA (Life Technologies) and 1 μg of pcPAN plasmid DNA was added to the cells. The cell-and-DNA mixture was placed in a 4-mm cuvette and placed on ice for 5 min. Electroporation was done in a Cell-porator device set at a 1,600 μF capacitance and 200 V. At 48 h postelectroporation, cells were selected with 2 mg/ml of G418 (Invivogen). Cells were transduced with pLVX-PAN lentivirus, and 48 h postransduction, 2 μg/ml puromycin was used to select cells that were successfully integrated. After selection, cells were grown and assessed for PAN expression by reverse transcription PCR (RT-PCR) and qPCR analysis. PAN RNA-containing cell lines were then used for MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] and growth curve experiments. For both MTT and growth curve experiments, 1 × 103 cells were plated in triplicate in a 96-well tissue culture plate. For growth curve experiments, cells were counted on various days (days 1, 4, 6, and 8) using trypan blue for dead-cell exclusion. The MTT assay was performed every day according to the manufacturer's instructions (Promega).
IL-18 ELISA.
THp-1 and PAN-THP1 cells (1 × 104) were plated in triplicate in a 96-well plate in 100 ml RPMI containing 10% FBS. Cells were either mock treated or primed with 5 ng/ml lipopolysaccharide (Sigma) for 1 h, followed by treatment with 2.5 mM ATP for 6 h at 37°C. Supernatants were harvested, and cytokine analysis was performed. An enzyme-linked immunosorbent assay (ELISA) for interleukin 18 (IL-18) was performed according to the manufacturer's instructions (R&D Systems).
RNA CLIP assay.
BCBL-1 cells (50 × 106) were treated with 0.3 mM sodium butyrate. Forty-eight hours after treatment, cells were harvested, washed once with 1× PBS, and fixed in 1% methanol-free formaldehyde for 10 min. Cells were pelleted, washed once with PBS, and quenched with 125 mM glycine for 5 min. After a final wash with PBS, cells were placed in in 2 ml RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitor cocktail (Sigma), RNase Out (Invitrogen), and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cells were sonicated, and the extract were centrifuged 800 × g for 5 min at 4°C to remove debris. RNA was precipitated by adding 300 μl lysate, 5 μl antibody to SUZ12 and EZH2 (Abcam), 50 μl protein G magnetic beads (Active Motif), and 1 μl RNase Out. This mixture was rotated overnight at 4°C. The input control was 98 μl of lysate mixed with 2 μl of 5 M NaCl and frozen at −80°C until the proteinase K digestion step the following day. After the overnight incubation, the beads were washed once with RIPA buffer (1 ml) and twice with Tris-EDTA (TE; 1 ml). The beads were resuspended in 150 μl elution buffer (1% SDS, 100 mM NaHCO3 [pH 9.0]) for 15 min. The elution step was repeated, the fractions were combined, 60 μl 1 M Tris-HCl (pH 6.8) was added to the elution complexes, proteinase K was added to 0.2 mg/ml to the samples and input, and the samples were incubated at 37°C for 60 min. The cross-links were reversed at 65°C for 18 h, the beads were pelleted, and supernatant was moved into 1 ml TRIzol LS (Invitrogen) and incubated for 5 min at room temperature. Chloroform (250 μl) was added to the TRIzol mixture, mixed by hand, and incubated for 15 min before centrifuging at 12,000 × g for 10 min at 4°C to separate the phases. The upper phase containing the RNA was removed, 1 volume of isopropanol was used to precipitate the RNA, and 1 μl of GlycoBlue (Ambion) was added to aid in visualizing the RNA pellet. After 15 min incubation at room temperature, the mixture was centrifuged at 12,000 × g for 15 min at 4°C and then washed with ice-cold 75% ethanol. The pellet was briefly allowed to air dry and then resuspended in 30 μl nuclease-free water. RNA samples were treated with Turbo DNA-free (Ambion) according to the manufacturer's instructions. A 5-μl portion of the RNA was then used in a Qiagen OneStep RT-PCR kit, using primers specific to an internal region within the PAN locus (forward, TAA TGT GAA AGG AAA GCA GCG CCC; reverse, TAA CAT TGA AAG AGC GCT CCC AGC). The no-RT control was subjected only to the PCR and not the reverse transcriptase step. The U1 primers were ATACTTACCTGGCAGGGGAG (forward) and CAGGGGAAAGCGCGAACGCA (reverse).
Chromatin isolation by RNA purification (ChIRP)-Seq.
Tiling of PAN RNA with biotin-labeled DNA probes retrieves specific PAN RNAs with bound proteins and DNA sequences. The original protocol was from the work of Chu et al. (13). All probes were biotinylated at the 3′ end with an 18-carbon spacer arm; probes were designed against the full-length PAN sequence using an online designer at http://www.singlemoleculefish.com and synthesized at the Protein and Nucleic Acid Facility (Stanford University). PAN and control oligonucleotides were previously published (5). Briefly, 20 × 106 TREx BCBL-1 Rta cells were treated with 1 μg/ml doxycycline to induce the expression of K-Rta and subsequent lytic reactivation of KSHV. Three days postinduction, cells were cross-linked with 1% glutaraldehyde in PBS for 10 min at room temperature. The glutaraldehyde was removed, and the cross-linking was quenched with 0.125 M glycine in PBS. Cells were washed twice with PBS and pelleted at 2,500 × g, and cell pellets were snap-frozen in liquid nitrogen and stored at −80°C. The cell pellets were quickly thawed in a 37°C water bath and resuspended in swelling buffer (0.1 M Tris-HCl [pH 7.0], 10 mM potassium acetate [KOAc], 15 mM MgOAc, freshly added 1% NP-40, 1 mM dithiothreitol [DTT], 1 mM PMSF, complete protease inhibitor [Sigma], and 0.1 unit/microliter Superase-In [Ambion]) for 10 min on ice. The cell suspension was then homogenized in a Dounce homogenizer with a B pestle, and nuclei were pelleted at 2,500 × g for 5 min. Nuclei were lysed in nuclear lysis buffer (50 mM Tris-HCl [pH 7.0], 10 mM EDTA, 1% SDS, and freshly added 1 mM DTT, 1 mM PMSF, complete protease inhibitor, and 0.1 unit/microliter Superase-In) on ice for 10 min and then subjected to sonication (Misonix sonicator with a microtip) to solubilize the chromatin and shear the DNA in the size range of 250 to 500 bp. A 100-μl portion of sheared chromatin was saved as “input” DNA, subjected to treatment with RNase-proteinase K, and extracted with an equal volume of phenol-chloroform-isoamyl alcohol before being precipitated with ethanol. The sheared chromatin was diluted in 2× hybridization buffer (750 mM NaCl, 1% SDS, 50 mM Tris-HCl [pH 7.0], 1 mM EDTA, 15% formamide, and freshly added 1 mM DTT, 1 mM PMSF, complete protease inhibitor, and 0.1 unit/microliter Superase-In). A total of 100 pmol of probes (specific for either PAN or LacZ RNA) was added to 3 ml of diluted chromatin and rotated at 37°C for 4 h. Streptavidin-magnetic C1 beads (Invitrogen) were washed three times in nuclear lysis buffer, blocked with 500 ng/μl yeast RNA (Ambion), 100 μg/ml sheared salmon sperm DNA (Ambion), and 1 mg/ml BSA (Sigma) for 1 h at room temperature, and washed three times again in nuclear lysis buffer before resuspension in its original volume. Blocked and washed beads were added (100 μl per 100 pmol of probes), and the whole reaction mixture was rotated at 37°C for 30 min. Using a magnet, the beads were precipitated and washed twice with 40 bead volumes of 2× SSC wash buffer (2× SSC, 0.5% SDS, and freshly added 1 mM DTT and 1 mM PMSF), then twice in 1× SSC wash buffer (1× SSC, 0.5% SDS, and freshly added 1 mM DTT and 1 mM PMSF), and finally twice in 0.1× SSC wash buffer (0.1× SSC, 0.5% SDS, and freshly added 1 mM DTT and 1 mM PMSF). After the last wash, all residual liquid was removed from the beads. The DNA was eluted from the beads using 3× the original volume of DNA elution buffer (50 mM NaHCO3, 1% SDS, 200 mM NaCl) with 100 μg/ml RNase A (Affymetrix) and 0.1 units/μl RNase H (Ambion). RNase elution was performed twice at 37°C for 20 min, and eluents from both steps were combined. Proteinase K was added (0.2 units/μl) and incubated at 65°C for 45 min. DNA was extracted with an equal volume of phenol-chloroform-isoamyl alcohol (Affymetrix) and precipitated with 100% ethanol at −80°C overnight. Eluted DNA was resuspended in 50 μl of TE and subjected to deep sequencing.
