

Abstract
Hepatitis C virus (HCV) proteins inhibit complement component expression, which may attenuate immunity against infection. In this study, we examined whether HCV regulates the membrane attack complex (MAC) via complement component C9. MAC is composed of C5b to C9 (C5b-9) and mediates cell lysis of invaded pathogens. Liver biopsy specimens from chronically HCV-infected patients exhibited a lower level of C9 mRNA expression than liver biopsy specimens from unrelated disease or healthy control human liver RNA. Hepatocytes infected with cell culture-grown HCV or expressing HCV core protein also displayed significant repression of C9 mRNA and protein levels. Promoter analysis suggested that the T cell factor-4 (TCF-4E) transcription factor is responsible for HCV core-mediated C9 promoter regulation. Sera from chronically HCV-infected patients displayed a lower level of C5b-9 and a reduced antimicrobial effect on model organisms compared to unrelated patient sera or sera from healthy volunteers. Together, these results for C9 regulation by HCV core protein coupled with functional impairment of the membrane attack complex underscore HCV-mediated attenuation of immune mechanisms.
INTRODUCTION
Hepatitis C virus (HCV) enters the body through direct blood exposure and primarily infecting hepatocytes in the liver. Up to 80% of people infected with HCV may become chronically infected. Most people with chronic HCV infection do not have symptoms and lead normal lives. People with chronic HCV infection often progress to cirrhosis and liver cancer. There is currently no vaccine for HCV, but available treatments can eradicate the virus and slow or stop disease progression in some chronically infected patients (1, 2).
The complement system, by virtue of its dual effector and priming functions, is a major host defense against pathogens. It promotes an inflammatory response, eliminates pathogens, and enhances immune responses. The complement system is involved in the pathogenesis of a variety of liver disorders, including liver injury and repair (3). The liver is responsible for biosynthesis of ∼90% of plasma complement components and their regulators such as CD35, CD46, CD55, and CD59 (3). Deficiencies in complement predispose patients to infection via ineffective opsonization, and defects in the membrane attack complex (MAC) mediate lysis activity (4–6). Therefore, insights into the mechanisms of complement regulation are crucial for understanding disease pathology and therapies. Activation of complement results in C3b deposition and formation of C5 convertases, which leads to insertion of C5b to C9 (C5b-9) into membranes (7). Gram-negative bacteria can be killed by appropriately inserted complexes consisting of C5b-9. However, Gram-positive bacteria and fungi possess thick cell walls and are intrinsically resistant to C5b-9-mediated lysis. Complement activation can also facilitate neutralization of viruses. Specific antibody and the early components of the classical pathway are often sufficient for neutralization. The alternative pathway in conjunction with IgG can also lyse cells infected with various DNA and RNA viruses.
MAC is typically formed on the surface of bacteria and enveloped virus as a result of the activation of the complement system. MAC-mediated formation of transmembrane channels disrupts the phospholipid bilayer of target cells, leading to cell lysis and death (8). The molecular mechanism of complement-mediated killing of Gram-negative bacteria requires damage to the energy-transducing inner membrane (IM), which results from the deposition of the MAC on the bacterial outer membrane. The MAC consists of the C5b-8 complex plus between 1 and 15 C9 molecules that can form a tubular complex (9). Briefly, freshly activated C5b binds to C6 to form a C5b-6 complex and then to C7, forming the C5b-7 complex. The C5b-7 complex binds to C8, which is composed of three chains (alpha, beta, and gamma), thus forming the C5b-8 complex. C5b-8 subsequently binds to C9 and acts as a catalyst in the polymerization of C9. Active MAC has a subunit composition of C5b-C6-C7-C8-C9 (n) (10).
Pathogenic microorganisms have evolved a number of mechanisms to usurp complement-mediated pathways in impairing immune activation and resolution of infection. In this study, we have examined the mechanism by which HCV proteins attenuate C9 expression and MAC-mediated microbicidal activity.
MATERIALS AND METHODS
Reagents.
Mouse monoclonal antibody (MAb) raised to human C9 (Santa Cruz Biotechnology), mouse MAb T cell factor-4 (TCF-4E) raised against amino acids 31 to 331 of human TCF-4 (Milipore), rabbit MAb to phospho-β-catenin at Ser 552 (Cell Signaling Technology), and anti-actin antibody (Sigma) were used in this study. Purified human complement component C9 (Quidel) was purchased and used for MAC-mediated bacterial or viral lysis assay.
Patient materials.
Paired serum samples and liver biopsy specimens from 12 chronically HCV-infected patients (11, 12), 6 nonalcoholic steatohepatitis (NASH) patients, and 2 non-HCV liver disease patients were used. Commercially available control liver RNAs (Clonetics, CloneTech, and Lonza) were procured and used in this study. Ten sera from healthy volunteers without evidence of HBV or HCV infection were used as controls. Sera and/or liver samples were collected from subjects with their written consent, and the protocol was approved by the Saint Louis University Internal Review Board. All samples were collected and sera prepared after storage on ice within a short time (2 to 3 h) to retain complement activity. Sera were aliquoted and stored at −70°C. Each aliquot was thawed and tested once for complement-related activity.
Generation of cell culture-grown HCV.