RNA-Seq.
A total of 5 × 105 BAC-containing 293L cells were plated in a 6-well plate. At 24 h after plating, the cells were transfected with either 5 μg of pXi ORF50-Flag or 3 mM sodium butyrate or mock treated. After treatment, the cells were incubated for an additional 72 h before total RNA was harvested. Total RNA was harvested using a Purelink RNA minikit (Life Technologies), followed by removal of genomic DNA using Turbo DNA-free (Life Technologies). RNA was quantified using a Nanodrop spectrophotometer, and 1 μg of total RNA was used with the TruSeq RNA library preparation kit (Illumina) according to the manufacturer's instructions. The RNA-Seq libraries were validated using a Bioanalyzer 2100 with a high-sensitivity DNA chip (Agilent) and quantified by qPCR (Kapa kit). RNA-Seq libraries were sequenced using a MiSeq (Illumina) 50-cycle reagent kit and flow cells. The resulting sequencing data were analyzed using CLC software to determine fold enrichment over untreated wild-type (WT) cells.
Transcriptome analysis of virion RNA.
Transcriptome analysis of virion RNA was performed after extraction of total virion RNA and next-generation (RNA-Seq) sequencing. Approximately 1 billion TRExBCBL1-RTA cells were induced with doxycycline (1 μg/ml) for 5 days. Culture supernatant was collected and filtered through a 0.45-μm filter to remove any cell debris before concentration of the virus by centrifugation at 25,000 rpm for 2 h at 4°C. Concentrated virions were resuspended in 1 ml of phosphate-buffered saline (PBS) and then layered onto a 30-to-50% sucrose gradient for centrifugation for 2 h at 70,000 × g. Purified virus formed an opaque ring, which was collected by puncturing the tube at the band. Sucrose-purified virions were diluted with PBS before centrifugation for 2 h at 25,000 × g.
Total RNA was isolated after treatment of the purified virions with micrococcal nuclease for 30 min at 37°C followed by quenching of the reaction with EGTA. RNA isolated by the TRIzol (Life Technologies) method was treated with DNase I to eliminate viral DNA, followed by inactivation of DNase before preparation of the library for transcriptome analysis. A 2-μg portion of total RNA from each of two independent virus preparations was used for making a cDNA library using a TrueSeq RNA sample preparation kit (v2; Illumina, Inc.). This kit generates fragment sizes of approximately 260 bp, which were confirmed by analyzing the mature libraries on a Bioanalyzer 2100 (Agilent). The quantities of the libraries were determined by qPCR using an Illumina Kapa library quantification kit (Kapa Biosystems). The libraries were sequenced using MiSeq (Illumina), and the sequences were mapped to the reference genome using the RNA-Seq analysis tool of CLC Genomic Workbench 5.5.1 (CLC Bio).
In order to identify the viral genes transcribed early during primary infection, PBMCs from a KSHV-negative donor were infected with purified virions at a multiplicity of infection (MOI) of 1. Noninternalized virions were removed by treating the cells with trypsin after 2 h postinfection. Cells were collected at 0 h, 4 h, 12 h, 24 h, and 48 h postinfection to extract total RNA for transcriptome analysis. Equal amounts of RNA (2 μg) from each time point were used for making cDNA libraries for RNA-Seq analysis. Viral and cellular RNA-Seq analysis was performed using CLC Genomic Workbench 5.5.1 (CLC Bio).
RESULTS
PAN RNA interacts with multiple regions of the KSHV and cellular genomes.
Our previously published results showed that PAN RNA interacts with the ORF50 promoter region (5). This is a significant finding, since one of the defining features for regulatory lncRNAs is a physical interaction with chromatin to mediate changes (activation/suppression) in gene expression. We wanted to investigate if PAN RNA interacted with additional regions of the KSHV genome. We would assume that given the broad regulatory nature and abundance of PAN RNA, other regions of the KSHV genome would be substrates for PAN RNA interactions.
We previously demonstrated an interaction of PAN RNA with the KSHV genome using a powerful and innovative method called chromatin isolation by RNA purification (ChIRP) (5). This method, illustrated in Fig. 1A, shows that by combining ChIRP with next-generation sequencing (ChIRP-Seq), it is possible to identify regions of DNA that are occupied by lncRNA. ChIRP-Seq was successfully used to identify the DNA targets occupied by several cellular lncRNAs (13). For KSHV ChIRP-Seq, we isolated PAN RNA bound to DNA from 3-day lytically induced TREx/BCBL-1 Rta cells using PAN RNA-specific biotinylated oligonucleotides. The isolated DNA was subjected to next-generation sequencing to identify regions of the KSHV genome occupied by PAN RNA. Next-generation sequencing was performed with an Illumina MiSeq instrument. This analysis allowed the identification of KSHV DNA sequences that were precipitated by the ChIRP procedure. We examined binding within 500 bp upstream of known KSHV genes and 1,000 bp upstream of cellular genes, assuming that these regions contained putative promoter and or regulatory regions; however, some intragenic binding regions were also identified (Fig. 1B). Peaks (PAN RNA genomic DNA binding sites) were identified using the ChIP-Seq analysis software within CLC Bio Genomics Workbench, which uses input DNA as a baseline and calculates enriched regions found in experimental samples. Control ChIRP-Seq experiments, where we used LacZ mRNA-specific oligonucleotides, showed no interaction with the KSHV genome.
Fig 1.

Occupancy of PAN RNA on the KSHV genome. (A) Chromatin isolation by RNA purification (ChIRP). Cells were cross-linked, sonicated, and hybridized to 2 sets (24 for PAN RNA) of nonoverlapping biotinylated tiling probes. RNA-protein-DNA complexes were immunoprecipitated with streptavidin magnetic beads. After a series of washes, isolated complexes were treated with RNases H and A to remove the RNA component, resulting in RNA-binding proteins and genomic DNA. Genomic DNA was identified using next-generation sequencing (ChIRP-Seq). (B) Schematic of the KSHV genome and the interaction of PAN RNA. The blue color indicates the DNA sequences identified that were precipitated with PAN RNA. Relative binding scores are shown in Table 1. (C) Consensus PAN RNA binding motifs within the KSHV genome based on ChIRP-Seq analysis. The height of the motif “block” is proportional to the −log (P value). All P values were less than 0.0001. Motifs were identified by MEME analysis and represented in Web Logo format. (D) Venn diagram representing a variety of cellular processes. Within each circle are genes that PAN RNA interacts with based on the results of the ChIRP-Seq.
The number of reads, which is indicative of the quantitative binding of PAN RNA at a specific KSHV locus, is reported in Table 1. ChIRP-Seq analysis confirmed the interaction of PAN RNA with the ORF50 promoter. Interestingly, this analysis indicated that PAN RNA occupies many regions of the KSHV genome, including an interaction with its own promoter locus, suggesting a feedback loop that involves augmenting its own expression (Fig. 1B and Table 1). ChIRP-Seq analysis also showed that PAN RNA occupied the oriLyt-L region, whereas the oriLyt-R region showed no specific PAN RNA interactions (Fig. 1B). This suggests that PAN RNA may regulate DNA replication.
Table 1.
Numbers of reads from ChIRP-Seq analysis of PAN RNA binding to the KSHV genome
| Gene promoter | No. of reads |
|---|---|
| ORF4 | 505 |
| ORF6 | 92 |
| ORF7 | 23 |
| ORF8 | 106 |
| ORF10 | 72 |
| ORF11 | 47 |
| K3 | 124 |
| oriLyt-L | 904 |
| PAN | 1,200 |
| ORF19 | 29 |
| ORF24 | 63 |
| ORF25 | 75 |
| ORF28 | 90 |
| ORF29 | 146 |
| ORF36 | 29 |
| ORF38 | 97 |
| ORF40 | 35 |
| ORF44 | 123 |
| ORF50 | 352 |
| K8 | 73 |
| ORF56 | 132 |
| ORF57 | 117 |
| K10 | 219 |
| K11 | 242 |
| ORF60 | 118 |
| ORF61 | 30 |
| ORF63 | 105 |
| ORF64 | 453 |
| ORF68 | 112 |
| ORF69 | 32 |
| K12 | 138 |
| ORF71 | 67 |
| ORF74 | 328 |
| K14 | 602 |
| K15 | 200 |
Based on the ChIRP-Seq analysis, we identified three major consensus PAN RNA binding motifs within the KSHV genome and five in the cellular genome (http://meme.ebi.edu.au/meme/cgi-bin/meme-chip.cgi) (Fig. 1C).
For cellular DNA interactions, PAN RNA interacted with promoter regions for genes that regulate a variety of cellular processes, including cell cycle, immune response, and energy production (Fig. 1D and Table 2). Overall, 90% of the total reads mapped to the cellular genome. The results of these experiments strongly suggest that PAN RNA is a major regulatory factor for both viral and cellular gene expression with the potential for mediating the expression of many KSHV genes and DNA replication.
Table 2.
PAN RNA cellular genome binding (ChIRP)
| Gene designation | Product |
|---|---|
| ABCA2 | ATP-binding cassette, subfamily A (ABC1), member 2 |
| ABR | Active BCR related |
| CARM1 | Coactivator-associated arginine methyltransferase 1 |
| CCDC109B | Coiled-coil domain containing 109B |
| CDKN2AIPNL | CDKN2A interacting protein N-terminal-like |
| CDKN2B | Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) |
| CLCN6 | Chloride channel, voltage sensitive 6 |
| COQ6 | Coenzyme Q6 |
| COQ7 | Coenzyme Q7 |
| DHFR | Dihydrofolate reductase |
| DHX35 | DEAH (Asp-Glu-Ala-His) box polypeptide 35 |
| DNA2 | DNA replication helicase |
| DNAJB3 | DnaJ (Hsp40) homolog, subfamily B, member 3 |
| DNAJC2 | DnaJ (Hsp40) homolog, subfamily C, member 2 |
| DNAJC16 | DnaJ (Hsp40) homolog, subfamily C, member 16 |
| DNAJC22 | DnaJ (Hsp40) homolog, subfamily C, member 22 |
| DNAJC28 | DnaJ (Hsp40) homolog, subfamily C, member 28 |
| ERK | Extracellular-signal-regulated kinases |
| FAHD2A | Fumarylacetoacetate hydrolase domain containing 2A |
| FBXO31 | F-box protein 31 |
| FGF2 | Fibroblast growth factor 2 (basic) |
| GIT2 | G protein-coupled receptor kinase interacting ArfGAP 2 |
| HDAC4 | Histone deacetylase 4 |
| Histone h3 | Histone H3 |
| Histone h4 | Histone H4 |
| HMCN1 | Hemicentin 1 |
| HOXA2 | Homeobox A2 |
| HOXB9 | Homeobox B9 |
| HSPA6 | Heat shock 70-kDa protein 6 (HSP70B') |
| HSPA12A | Heat shock 70-kDa protein 12A |
| IL-34 | Interleukin 34 |
| INPP5E | Inositol polyphosphate-5-phosphatase, 72 kDa |
| IQSEC1 | IQ motif and Sec7 domain 1 |
| IRAK1 | Interleukin-1 receptor-associated kinase 1 |
| KDM1A | Lysine (K)-specific demethylase 1A |
| KDM2B | Lysine (K)-specific demethylase 2B |
| LRRC6 | Leucine rich repeat containing 6 |
| LSG1 | Large subunit GTPase 1 |
| LZTR1 | Leucine-zipper-like transcription regulator 1 |
| MAEA | Macrophage erythroblast attacher |
| MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 |
| MAPK14 | Mitogen-activated protein kinase 14 |
| MECOM | MDS1 and EVI1 complex locus |
| MRPS33 | Mitochondrial ribosomal protein S33 |
| MRPS18C | Mitochondrial ribosomal protein S18C |
| MST1 | Macrophage stimulating 1 (hepatocyte growth factor-like) |
| MTRF1L | Mitochondrial translational release factor 1-like |
| MUC12 | Mucin 12, cell surface associated |
| MYADM | Myeloid-associated differentiation marker |
| NALP | NOD-like receptor |
| NCAM1 | Neural cell adhesion molecule 1 |
| NDUFA13 | NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 13 |
| NFIX | Nuclear factor I/X (CCAAT-binding transcription factor) |
| NLRP9 | NLR family, pyrin domain containing 9 |
| NUDT6 | Nudix (nucleoside diphosphate-linked moiety X)-type motif 6 |
| ORMDL2 | ORM1-like 2 (Saccharomyces cerevisiae) |
| PAX2 | Paired box 2 |
| PAX5 | Paired box 5 |
| PAX6 | Paired box 6 |
| PAX8 | Paired box 8 |
| PBX1 | Pre-B-cell leukemia homeobox 1 |
| RAD52 | DNA repair |
| RNF213 | Ring finger protein 213 |
| SP2 | Sp2 transcription factor |
| STAC | SH3 and cysteine rich domain |
| TFDP2 | Transcription factor Dp-2 (E2F dimerization partner 2) |
| TGFB1 | Transforming growth factor, beta 1 |
| TIMM21 | Translocase of inner mitochondrial membrane 21 |
| TMEM43 | Transmembrane protein 43 |
| TMEM63B | Transmembrane protein 63B |
| TMUB2 | Transmembrane and ubiquitin-like domain containing 2 |
| TNF | Tumor necrosis factor |
| TRAM2 | Translocation-associated membrane protein 2 |
| TRIM68 | Tripartite motif containing 68 |
| UBC | Ubiquitin C |
| UTP23 | UTP23, small-subunit (SSU) processome component |
| VEGF | Vascular endothelial growth factor |
| ZBTB16 | Zinc finger and BTB domain containing 16 |
| ZC3H7B | Zinc finger CCCH-type containing 7B |
| ZDHHC8 | Zinc finger, DHHC-type containing 8 |
| ZNF142 | Zinc finger protein 142 |
| ZNF330 | Zinc finger protein 330 |
| ZNF586 | Zinc finger protein 586 |
| ZNF772 | Zinc finger protein 772 |
Lack of expression of PAN RNA results in an almost complete abrogation of viral gene expression.