Human hepatoma cells were used to grow HCV genotype 2a (clone JFH1), as previously described (13). Cell culture-grown HCV released in culture supernatant was filtered through a 0.45-μm-pore-size cellulose acetate membrane (Nalgene), divided into aliquots, and stored at −70°C for single use. RNA (IU/ml) was quantified by real-time PCR (Prism 7500 real-time thermocycler; ABI) with the use of HCV analyte-specific reagents (ASR; Abbott) in the Pathology Clinical Laboratory at Saint Louis University. Infectious virus titers from cell culture supernatant were measured as fluorescence focus-forming units (FFU) using a NS5A-specific monoclonal antibody. For virus infection, hepatocytes were incubated with 0.4 ml of cell culture-grown HCV at a multiplicity of infection (MOI) of 0.5. After virus adsorption for ∼8 h, cells were washed, incubated with fresh medium for 4 days, and used in subsequent experiments.
SC5b-9 ELISA.
HCV-infected patient sera were used for SC5b-9 complex estimation by a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Quidel, CA) following the manufacturer's manual.
Luciferase reporter assay.
HepG2 cells were transfected together with an HCV protein expression plasmid and a C9 luciferase reporter construct: pGL3/C9 (region −789 to +40), pGL3/C9 (−677 to +40), or pGL3/C9 (−166 to +40). Gamma interferon (IFN-γ) was added to the cells 24 h after transfection, and the mixture was incubated for 18 h. Cells were lysed by a single freeze-thaw cycle after addition of lysis buffer (Promega). Cell lysates prepared after clarification by centrifugation were subjected to luciferase reporter assay using a luminometer (Glomax; Promega).
Bactericidal assay.
A complement activation assay using Gram-negative bacteria was performed with minor modification of a previous study (14). Briefly, Escherichia coli DH5α (∼1 × 104 cells) was incubated with 100 μl of a 1:500 dilution of HCV-infected patient sera or normal human sera (NHS) at 37°C for 60 min. After washing, serial 10-fold dilutions of bacterial suspensions into saline solution were made, and 100 μl was spread on LB plates. After 16 h of incubation at 37°C, the CFU were counted. The experiment was performed in triplicate and repeated twice.
Impairment of VSV plaque formation.
A known titer of vesicular stomatitis virus (VSV) was exposed to HCV patient sera, non-HCV-related liver disease sera, or normal human sera in the presence or absence of exogenous C9 added to reach a physiological concentration. VSV was separately incubated with a monoclonal antibody to VSV surface glycoprotein G (procured from ATCC) or to non-VSV-specific antibody (anti-gp41 MAb Chessie 8) prior to addition of sera from chronically HCV-infected patients (1:20 dilution) or normal human subjects (1:20 dilution) and further incubated for 45 min at 37°C prior to addition onto a Huh7 cell monolayer for virus plaque formation as previously described (15). HCV treated in the absence of serum was used as 0% virolysis, and the percentage of virolysis in the presence of serum antibodies was calculated. Serum from HCV-infected patients and normal human control serum at the indicated dilution were incubated with a predetermined number of VSV PFU and incubated for 45 min at 37°C prior to addition onto a Huh7 cell monolayer for virus plaque formation.
C9 deposition assay.
C9 deposition on Gram-negative bacterial cell surfaces was performed based on the previous study with modification (16). Briefly, E. coli DH5α (∼1 × 107 cells) was incubated with HCV-infected patient sera or NHS (20% sera in a final volume of 100 μl) at 37°C for 60 min. After a wash with phosphate-buffered saline (PBS), opsonized log-phase bacteria were plated onto 96-well enzyme immunoassay (EIA) plates at ∼5 × 106 per well and adhered to them by dry desiccation. Nonspecific binding sites were blocked with 200 μl of 0.5% bovine serum albumin (BSA)–PBS and incubated for 30 min at 37°C. After a wash, the plates were sequentially incubated at room temperature with 100 μl of 1:100 (vol/vol) C9 antibody for 60 min, 100 μl of 1:1,000 horseradish peroxidase (HRP)-conjugated secondary antibody for 30 min, and, finally, 100 μl of substrate solution for visualizing HRP activity. The reaction was stopped with 50 μl of 10% H2SO4, and the color reaction was detected at an optical density at 490 nm (OD490).
Real-time PCR.
C9 mRNA quantitation was performed by real-time PCR analysis using specific TaqMan primers and probes (Applied Biosystems). RNA was isolated from liver biopsy specimens as well as from Huh7 cells infected with HCV or transiently transfected with plasmids expressing specific HCV genomic regions by using TRIzol (Invitrogen). cDNA synthesis was done using random hexamers and a SuperScript III first-strand synthesis kit (Invitrogen). C9 mRNAs were evaluated using specific oligonucleotide primers (Applied Biosystems gene expression assays [Hs00156205_m1]) after normalization with 18S gene expression (Hs03928992-g1).
Western blotting.
Proteins in cell lysates were resolved by SDS-PAGE, transferred onto a nitrocellulose membrane, and blotted with the appropriate primary antibody. Positive signals were detected using a peroxidase-conjugated secondary antibody. Protein bands were visualized using an enhanced chemiluminescence detection kit (Super Signal West Pico; Thermo Chemical Company). Cellular actin was detected similarly for comparison of the protein loads in the different lanes.