Since the ChIRP-Seq data showed that PAN RNA interacted with a number of regions of the KSHV genome, we wanted to evaluate the expression of individual genes in the absence of PAN RNA. We previously showed that the recombinant BACmid BAC36CRΔPAN, which had the PAN RNA locus deleted, was defective for production of virus. BAC36CRΔPAN showed a decrease in expression of K-Rta, and the overexpression of K-Rta could not compensate for the lack of PAN RNA expression (5). We examined viral gene expression across the entire KSHV genome in WT BAC36CR- or BAC36CRΔPAN-containing cell lines transfected with a plasmid expressing K-Rta. Transfected cell lines were incubated for 3 days, RNA was extracted, and gene expression was measured using RNA-Seq. RNA transcript accumulation from transfected BAC36CRΔPAN-containing cell lines was compared to that in transfected WT BAC36CR-containing cell lines. Evaluation of KSHV gene expression showed that almost the entire KSHV genome is transcriptionally suppressed in the absence of PAN RNA (Fig. 2). Interestingly, several genes—K15, K12, ORF20, ORF31, ORF53, ORF67A, and ORF75—were upregulated compared to WT BAC genes (Fig. 2). ORF75 was expressed at a level that was approximately 50-fold higher in the absence of PAN RNA than what was observed in induced WT BAC (Fig. 2). LANA mRNA was present at the same level as WT BAC36CR, suggesting that the lack of PAN RNA does not affect LANA expression (Fig. 2). These data suggest that PAN RNA, through a physical interaction with the KSHV genome, mediates the activation of most of the KSHV transcription program during lytic infection and in some cases may negatively regulate KSHV gene expression.
Fig 2.

Disruption of KSHV transcription program in the absence of PAN RNA. RNA-Seq analysis was performed by transfecting a K-Rta expression plasmid into cell lines containing either BAC36CRΔPAN or WT BAC36CR. Expression of K-Rta in transfected cell lines is shown by Western blotting (inset). Values are fold change versus BAC36 plus K-Rta. RNA-Seq evaluations were done in triplicate, and error bars show standard deviations of the means.
PAN RNA is an abundant transcript present within the virion and at 4 h postinfection of PBMCs.
Our data suggest that PAN RNA is essential for virus replication and that it is the major regulatory factor controlling virus gene expression. We next wanted to evaluate the amount of PAN RNA present within the KSHV virion and the amount of PAN RNA that is present early after infection. Previous studies have shown that PAN RNA is a component of the virion (14). For our studies, we used RNA isolated from purified virus and performed next-generation sequencing (virion RNA-Seq). Also, we infected cells and evaluated the amount of virus-encoded RNA transcripts present at 4 h postinfection. The goal was to determine the amount of input (virion) PAN RNA compared to the amount produced early in infection. Although small amounts of many KSHV-encoded RNA transcripts were detected within virions, three transcripts stand out as the most abundant, PAN, K12, and K15 RNA (Fig. 3A). These data are consistent with what was previously reported in that PAN RNA was present within the KSHV virion at very high levels (14). At 4 h postinfection, PAN RNA was the most abundant transcript found in the virus transcriptome (Fig. 3B). In addition, transcripts encoding ORF58/59 and K12 were detected (Fig. 3B). Hence, PAN RNA, either originating from virus input or the result of early transcription, is the most abundant virus-encoded transcript. These data suggest that PAN RNA has the ability to regulate virus and host cell gene expression at the time of infection as well as during early time points after initial infection.
Fig 3.

PAN RNA is highly abundant in KSHV virions and at early times postinfection. (A) RNA-Seq analysis of KSHV purified virions. KSHV virions were purified from TREx/BCBL-1 Rta cells treated with doxycycline. Virions were prepared by sucrose gradient purification followed by DNase I treatment. Purified virions were lysed and DNase I treated, and RNA was extracted and used to prepare a next-generation sequencing library. (B) RNA-Seq analysis 4 h after infection of PBMCs. Cells were infected, and RNA was isolated and used to prepare a library for next-generation sequencing. Shown is a peak map of next-generation sequencing reads where the peak heights correspond to the relative abundance of transcripts. The peak maps are averages from 3 separate experiments. Numbers in parentheses are the numbers of reads for several selected transcripts. Also shown are the 8 next most abundant RNAs and reads.
PAN RNA is expressed in uninduced BCBL-1 cells.
Since we observed a high abundance of PAN RNA upon infection of primary cells, we wanted to evaluate the expression of PAN RNA under latent conditions in BCBL-1 cells. We performed RNA-Seq to evaluate the expression level of PAN RNA in uninduced, induced, and sodium phosphonoformate tribasic hexahydrate (PFA)-treated BCBL-1 cells. The goal of these experiments was to determine if PAN RNA is present in latently infected cells. Since we acknowledge that a low level of spontaneous lytic reactivation occurs in a population of uninduced BCBL-1 cells, we also evaluated PAN RNA expression in BCBL-1 cells treated with PFA, an inhibitor of herpesvirus DNA polymerase and lytic DNA replication. As a control, we also induced lytic DNA replication by treatment with n-butyrate. Highly sensitive RNA-Seq showed that PAN RNA is expressed in uninduced/latently infected BCBL-1 cells (Fig. 4A). This suggests that PAN RNA is present during latent infection. Treatment with n-butyrate resulted in a 20-fold increase in PAN RNA accumulation (Fig. 4B). Also, treatment with PFA, an inhibitor of lytic DNA replication, decreased PAN RNA accumulation only slightly compared to no induction in BCBL-1 cells (Fig. 4C). These data suggest that PAN RNA and activities associated with it, namely, regulation of gene expression, can occur in the absence of lytic DNA replication. Further, PAN RNA is present during a nonlytic, latent infection in BCBL-1 cells.
Fig 4.
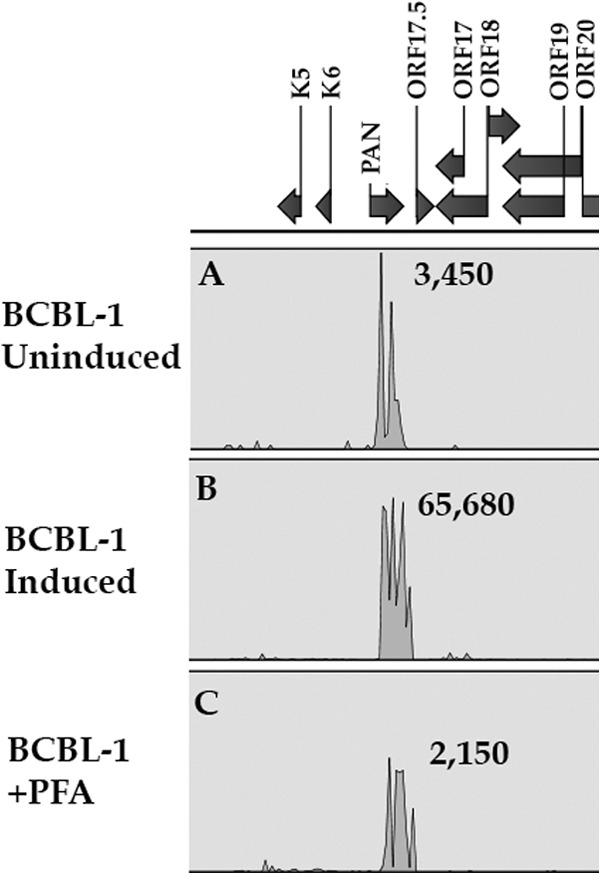
PAN RNA is expressed in uninduced BCBL-1 cells. RNA was harvested from BCBL-1 cells that were untreated, treated with n-butyrate, or incubated with PFA. Next-generation sequencing libraries were prepared, and RNA-Seq was performed. Shown is a peak map of reads for the PAN RNA locus from each sample. The peak maps are averages from three independent experiments.
PAN RNA is present in the absence of K-Rta.
Since the early infection data from PBMCs suggested that PAN RNA was highly abundant when K-Rta expression was low and PAN RNA was present in uninduced, latently infected BCBL-1 cells, we wanted to investigate if PAN RNA expression was completely dependent upon K-Rta. In addition to data from PBMCs and BCBL-1 cells, the expression of PAN RNA in the absence of K-Rta would establish PAN RNA as a latent as well as lytic transcript. Hence, many of the activities associated with PAN RNA, as a regulatory lncRNA, could occur during viral persistence (latency) as well as in the lytic phase of replication. To this end, we wanted to determine if PAN RNA expression, as well as other viral gene expression, could occur in the absence of K-Rta. Therefore, we evaluated KSHV transcription in the context of the viral genome in the absence of K-Rta expression. We previously generated a recombinant KSHV BACmid with a deletion in the ORF50 locus (15). This virus was unable to reactivate or produce infectious virus. We now wanted to evaluate gene expression patterns from the deleted ORF50 KSHV BAC using high-resolution next-generation sequencing. HEK293 cells were transfected with BAC36ΔORF50, and cell lines were selected. Three independent cells lines were isolated, RNA was harvested, and next-generation sequencing libraries were generated. RNA-Seq was performed, and expression profiles from the ORF50 deletion-containing BAC were compared to those of WT BAC36. RNA-Seq revealed that, even in the absence of K-Rta expression, several genes were still expressed, including PAN RNA (Fig. 5). These data are very interesting and show that many KSHV genes are not dependent on K-Rta transactivation. This is the first evaluation of KSHV gene expression in the absence of K-Rta. Collective data presented here suggest that PAN RNA is a ubiquitous transcript and its expression is not dependent upon K-Rta or lytic DNA replication.
Fig 5.
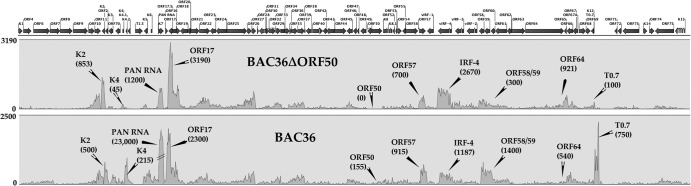
PAN RNA is expressed in the absence of K-Rta. Gene expression in the presence or absence of K-Rta expression was analyzed by RNA-Seq. WT BAC or BAC36CRΔPAN was transfected into HEK293 cells, and cell lines were selected. RNA was harvested, and next-generation sequencing libraries were prepared. Shown are peak maps where the read numbers reflect the abundance of RNA transcripts normalized to GAPDH expression. Three separate cell lines were selected for each BAC, and the collective averaged data are shown. Numbers in parentheses are numbers of reads for several selected transcripts.
PAN RNA interacts with protein components within Polycomb repressive complex 2 (PRC2).
One mechanism for repression of gene expression by lncRNAs is by the interaction of chromatin-modifying complexes to mediate changes in histone modifications. We postulate that PAN RNA is a multifunctional transcript that can activate or repress gene expression. This assumption is consistent with previously published observation that several genes associated with immune modulation were suppressed (6). Also, it was recently demonstrated that EZH2 plays a role in KSHV cancer angiogenesis (16). Hence, we wanted to test the possibility that PAN RNA interacts with protein components comprising the PRC2.
We performed RNA cross-linking immunoprecipitation (rCLIP) assays using TREx/BCBL-1 Rta treated with doxycycline (DOX) to induce lytic replication. rCLIP assays were performed using antibodies specific for SUZ12 or EZH2. After an RT step, we then used PCR primers specific for PAN RNA or unrelated RNAs (ORF45 and U1 RNA). As additional controls, we used an isotype-specific antibody, beads alone, and omission of reverse transcriptase. The rCLIP assay showed that PAN RNA does interact with PRC2 proteins (Fig. 6A). Control samples showed no PCR amplification products (Fig. 6A, ORF45 and U1). Hence, these data strongly suggest that PAN RNA has the capacity either to repress gene expression by interacting with PRC2 to mediate the trimethylation of H3K27 or to activate gene expression by interacting with UTX and JMJD3 and the methyltransferase MLL2 to mediate the removal of the H3K27me3 mark and simultaneously mark for activation (Fig. 6B).
Fig 6.

PAN RNA interacts with Polycomb repression complex proteins EZH2 and SUZ12. (A) rCLIP assays were performed using TREx/BCBL-1 Rta cells treated with DOX. Protein-PAN RNA complexes were immunoprecipitated using antibodies specific for EZH2 or SUZ12. Following the generation of cDNA, PCR primers specific for either PAN, ORF45, or U1 mRNA sequences were used to detect immunoprecipitated transcripts. Isotype control antibodies and beads alone were used as controls. Also, reverse transcriptase was omitted from the PAN RNA PCR sample (noRT). (Inset) Western blot of expression of SUZ12 and EZH2. (B) Schematic suggesting PAN RNA has the capacity either to repress gene expression by interacting with the PRC2 complex to methylate histones or to activate gene expression by interacting with methyltransferases.
PAN RNA mediates changes in cellular gene expression programs in BJAB cells.
Since we determined that PAN RNA interacts with both PCR2 proteins as well as demethylases, it is possible that PAN RNA either activates or represses gene expression. We previously demonstrated that PAN RNA could dysregulate the accumulation of transcripts that encode proteins that are involved in immune response. Hence, one goal for our studies is to understand the global influence of PAN RNA on cellular gene expression. We examined the transcriptome for BJAB cells that constitutively express PAN RNA. PAN RNA-expressing BJAB (BJAB-PAN) and control BJAB cell lines were generated using a lentivirus vector that expressed PAN RNA. This lentivirus, along with a control lentivirus that expressed enhanced green fluorescent protein (EGFP), was used to transduce BJAB cells. Cell lines were selected and confirmed to express PAN RNA or EGFP using RT-PCR and qPCR. Three independent cell lines were expanded and subjected to RNA-Seq analysis.
Total cellular RNA was harvested and the transcriptome for PAN RNA-expressing cells and control cells was elucidated (RNA-Seq) using next-generation sequencing (HiSeq 2000; 100 million reads for each sample). mRNA accumulation was quantitated for BJAB-PAN cells using the BJAB-EGFP transcriptome as a control. We used a cutoff of more than a 5-fold change in transcript levels compared to the control. Statistical analysis was performed using the t test described by Kal et al., with a P value of <0.001 (17). The goal of the study was to identify changes in global gene expression in the presence of PAN RNA. RNA-Seq analysis identified 115 transcript levels that were up- or downregulated by PAN RNA expression in BJAB cells 5-fold or more. The RNA-Seq analysis generated many possible cellular networks of protein pathways that could potentially be affected by the expression of PAN RNA. However, one of the most striking results of this analysis was that cellular transcription programs regulating proteins involved in cell cycle and immune function were highly dysregulated.
Cellular gene expression networks were generated by importation of CLC Bio Genomics Workbench data into Ingenuity IPA software (Ingenuity Systems). Figure 7 shows two putative network pathways where transcripts were up- or downregulated in the presence of PAN RNA expression. One network pathway revolves around an approximate 19-fold decrease in the expression of cyclin-dependent kinase inhibitor 2B (CDKN2B) and a 9-fold decrease in cyclin-dependent kinase inhibitor 1A (CDKN1A) (Fig. 7A). CDKN2B is often deleted or mutated in many tumors and forms a complex with CDK4 or CDK6. CDKN2B prevents the activation of the CDKs by cyclin D. CDKN2B inhibits cell growth by regulating the progression of the cell through G1. CDKN1A interacts with CDK2, CDK4, and PCNA (and other factors) to control the cell cycle. Within this same pathway, several other genes were downregulated, including ATF3 (expression decreased 15-fold), tumor necrosis factor receptors, and IRF6 (expression increased 18-fold) (Fig. 7A). The dysregulation of this pathway by PAN RNA expression strongly suggests changes in cell cycle, cell growth, and immune function.
Fig 7.