Statistical analysis.
Results were expressed as the means ± standard deviations (SD), and statistical analyses were performed using a two-tailed unpaired Student t test or one-way analysis of variance (ANOVA) in GraphPad Prism, version 5 (GraphPad, La Jolla, CA). A P value of <0.05 was considered statistically significant.
RESULTS
Downregulation of C9 in HCV-infected patient samples.
In order to investigate the role of HCV infection in C9 expression, the C9 mRNA status was measured in liver biopsy samples. For this, RNA was extracted from 12 liver biopsy samples of HCV genotype 1a-infected patients; 8 non-HCV-infected liver disease samples were included in this study for comparison. Among them, 6 were NASH patient samples and 2 were from other non-HCV liver disease patients. C9 mRNA expression was measured by real-time PCR. All HCV-infected liver biopsy specimens exhibited a significant reduction in C9 mRNA expression compared to RNAs from commercially available control human liver or unrelated diseases (Fig. 1A). Interestingly, the C9 mRNA level in non-HCV-disease-bearing patient samples was significantly higher than that seen with healthy control liver RNA except one liver sample (no. 321). This result suggests that HCV specifically mediates C9 depression in infected patients. Our results exhibited C9 repression irrespective of the fibrotic stage or grade in this limited number of samples or of whether rheumatoid factor was present in the blood of chronically HCV-infected patients (11). Among the membrane attack complex (MAC) components, only C5 and, in particular, C9 mRNA levels were significantly decreased in HCV-infected liver biopsy specimens (Fig. 1B). Other MAC components, such as C6 and C8, did not show a significant reduction, and C7 was undetectable in this experiment. The level of mRNAs encoding other plasma proteins acting on the complement cascade was also analyzed. The mRNA levels of CD46, CD55, and CD59 were increased in HCV-infected liver biopsy specimens (data not shown). Next, we analyzed the C5b-9 level in HCV-infected patient sera and compared these with sera from healthy volunteers. Results from 12 chronically HCV-infected patients and 10 healthy volunteers were compared, with an approximately 10-fold decrease of C5b-9 level seen in infected patients (Fig. 1C). Thus, a correlation with generalized liver disease progression and C9 repression did not appear from examination of this limited sample number.
Fig 1.

Repression of C9 mRNA by HCV. (A) Real-time PCR analysis for C9 mRNA expression in 12 chronically HCV genotype 1a-infected liver biopsy specimens (no. 167, 170, 158, 176, 183, 164, 173, 180, 186, 165, 171, and 181), 6 NASH patient liver biopsy specimens (no. 293, 321, 328, 330, 335, and 338), and 2 non-HCV liver disease patient specimens (no. 295 and 339) compared to control human liver RNA. The disease state of patients is shown at the bottom for each liver specimen, excepting one of unknown status with a NASH liver specimen (marked with a crosshatch symbol [#]). (B) Relative mRNA expression of the MAC components in 4 chronically HCV genotype 1a-infected liver biopsy specimens compared to healthy control liver RNA. (C) Comparison of serum C5b-9 levels in the same group of infected patient sera and 10 HCV-negative healthy controls using a commercially available ELISA kit (Quidel). (A to C) *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to the control). NS, not significant.
Decrease in C9 expression during HCV infection or core protein expression in a cell culture model.
We analyzed C9 mRNA expression in hepatocytes infected with cell culture-grown HCV. A significant reduction of C9 mRNA expression was observed in HCV genotype 2a-infected Huh7 cells compared to mock-infected hepatocytes (Fig. 2A). The results clearly indicated that HCV infection regulates the C9 complement component at the transcriptional level. To identify which viral protein is primarily responsible for C9 downregulation, we performed further examination using HCV genomic regions, and the results suggested that HCV core exhibited an over 95% reduction of C9 mRNA expression (Fig. 2B). HCV NS2 and NS5A proteins also reduced C9 mRNA to a lesser degree (∼80%), while NS3/4A did not exert an effect on C9 mRNA expression. Expression of C9 protein in HCV genotype 2a-infected Huh7.5 cells was dramatically decreased compared to that in uninfected Huh7.5 cells (Fig. 2C). Expression of C9 at the protein level was also reduced in HCV core-expressing hepatocytes (Fig. 2D). Together, these results suggested that HCV infection, especially that involving HCV core protein, inhibits C9 complement expression in hepatocytes, which is also reflected in the results from paired chronically infected patient liver biopsy specimens and sera.
Fig 2.

HCV infection decreases C9 expression. (A) Real-time PCR analyses of HCV genotype 2a-infected Huh7 cells and mock-infected control cells suggest that virus infection also represses C9 mRNA expression. (B) Huh7 cells transfected with HCV core, NS2, NS3/4A, or NS5A protein-encoding plasmids display C9 mRNA regulation by real-time PCR. Vec, vector. (C) The repression of C9 protein was examined with HCV genotype 2a-infected or uninfected hepatocyte lysates by Western blotting. (D) HCV core-mediated repression of C9 protein shown by Western blotting. Vector-transfected (control) and HCV core-transfected Huh7 cells were treated with or without 1,000 U/ml IFN-γ for 2 days. Cell lysates were subjected to C9 analysis using specific antibody. Actin was used as a loading control in Western blotting experiments. mRNA levels in real-time PCR were normalized to endogenous 18S RNA. (A to E) **, P < 0.01; ***, P < 0.001 (compared to mock or vector only).
HCV core protein suppresses C9 promoter activation.
We examined whether HCV inhibits C9 promoter regulation, which is an important process for the terminal component expression of MAC. For this, a luciferase-tagged C9 promoter construct (region −768 to +40) was cotransfected with HCV protein-expressing plasmid in HepG2 cells. C9 promoter activity was significantly inhibited by HCV core and, to a lesser extent, by NS2 and NS5A, while NS3/4A protein expression did not inhibit C9 promoter activity (Fig. 3A).
Fig 3.

Inhibition of C9 promoter activity by HCV genomic regions. (A) A C9 promoter luciferase construct (pGL3/C9 −786 to +40) was examined after cotransfection with an empty vector control, HCV core, NS2, NS3/4A, or NS5A in HepG2 cells for promoter activity. (B) C9 promoter luciferase activity was measured in the presence or absence of the HCV core genomic region transfected into HepG2 cells following stimulation with 1,000 U/ml IFN-γ, 40 ng/ml TNF-α, 20 ng/ml IL-1β, or 40 ng/ml IL-6. (C) IFN-γ-mediated dose-dependent stimulation of C9 promoter activity is shown after 18 h of treatment. **, P < 0.01; ***, P < 0.001 (for comparisons to empty vector-transfected or cytokine-treated cell results).
Next, we examined the effect of cytokines on the regulation of C9 promoter activity in HepG2 cells with or without HCV core protein. C9 promoter activity was significantly increased by IFN-γ but not by tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and IL-6 compared to untreated cell results (Fig. 3B). The IFN-γ-mediated increase of C9 promoter activity was inhibited by HCV core protein expression. C9 promoter activity displayed a dose-dependent increase with IFN-γ in empty vector-transfected cells, but expression of HCV core in treated cells led to suppression of promoter activity at all concentrations of IFN tested (Fig. 3C). Thus, our results suggested that the C9 promoter is regulated by IFN-γ-related transcription factors, and this could be analyzed in subsequent experiments.
HCV core protein suppresses the TCF-4E transcription factor for inhibition of C9 promoter activation.
Promoter analysis was performed using putative transcription factor prediction software (TRANSFAC and PROMO). The cyclic AMP (cAMP) response element-binding protein/activating transcription factor (CREB/ATF) and T cell factor-4 (TCF-4E) binding sites were found (Fig. 4A). Based on the CREB and TCF-4E binding sites, we constructed C9 promoter deletion mutants: C9 (−786 to +40), C9 (−677 to +40), and C9 (−164 to +40). C9 promoter activity was increased by IFN-γ in C9 (−786 to +40)-transfected cells, while it was totally inhibited after deletion of the CREB/ATF and TCF-4E binding sites in a different C9 (−164 to +40) construct, indicating that CREB or TCF-4E may be responsible for the IFN-γ-mediated C9 promoter activity (Fig. 4A and B). Interestingly, luciferase activity was increased in C9 (−677 to +40)-transfected cells at ∼3-fold in IFN-γ-untreated cells and ∼2-fold in IFN-γ-treated cells compared to intact C9 (−786 to +40)-transfected cells. This result suggests that an unknown inhibitory element may bind to some of the regions between −786 and −677 of the C9 promoter but that the putative binding sites for IRF-1 and USF-1 are not responsible. IFN-γ-dependent or -independent C9 promoter activity was inhibited in HCV core-expressing cells, indicating that HCV impairs the activity of an unidentified transcription factor (Fig. 4C).
Fig 4.

TCF-4E transcription factor-mediated C9 promoter activation is inhibited by HCV core protein. (A) Schematic view of the full-length C9 promoter containing the −786 to +40 region, the IRF-1/USF-1 deletion mutant containing the −677 to +40 region, and the TCF-4E and CREB deletion mutant containing the −164 to +40 region tagged with luciferase reporter gene at the 3′ end. (B) Luciferase activities of C9 promoter constructs, pGL3/C9 (−786 to +40), pGL3/C9 (−677 to +40), and pGL3/C9 (−164 to +40), transfected into HepG2 cells are shown with or without IFN-γ (1,000 U/ml) treatment. (C) HCV core protein-mediated pGL3/C9 (−677 to +40) promoter regulation with or without IFN-γ treatment. *, P < 0.05; **, P < 0.01; ***, P < 0.001 [for comparisons to the results determined for pGL3/C9 (−786 to +40) transfectants or vector transfectants]. (D) The expression of TCF-4E and the phosphorylation of β-catenin were examined using HCV genotype 2a-infected or uninfected hepatocyte lysates by Western blotting. (E) Vector-transfected control and HCV core-transfected HepG2 cells were treated with or without IFN-γ (1,000 U/ml) for 2 days, lysed, and subjected to Western blot analysis for TCF-4E expression and β-catenin phosphorylation using specific antibody. Actin was used as a loading control in the experiments whose results are shown in panels D and E.