Dysregulation of cellular networks in the presence of PAN RNA expression. (A) PAN RNA expression in BJAB cells affects transcription of genes controlling cell growth regulators and immune function. (B) PAN RNA expression in BJAB cells affects transcription of genes controlling inflammatory response regulators. Pathway analysis was generated using IPA software. Transcript number was calculated using a paired t test of RPKM values and 3 biological replicates. Green indicates a decrease in transcripts; red indicates an increase in transcripts; white signifies factors within a pathway but not directly affected. Genes shown in networks are listed in Table 3.
A second identified cellular network pathway suggests that PAN RNA influences inflammatory response with the downregulation of several transcription factors, IRF7, STAT4, FOSB, NLRP3, NOD2, and ELF3. Also downregulated were transcripts encoding several tumor necrosis factor receptors (TNR), chemokines CLC2, CLC3, CLC4, and CXCL11, and interleukin 18 receptor (IL18R1) (Fig. 7B). Interestingly, RNA-Seq also identified an increase in transcription for the potassium voltage gated channel (KCNQ1; 8-fold increase) and an increase in the tumor necrosis factor ligand (TNFSF15; 8-fold increase). Table 3 shows the list of transcripts for factors shown in the networks and the fold changes in PAN RNA-expressing cells versus control cells. These data support our original observation that PAN RNA can regulate immune response factors. Also, the data suggest that PAN RNA can broadly affect immune response and regulate the production of inflammatory cytokines, and they point to a complex regulatory dynamic controlled by PAN RNA, which was also observed with other previously characterized human lncRNAs.
Table 3.
Fold changes in transcript accumulation in PAN-expressing cells versus control
| Symbol | Entrez gene product | Fold change | P value |
|---|---|---|---|
| TNFRSF13B | Tumor necrosis factor receptor | −109.247 | 8.84E-13 |
| IL18R1 | Interleukin 18 receptor 1 | −50.398 | 3.47E-12 |
| EGR2 | Early growth response 2 | −29.761 | 6.13E-13 |
| PROC | Protein C (inactivator of coagulation factors Va and VIIIa) | −27.868 | 5.78E-14 |
| TNFRSF1B | Tumor necrosis factor receptor superfamily, member 1B | −24.004 | 6.63E-13 |
| CSF1 | Colony-stimulating factor 1 | −23.527 | 8.93E-11 |
| ARRDC4 | Arrestin domain containing 4 | −22.599 | 1.64E-14 |
| TREM1 | Triggering receptor expressed on myeloid cells 1 | −22.308 | 9.42E-11 |
| CDKN2B | Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) | −18.880 | 9.10E-15 |
| ATF3 | Activating transcription factor 3 | −15.733 | 5.64E-14 |
| PTGIR | Prostaglandin I2 (prostacyclin) receptor (IP) | −13.635 | 3.73E-11 |
| NR4A3 | Nuclear receptor subfamily 4, group A, member 3 | −12.802 | 4.83E-11 |
| TFF1 | Trefoil factor 1 | −10.087 | 7.78E-12 |
| NOD2 | Nucleotide-binding oligomerization domain containing 2 | −10.084 | 7.83E-11 |
| NGFR | Nerve growth factor receptor | −10.032 | 4.20E-14 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | −8.797 | 1.29E-14 |
| IRF7 | Interferon-regulatory factor 7 | −8.768 | 2.64E-14 |
| ZBP1 | Z-DNA binding protein 1 | −8.512 | 2.26E-11 |
| CARD9 | Caspase recruitment domain family, member 9 | −8.329 | 5.04E-14 |
| NT5E | Ecto-5′-nucleotidase (CD73) | −7.848 | 8.28E-11 |
| RGS1 | Regulator of G-protein signaling 1 | −7.812 | 4.13E-13 |
| BAG3 | BCL2-associated athanogene 3 | −7.529 | 1.40E-14 |
| STAT4 | Signal transducer and activator of transcription 4 | −6.892 | 6.11E-12 |
| ULBP2 | UL16-binding protein 2 | −6.780 | 2.76E-13 |
| MT1F | Metallothionein 1F | −6.359 | 1.10E-11 |
| DUSP1 | Dual-specificity phosphatase 1 | −6.285 | 3.38E-14 |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B | −6.275 | 1.27E-13 |
| KRT17 | Keratin 17 | −6.101 | 2.38E-10 |
| PHLDA2 | Pleckstrin homology-like domain, family A, member 2 | −5.950 | 1.10E-10 |
| DUSP5 | Dual-specificity phosphatase 5 | −5.860 | 1.26E-12 |
| CCL3 | Chemokine (C-C motif) ligand 3 | −5.746 | 5.53E-13 |
| CXCL11 | Chemokine (C-X-C motif) ligand 11 | −5.741 | 8.81E-12 |
| NLRP3 | NLR family, pyrin domain containing 3 | −5.602 | 1.03E-09 |
| NEU1 | Sialidase 1 (lysosomal sialidase) | −5.554 | 6.43E-12 |
| UBD | Ubiquitin D | −5.346 | 2.69E-10 |
| PPP1R15A | Protein phosphatase 1, regulatory subunit 15A | −5.173 | 5.71E-12 |
| CCL4 | Chemokine (C-C motif) ligand 4 | −5.132 | 1.01E-13 |
| TNFRSF6B | Tumor necrosis factor receptor superfamily, member 6b, decoy | −5.092 | 2.79E-11 |
| ELF3 | E74-like factor 3 (ETS domain transcription factor, epithelial-specific) | −5.055 | 1.46E-12 |
| HES1 | Hairy and enhancer of split 1 (Drosophila) | −5.002 | 1.57E-13 |
| CCL22 | Chemokine (C-C motif) ligand 22 | −5.000 | 2.32E-11 |
| CD1A | CD1a molecule | 6.341 | 9.89E-11 |
| TNFSF15 | Tumor necrosis factor (ligand) superfamily, member 15 | 8.176 | 1.59E-11 |
| KCNQ1 | Potassium voltage-gated channel, KQT-like subfamily, member 1 | 8.355 | 7.02E-14 |
| NOTCH3 | Notch 3 | 10.217 | 2.46E-09 |
| PBX1 | Pre-B-cell leukemia homeobox 1 | 16.064 | 2.83E-11 |
| IRF6 | Interferon-regulatory factor 6 | 18.042 | 4.54E-11 |
| P2RX7 | Purinergic receptor P2X, ligand-gated ion channel, 7 | 42.302 | 7.69E-11 |
Cells expressing PAN RNA display an enhanced growth phenotype and reach greater densities in culture than control cells.
Since the RNA-Seq data implicated PAN RNA as a factor controlling cell growth, we investigated if PAN RNA expression impacts cell growth and viability. We generated several cell lines in different cell types that expressed PAN RNA. Cell lines were generated using either a lentivirus or a plasmid that expressed the PAN RNA. These cell lines—BJAB-PAN, Jurkat-PAN, THP-PAN, and primary retinal pigmented epithelium (RPE) cells—were selected and grown in culture. As a control, cell lines were generated that were transfected with empty plasmid or transduced lentivirus vector. Expression of PAN RNA was confirmed using reverse transcriptase PCR as well as qPCR assays (data not shown). PAN RNA-expressing and control cell lines were seeded in triplicate and counted over an 8-day period (for BJAB and Jurkat cells). In all cases, cells expressing PAN RNA appeared to have decreased doubling times and reached a higher growth density than control cell lines (Fig. 8). MTT assays showed that cells were healthy and cell viability was high for each cell line expressing PAN RNA (Fig. 8).
Fig 8.

Enhanced cell growth and viability in PAN RNA-expressing cell lines. Cells of the PAN RNA-expressing lines BJAB, Jurkat, and RPE were grown in culture for 5 to 8 days. Cell number and cell viability (MTT assays) were evaluated at 1, 4, 6, and 8 days (5 days for RPE). Each data point was generated from three separate replicates for each time point.