HCV core protein is not likely to downregulate CREB for C9 inhibition, although IFN-γ activates cAMP/protein kinase A (PKA)/CREB independently of the Jak-Stat signaling pathway (17). In fact, HCV core protein is known to upregulate CREB phosphorylation in hepatocarcinoma cells (18, 19). However, we observed an inhibition of C9 promoter activity by core protein, indicating that CREB does not directly regulate C9 expression. Thus, we excluded an examination of CREB for downregulation of the C9 promoter by core protein. Instead, we analyzed the transcription factor TCF-4E expression level for HCV core-mediated C9 reduction. The C9 promoter region contains 4 TCF-4E binding sites (Fig. 4A). After binding with TCF-4E to its binding site (TTTTGTT), β-catenin and coactivator p300 are associated and initiate transcription. Western blot analysis using total cell lysates from HCV genotype 2a-infected Huh7.5 cells revealed a reduced TCF-4E protein level (Fig. 4D). TCF-4E expression was also decreased in core-expressing Huh7 cells (Fig. 4E). The inhibition of TCF-4E was not affected by IFN-γ, although it was slightly increased by the presence of IFN-γ in empty vector-transfected cells. The phosphorylation of β-catenin, however, was increased in HCV genotype 2a-infected hepatoma cells. Core protein expression in cells also upregulated β-catenin phosphorylation, which was further increased by the presence of IFN-γ in both vector control and HCV core-expressing cells. The results suggest that HCV core-mediated inhibition of C9 promoter activity is specifically mediated by TCF-4E reduction and not by β-catenin, although the two factors associate to initiate transcription of certain genes.
Impairment of complement-mediated VSV neutralization by HCV-infected patient sera.
We have observed poor HCV neutralization by human antibodies to viral envelope glycoproteins (20–22). The newly synthesized HCV genome buds into the endoplasmic reticulum (ER) lumen to form virus particles. Thus, the ER plays a critical role in the formation of HCV envelope and viral maturation. The ER also plays a critical part in the synthesis and modification of glycosylphosphatidylinositol-anchored proteins (GPI-Aps) such as CD55 and CD59 in eukaryotic cells. Thus, it is possible for HCV to encounter and obtain CD55 or CD59 in the ER and other cellular organelles (23). Therefore, working with a susceptible model was necessary to determine the qualitative nature of the complement in chronically HCV-infected patient sera. For this, we used VSV as a model virus since it is easy to grow in large quantities and infects diverse cell types. VSV was incubated with normal human sera (NHS), NASH patient sera, non-HCV liver disease patient sera, or chronically HCV-infected patient sera in the presence or absence of VSV G-specific antibody. For each set of experiments, six individual patient or normal sera were used. To examine classical pathway-mediated VSV neutralization, VSV was first incubated with serial dilutions of anti-VSV G antibody prior to the addition of a 1:20 (5%) final dilution of NHS, NASH patient sera, non-HCV liver disease patient sera, or HCV-specific patient sera (Fig. 5A). When non-VSV-specific antibody was treated with sera, VSV neutralization was not affected (Fig. 5B). The result suggests that VSV neutralization in human sera is mediated by VSV-specific antibody.
Fig 5.

Serum complement from HCV-infected patients impairs VSV infectivity. VSV was incubated with normal human sera, HCV-infected patient sera, or non-HCV liver disease patient sera in the presence or absence of VSV G-specific antibody, and the effect is shown as percent reduction of virus infectivity. Six individual sera were used for each set of experiments. (A) VSV was incubated with serial dilutions of antibody to VSV G prior to the addition of a 1:20 (5%) final dilution of HCV-specific patient sera (white bars), non-HCV liver disease patient sera (dark gray bars), or sera from healthy individuals (light gray bars). (B) Lack of complement-mediated neutralization of VSV reacted with non-VSV-specific antibody prior to a 1:20 dilution of human serum. Anti-gp41 MAb Chessie 8 (epitope PDRPEG) was used for this experiment. (C) Percent reductions of VSV infectivity were compared between these three groups of human sera after incubation with serial dilutions of HCV patient sera (light gray bars), non-HCV liver disease patient sera (dark gray bars), or sera from healthy individuals (white bars). (D) The classical pathway blocker, Mg-EGTA (final concentration of 10 mM MgCl2 and 10 mM EGTA), was added to HCV-positive patient serum, and the mixture was preincubated for 30 min at 37°C. A fixed number of VSV in duplicate samples were incubated with Mg-EGTA-treated HCV-positive sera at a 1:5 or 1:10 final dilution. Gray and white bars indicate Mg-EGTA-treated and Mg-EGTA-untreated HCV patient sera, respectively. (A and C) *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to control results).
For an alternative pathway, VSV was incubated with serial dilutions of NHS, NASH sera, non-HCV liver disease patient sera, or HCV-infected patient sera without anti-VSV G antibody (Fig. 5C). When we compared healthy human experimental groups, it became clear that HCV-infected patients displayed a significant decrease in their ability to effect both alternative and antibody-mediated virolysis. On the other hand, HCV-unrelated patient serum-mediated VSV neutralization was modestly higher than the neutralization by healthy control sera and (significantly) much higher than that seen with HCV-infected patient sera. This result indicated that there may be a potentially significant lesion in the complement function of HCV patient sera. To more clearly determine the complement activation pathway involved in serum-mediated killing of VSV, HCV-positive sera were preincubated with Mg-EGTA, a blocker of the classical and lectin pathway. The result suggested no major difference between Mg-EGTA-treated and untreated sera in VSV neutralization activity, indicating that serum-mediated VSV neutralization is mediated by the alternative pathway (Fig. 5D). Taken together, these results suggest that complement-mediated neutralization of VSV is impaired specifically by HCV-infected sera.
Exogenous C9 increased VSV neutralization by HCV-infected patient sera.
To investigate the relationship between inhibition of C9 by HCV and VSV clearance, we also examined virocidal effects in the presence or absence of exogenous C9 with sera from chronically HCV-infected patients and from healthy volunteers. HCV sera or NHS as a control were incubated with a known titer of VSV (∼100 PFU) in the presence of antibody to VSV G glycoprotein and with a fixed dilution (1:20) of NHS or HCV patient sera (as a source of serum complement). Alternatively, HCV patient sera were incubated at serial dilutions to analyze the effect HCV may have on the neutralization mediated by serum alone. Purified complement C9 (Quidel) was added to each group at a 60 μg/ml final concentration to mimic the levels available in healthy subjects. When neutralization occurred in the presence of VSV-specific antibody, the exogenous addition of purified C9 did not affect the ability of NHS to neutralize VSV (Fig. 6A), while HCV patient sera exhibited an increased neutralization activity against VSV in the presence of C9 (Fig. 6B). The presence of NHS alone resulted in no elevation in VSV neutralization after exogenous C9 addition at the indicated dilutions (Fig. 6C), but HCV patient sera exhibited an increase in VSV neutralization activity resulting from C9 addition (Fig. 6D). There were considerable variations in VSV plaque neutralization caused by HCV-infected patient sera; however, the results were close to statistical significance. The results suggested that the presence of exogenous C9 displayed a positive effect upon the neutralizing ability of HCV patient sera that was not a characteristic of NHS. Thus, C9 modulation by HCV may have a direct effect upon the ability of infected subjects to raise an innate immune response to invasive pathogens by modulating MAC activity.
Fig 6.

Exogenous C9 increases complement-mediated VSV neutralization by HCV-infected patient sera. A predetermined titer of VSV was incubated with HCV-infected patient sera (n = 6) and HCV-negative normal human sera (n = 6) in the presence or absence of purified exogenous C9 at a physiological concentration (60 μg/ml final concentration). (A and B) VSV was incubated with a monoclonal antibody to VSV G surface glycoprotein at the indicated dilution prior to addition of normal human sera at a 1:20 dilution (A) or of chronically HCV-infected patient sera (B), and the mixture was incubated for 45 min at 37°C prior to addition onto a Huh7 cell monolayer for virus plaque formation. (C and D) Normal human sera (C) or HCV-infected patient sera (D) at the indicated dilution were incubated with a predetermined number of VSV PFU and incubated for 45 min at 37°C prior to addition onto a Huh7 cell monolayer for virus plaque formation. Gray bars indicate the presence of exogenous C9, and white bars indicate untreated sera. *, P < 0.05 (compared to control results). The other values are shown in the figure.
Impairment of complement-mediated bacterial killing by HCV-infected patient sera.
Complement C9 is one of the components of MAC and forms a pore on the bacterial cell surface (10). We examined whether HCV can interfere with MAC-mediated Gram-negative bacterial lysis. C9 deposition by HCV-infected patient sera appeared to be much lower than that by healthy sera (Fig. 7A). The kinetics of complement-mediated bacterial killing by sera from HCV-infected patients or healthy individual donors was determined by measuring CFU after opsonization of bacteria in 0% to 5% serum for 60 min. The loss of bacterial killing by HCV patient sera was most discernible at a 0.2% serum concentration, while healthy control serum maintained bacterial killing activity at the same concentration (data not shown). Results from 6 HCV-infected patient sera displaying reduced complement-mediated bacterial killing compared to healthy control sera are shown (Fig. 7B). NASH is more prevalent in patients showing hepatic C9 or activated C3 deposition (24). Since we had already observed a high level of C9 in NASH patient samples, assays using bacteria were not performed for further comparison with matched liver disease clinical specimens. The results indicate that HCV-infected patient sera have a decreased level of MAC activity.
Fig 7.
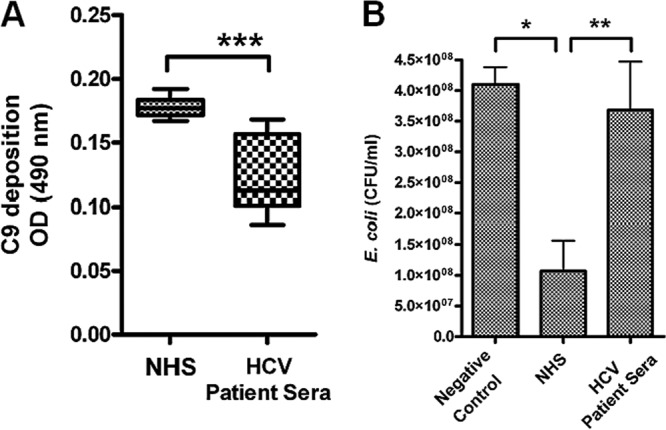
Chronically HCV-infected patient sera exhibit impaired complement-mediated bacterial killing. (A) HCV-infected patient sera (n = 6) and HCV-negative human sera (n = 4) were subjected to analysis for comparison of levels of C9 deposition on the E. coli surface. (B) The same sets of sera were also analyzed for comparison of levels of complement-mediated bacterial killing. Normal bacterial growth in the absence of serum was included as a negative control. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to control results).