We also investigated the effects of PAN RNA expression on normal (untransformed) cells in culture. We transduced primary RPE cells with the PAN RNA-expressing lentivirus or empty lentivirus vector. Cells were grown in culture for 5 days, and cell viability was evaluated at 1, 2, 3, 4, and 5 days. Viability of RPE-PAN cells was significantly higher than that of control cells (Fig. 8). Cells counts at 5 days were 1.2 × 106 for RPE-PAN and 172,000 for RPE (each culture contained 5,000 cells on day 0). Hence, even in normal cells, PAN RNA expression apparently increased cell viability and decreased cell doubling times. These data strongly suggest that PAN RNA can mediate changes in cellular gene expression that lead to enhanced cell growth and increased survival of cells in culture.
PAN RNA expression inhibits the expression of inflammatory cytokines.
RNA-Seq results show that NLRP3 and NOD2 mRNA levels were reduced approximately 6- and 11-fold, respectively (Fig. 7). Also, tumor necrosis factor receptors (TNRFS1B and TNRFS13B) were reduced 25- and 100-fold, respectively. Consistent with the regulation of immune response, ELF3 was also downregulated in PAN RNA-expressing cells. ELF3 expression is regulated by IL1B. Expression of STAT4, a factor involved in mediating responses to IL-12, was reduced approximately 7-fold. These data strongly suggest that PAN RNA is capable of globally suppressing host genes that regulate inflammatory response and release of inflammatory cytokines.
We examined THP1-PAN and control THP1 cells (15,000 cells each) that had been treated with lipopolysaccharide (LPS) and subsequently stimulated with ATP for their ability to induce IL-18 production. If PAN RNA expression affected the release of IL-18, we would expect a decrease in the measurable amount of IL-18 in the supernatant. Results show that the release of IL-18 in THP1 PAN cells was suppressed (Fig. 9). Hence, these results strongly suggest that PAN RNA also acts to increase survival of the host cell by suppressing the expression of inflammatory cytokines.
Fig 9.
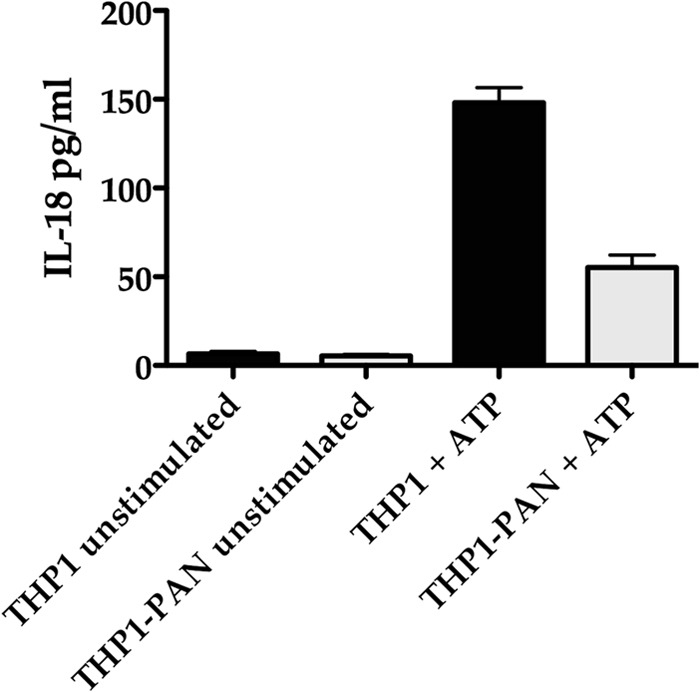
PAN RNA expression inhibits release of IL-18. THP-1 cells expressing PAN RNA and THP-1 control cells transduced with empty lentivirus were treated with LPS for 6 h and then incubated with ATP. IL-18 release was measured by ELISA and normalized to LDH in the supernatant. Error bars show standard deviations for 3 biological replicates.
DISCUSSION
Regulation of viral and cellular gene expression in KSHV infected cells is complex. Although viral protein factors have the capacity to mediate changes in gene expression, increasing and significant evidence suggests that lncRNAs are major regulators of gene expression in cells (18–21). lncRNAs can mediate global changes in gene expression and affect many different transcription profiles. For KSHV, PAN RNA is the most abundant transcript in infected cells (4). Although it is assumed to be primarily a lytic transcript, data presented in this report show that PAN RNA is highly abundant within virions and is the most abundant transcript present at 4 h postinfection. Also, recent studies showed that PAN RNA may also be present in KSHV-infected cell lines that are uninduced, arguing that the transcript is produced during latency (8). Despite the abundant presence of PAN RNA at 4 h after infection of primary PBMCs, we did not observe a large increase in accumulation of K-Rta mRNA. This is perhaps due to the multifunctional nature of PAN RNA, as PAN plays more of a regulatory role early in infection, controlling host cell gene expression. Other studies have examined gene expression patterns during primary infection (22). We have now extended these studies using highly sensitive RNA-Seq. These early PAN RNA-related regulatory events may serve to change the cellular environment such that viral persistence can occur. One obvious question from the work presented here is the following: if PAN RNA is a lytic factor, then why would the transcript have regulatory properties that affect cell survival? The primary cell infection data coupled with the fact that PAN RNA is present in both uninduced and PFA-treated BCBL1 argue that PAN is a ubiquitous transcript present during latent infection (or in the absence of lytic infection). Also, using the ORF50-deletion-containing BAC, we show that the expression of K-Rta is not required for the expression of PAN RNA, further evidence that PAN RNA is present during latency. Hence, PAN RNA has the capacity to regulate cellular gene expression, specifically the genes controlling cell cycle, inflammation, and immune response at all phases of infection. The K-Rta mutant virus gene expression data challenges the dogma that K-Rta expression is essential for widespread viral gene expression. In the context of PAN RNA, we demonstrate that its expression can occur in the absence of lytic replication or the expression of K-Rta.
We demonstrate that PAN RNA interacts with many regions of the KSHV and cellular chromosome and that almost the entire transcription program of KSHV is dependent on PAN RNA expression. We show that PAN RNA interacts with many promoter regions for cellular genes, which is consistent with the RNA-Seq data showing that PAN RNA expression can influence the changes in expression of several cellular factors. These data are also consistent with the general activity of lncRNAs, which have the capacity to globally impact gene expression. The MEME data from PAN RNA interaction with cellular DNA suggest that, at least in some cases, PAN RNA uses motifs of known transcription factor binding sites. This is interesting and suggests that PAN RNA may target specific genes that are activated by particular cellular transcription factors. For example, one motif identified was the forkhead box D3 (FOXD3) transcription factor binding site. FOXD3 is a transcriptional repressor, and hence PAN RNA may silence gene expression by occupying this site within cellular DNA promoters (23). Our data strongly suggest that PAN RNA functions as the trigger for mediating the release of H3K27me3 suppression on the KSHV genome through an interaction with the demethylases UTX and JMJD3. This is consistent with an earlier study demonstrating that most of the KSHV genome is silenced through epigenetic control, which can be released by the overexpression of H3K27me3-specific demethylases (24). New data, presented in this report, also show that PAN RNA interacts with PRC2 and therefore has the capacity to suppress as well as activate gene expression.
In the context of gene repression, we also explored the role of PAN RNA in suppression of specific cellular gene expression. Using an experimental PAN RNA expression protocol where BJAB cells were transduced with a lentivirus that allowed constitutive expression of PAN RNA, we show that transcription programs involving cell cycle, immune response, and inflammation are disrupted. Although it is acknowledged that in KSHV-infected cells, PAN RNA associates with ORF57, which stabilizes the transcript (25–27), we wanted to evaluate the effects of PAN RNA in the absence of any other KSHV factors. PAN RNA expression was confirmed by RNA-Seq and qPCR; hence, expression of PAN RNA itself in the absence of ORF57 is a stable transcript. The dysregulation of cell cycle protein expression results in an enhanced growth phenotype in several cell types. This is the first report of the ability of a virally encoded lncRNA to affect cellular growth characteristics. We consistently observed the ability of PAN RNA-expressing cells to reach greater cell densities than control cells. Other lncRNAs were also demonstrated to mediate cell cycle effects (28, 29). The ability of PAN RNA to regulate cell cycle and survival appears to be complex and due to a global dysregulation of several gene expression programs and not a result of a targeted up- or downregulation of one protein or pathway. The multifunctional activity of lncRNAs, which includes both suppression and activation of gene expression, is a new and significant feature of lncRNAs (30).