DISCUSSION
In the current study, we examined the role of HCV in C9 regulation and C9-mediated MAC function. Hepatocytes expressing HCV core protein displayed a significant repression of C9 promoter activity. Repression of C9 was evident from the mRNA and protein levels in HCV core-expressing cells. Further, C9 mRNA and protein levels were reduced in hepatocytes infected with cell culture-grown HCV. Liver biopsy specimens from chronically HCV-infected patients also exhibited a lower level of C9 mRNA expression than healthy control or NASH patient liver biopsy specimens. Indeed, NASH patient liver biopsy specimens displayed a higher expression level of C9 than healthy control liver biopsy specimens. An enhancement of C9 mRNA levels in NASH subjects has not been reported; however, the enhanced deposition of C9 on hepatocytes has been identified (24). Promoter analysis suggested that the TCF-4E transcription factor is primarily responsible for core-mediated C9 promoter regulation. MAC is composed of C5b to C9 and mediates cell lysis of invaded pathogens. Sera from chronically HCV-infected patients displayed a lower C5b-9 level and a reduced antimicrobial effect on model organisms compared to sera from healthy volunteers.
C9 is involved in various complement-related diseases (7). Killing with a serum-sensitive strain of E. coli was much less efficient than the killing seen with normal serum. Predisposition to meningococcal disease is strongly linked to the efficiency of complement-dependent bactericidal activity. An epidemiological study provided strong evidence for a relationship between C9 deficiency and meningococcal sepsis and meningitis (25). An Arg95 stop mutation of exon 4 in the complement C9 gene is common in individuals in Japan with C9 deficiency, and the lack of the membrane attack complex due to this mutation predisposed patients to pathognomonic glomerulonephritis (26). Morgan et al. studied the concentration of the terminal component of complement (C9) in cerebrospinal fluid (CSF) and plasma from 35 patients with multiple sclerosis and 55 controls with other neurological diseases. There was a highly significant reduction in the CSF C9 concentration in patients with multiple sclerosis compared with controls (27). On the other hand, the level of complement component 9, a final component to form MAC, is significantly increased whereas CD59 protein levels are significantly decreased in the frontal cortex and hippocampus of Alzheimer's disease (AD) patients compared to nondemented elderly (ND) patients. This result suggests that CD59 deficits and C9 induction may play a role in the neuritic losses characteristic of AD (28). In most cases, it is believed that C9 reduction is related to the inhibition of MAC-mediated cell killing. However, in certain cases, serum lacking C9 can kill serum-sensitive Neisseria, unlike sera deficient in the other terminal C components (29). On the other hand, multiple researchers have shown that C9 polymers with a tubular ultrastructure resembling the MAC complex of complement can be formed spontaneously under certain conditions (30, 31). Dankert and Esser (32) reported C9-mediated bacterial killing in the absence of C5b-8. Thus, addition of C9 to serum samples may increase bacterial or virus killing activity. In our study, however, C9 component itself did not increase bactericidal activity. In an attempt to compensate for the bactericidal status of the serum, we added purified C9 protein to the serum. Although additional C9 protein modestly increased bacterial killing, the bactericidal state did not change significantly (data not shown). Bactericidal activity by multimeric C9 of the C5b-9 complex is seen only in the presence of specific antibody to Neisseria gonorrhoeae (33). However, in our study, we did not use a specific bacterial antibody. On the other hand, virocidal activity using VSV was also moderately upregulated after addition of purified C9 together with a specific VSV antibody, indicating that specific antibody is required for MAC activation. In addition to specific antibody, MAC formation also affects the bactericidal activity. Serum-mediated killing of N. meningitidis is increased by addition of anti-PorA antibody, but MAC formation is not changed, which suggests that efficient meningococcal lysis is mediated by a proper MAC insertion but not by the amount of MAC formation (34). Thus, both an increase in MAC formation caused by a specific antibody and proper insertion of MAC into the bacteria cell wall or virus envelope are required for the acceleration of bacterial or virus killing activity.