PAN RNA expression also dysregulates the expression of cellular factors that contribute to the expression of inflammatory cytokines. Earlier reports demonstrated that the KSHV-encoded protein ORF63 can inhibit the release of inflammatory cytokines by interacting with the components of the inflammasome (31). PAN RNA expression downregulates the transcription of the inflammasome components NOD2, NLRP3, and Card9 (32). Transcripts encoding these proteins were reduced 5- to 10-fold in cells expressing PAN RNA. In order to follow up and confirm these RNA-Seq data, we evaluated the ability of THP-1 cells to release inflammatory cytokines in the presence of PAN RNA. Interestingly, there was a decrease in mRNA accumulation of the IL-18 receptor IL18R1 of approximately 50-fold. Hence, a decrease in the expression of inflammasome components along with a decrease in expression of the IL-18 receptor underscores the global targeting of suppression of specific gene expression pathways by PAN RNA. Our data show that PAN RNA suppresses the release of inflammatory cytokines, which could also contribute to increased cell survival in KSHV-infected cells.
RNA-Seq analysis also identified a decrease in expression of IRF7 mRNA in PAN RNA-expressing BJAB cells. IRF7 is a key factor in interferon mediated antiviral response, and several reports have described the ability of KSHV proteins to interfere with IRF7 activity (33–36). PAN RNA globally dysregulates immune response, apparently by at least two mechanisms. PAN RNA has the ability to directly interact with IRF4 (6) as well as affecting gene expression of other factors involved in immune response, as reported here.
Because of its high abundance in KSHV virions and infected cells, PAN RNA has the ability to act as a major regulator of viral and cellular gene expression. We now show that PAN RNA primarily affects gene expression of factors that affect cell growth and survival, immune response, and inflammation. PAN RNA is a potent regulator of the entire viral transcription cascade as well as globally deregulating specific cellular transcription programs. Based on our previous results and the data shown here, one possible mechanism for the observed effects on cellular gene expression is through interaction with both specific demethylases as well as with PRC2. Hence, PAN RNA has the ability to up- or downregulate gene expression. As with many previously characterized lncRNAs, PAN RNA can act as a molecular scaffold to mediate epigenetic changes in histone modifications. Although the mechanism for specific targeting of PAN RNA promoters is unknown, we have identified several consensus DNA sequence motifs associated with PAN RNA binding. Further study is necessary to fully understand the exact mechanisms involved in coordination of the interaction of PAN RNA with specific chromatin.
Footnotes
Published ahead of print 6 March 2013
REFERENCES
- 1. Martin JN. 2011. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 and Kaposi sarcoma. Adv. Dent. Res. 23:76–78 [DOI] [PubMed] [Google Scholar]
- 2. Uldrick TS, Whitby D. 2011. Update on KSHV epidemiology, Kaposi sarcoma pathogenesis, and treatment of Kaposi sarcoma. Cancer Lett. 305:150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang PJ, Shedd D, Gradoville L, Cho MS, Chen LW, Chang J, Miller G. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sun R, Lin SF, Gradoville L, Miller G. 1996. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 93:11883–11888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 8:e1002680 doi:10.1371/journal.ppat.1002680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rossetto CC, Pari GS. 2011. Kaposi's sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J. Virol. 85:13290–13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borah S, Darricarrere N, Darnell A, Myoung J, Steitz JA. 2011. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog. 7:e1002300 doi:10.1371/journal.ppat.1002300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dresang LR, Teuton JR, Feng H, Jacobs JM, Camp DG, 2nd, Purvine SO, Gritsenko MA, Li Z, Smith RD, Sugden B, Moore PS, Chang Y. 2011. Coupled transcriptome and proteome analysis of human lymphotropic tumor viruses: insights on the detection and discovery of viral genes. BMC Genomics 12:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043 [DOI] [PubMed] [Google Scholar]
- 11. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Vlag J, Otte AP. 1999. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23:474–478 [DOI] [PubMed] [Google Scholar]
- 13. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44:667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bechtel J, Grundhoff A, Ganem D. 2005. RNAs in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:10138–10146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He M, Zhang W, Bakken T, Schutten M, Toth Z, Jung JU, Gill P, Cannon M, Gao SJ. 2012. Cancer angiogenesis induced by Kaposi's sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 72:3582–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kal AJ, van Zonneveld AJ, Benes V, van den Berg M, Koerkamp MG, Albermann K, Strack N, Ruijter JM, Richter A, Dujon B, Ansorge W, Tabak HF. 1999. Dynamics of gene expression revealed by comparison of serial analysis of gene expression transcript profiles from yeast grown on two different carbon sources. Mol. Biol. Cell 10:1859–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moran VA, Perera RJ, Khalil AM. 2012. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 40:631–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saxena A, Carninci P. 2011. Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays 33:830–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang KC, Chang HY. 2011. Molecular mechanisms of long noncoding RNAs. Mol. Cell 43:904–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wapinski O, Chang HY. 2011. Long noncoding RNAs and human disease. Trends Cell Biol. 21:354–361 [DOI] [PubMed] [Google Scholar]
- 22. Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katoh M, Katoh M. 2004. Human FOX gene family (review). Int. J. Oncol. 25:1495–1500 [PubMed] [Google Scholar]
- 24. Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013 doi:10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Massimelli MJ, Kang JG, Majerciak V, Le SY, Liewehr DJ, Steinberg SM, Zheng ZM. 2011. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5′ end to interact with viral ORF57 and cellular PABPC1. Int. J. Biol. Sci. 7:1145–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sahin BB, Patel D, Conrad NK. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 6:e1000799 doi:10.1371/journal.ppat.1000799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sei E, Conrad NK. 2011. Delineation of a core RNA element required for Kaposi's sarcoma-associated herpesvirus ORF57 binding and activity. Virology 419:107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berteaux N, Lottin S, Monte D, Pinte S, Quatannens B, Coll J, Hondermarck H, Curgy JJ, Dugimont T, Adriaenssens E. 2005. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 280:29625–29636 [DOI] [PubMed] [Google Scholar]
- 29. Lottin S, Adriaenssens E, Dupressoir T, Berteaux N, Montpellier C, Coll J, Dugimont T, Curgy JJ. 2002. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis 23:1885–1895 [DOI] [PubMed] [Google Scholar]
- 30. Flynn RA, Chang HY. 2012. Active chromatin and noncoding RNAs: an intimate relationship. Curr. Opin. Genet. Dev. 22:172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B. 2011. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331:330–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10:417–426 [DOI] [PubMed] [Google Scholar]
- 33. Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. 2007. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81:8282–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liang Q, Fu B, Wu F, Li X, Yuan Y, Zhu F. 2012. ORF45 of Kaposi's sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKε and TBK1 as an alternative substrate. J. Virol. 86:10162–10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu FX, King SM, Smith EJ, Levy DE, Yuan Y. 2002. A Kaposi's sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. U. S. A. 99:5573–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu FX, Sathish N, Yuan Y. 2010. Antagonism of host antiviral responses by Kaposi's sarcoma-associated herpesvirus tegument protein ORF45. PLoS One 5:e10573 doi:10.1371/journal.pone.0010573 [DOI] [PMC free article] [PubMed] [Google Scholar]