The C9 complement component is significantly reduced in chronically HCV-infected liver biopsy specimens regardless of disease state, while many hepatocyte-derived complement components (C6, C8, factor B, MASP1, and MBL) and many unrelated genes remain unaffected. This implies an HCV-specific effect, not a global effect from liver disease. The change in complement level (C4, C3, or C9) in liver biopsy specimens did not correlate with the liver fibrosis stage or with rheumatoid factor, serum albumin, and alkaline phosphatase levels (11, 12). Since the fibrosis stage represents the amount of fibrosis and scarring resulting in reduced relative numbers of hepatocytes, it is possible that advanced fibrosis or cirrhosis may decrease the liver-derived C9 level. However, a report in the literature suggested that plasma concentrations of C9 were not decreased among alcoholic cirrhosis patients (35). Further, we have not found C9 downregulation corresponding to the fibrosis stage for non-HCV-related patients. Thus, the results suggest that a viral factor(s) is contributing to the suppression of complement synthesis in the liver. Associations with persistent infection, including cytomegalovirus (CMV) infection, cryptococcus infection, and tuberculosis, were found to be more common in HCV-infected patients than in non-HCV-infected controls (36). Systemic autoimmune diseases also occur in patients with HCV infection (37). Further, HAV or HBV vaccination response rates are reduced in patients with chronic HCV infection (38–40). These observations suggest that chronic HCV infection may generate an overall state of immune suppression. The role of a specific complement component(s) in HCV-associated disease progression should be further clarified in the future. HCV-mediated immune suppression may include the acquisition of proteins belonging to the family of complement regulators, such as CD46, CD55, and CD59 (41–43), and the interaction with regulatory factors, such as factor H (44). Opsonized virus may bind to complement receptor-positive cells to infect them more efficiently or remain bound on the surface of such cells. Direct demonstration of the presence of CD46, CD55, and CD59 on the viral surface was shown by neutralization antibody-induced complement-mediated virolysis of human immunodeficiency virus (HIV) (42, 43) or HCV (23) particles. On the basis of the results of our previous study and the current study, we suggest that HCV can escape innate immune system function through the inhibition of complement component synthesis. Therefore, impairment of specific complement component expression by HCV results in the failure of the complement-mediated innate immune response.
ACKNOWLEDGMENTS
We thank John Atkinson, Robert B. Belshe, and Mike Diamond for helpful suggestions and Lin Cowick for preparation of the manuscript.
This work was supported by research grant U54-AI057160 to the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Diseases Research from the National Institutes of Health and by the Internal Blue Ribbon Fund from Saint Louis University.
Footnotes
Published ahead of print 13 March 2013
REFERENCES
- 1. Klenerman P, Gupta PK. 2012. Hepatitis C virus: current concepts and future challenges. QJM 105:29–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrovic D, Dempsey E, Doherty DG, Kelleher D, Long A. 2012. Hepatitis C virus–T-cell responses and viral escape mutations. Eur. J. Immunol. 42:17–26 [DOI] [PubMed] [Google Scholar]
- 3. Qin X, Gao B. 2006. The complement system in liver diseases. Cell. Mol. Immunol. 3:333–340 [PubMed] [Google Scholar]
- 4. Joller N, Weber SS, Oxenius A. 2011. Antibody-Fc receptor interactions in protection against intracellular pathogens. Eur. J. Immunol. 41:889–897 [DOI] [PubMed] [Google Scholar]
- 5. Wagner E, Frank MM. 2010. Therapeutic potential of complement modulation. Nat. Rev. Drug Discov. 9:43–56 [DOI] [PubMed] [Google Scholar]
- 6. Zipfel PF, Skerka C. 2009. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 9:729–740 [DOI] [PubMed] [Google Scholar]
- 7. Ram S, Lewis LA, Rice PA. 2010. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin. Microbiol. Rev. 23:740–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peitsch MC, Tschopp J. 1991. Assembly of macromolecular pores by immune defense systems. Curr. Opin. Cell Biol. 3:710–716 [DOI] [PubMed] [Google Scholar]
- 9. Müller-Eberhard HJ. 1986. The membrane attack complex of complement. Annu. Rev. Immunol. 4:503–528 [DOI] [PubMed] [Google Scholar]
- 10. Tegla CA, Cudrici C, Patel S, Trippe R, III, Rus V, Niculescu F, Rus H. 2011. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol. Res. 51:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2011. Transcriptional repression of C4 complement by hepatitis C virus proteins. J. Virol. 85:4157–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus proteins inhibit C3 complement production. J. Virol. 86:2221–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, Ray RB. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 80:4633–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rautemaa R, Rautelin H, Puolakkainen P, Kokkola A, Kärkkäinen P, Meri S. 2001. Survival of Helicobacter pylori from complement lysis by binding of GPI-anchored protectin (CD59). Gastroenterology 120:470–479 [DOI] [PubMed] [Google Scholar]
- 15. Lagging LM, Meyer K, Owens RJ, Ray R. 1998. Functional role of hepatitis C virus chimeric glycoproteins in the infectivity of pseudotyped virus infectivity. J. Virol. 72:3539–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Egan AM, Gordon DL. 1996. Burkholderia pseudomallei activates complement and is ingested but not killed by polymorphonuclear leukocytes. Infect. Immun. 64:4952–4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu L, Wang Y, Fan Y, Li CL, Chang ZL. 2004. IFN-gamma activates cAMP/PKA/CREB signaling pathway in murine peritoneal macrophages. J. Interferon Cytokine Res. 24:334–342 [DOI] [PubMed] [Google Scholar]
- 18. Christen V, Treves S, Duong FH, Heim MH. 2007. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 46:558–565 [DOI] [PubMed] [Google Scholar]
- 19. Hassan M, Selimovic D, Ghozlan H, Abdel-kader O. 2009. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 49:1469–1482 [DOI] [PubMed] [Google Scholar]
- 20. Meyer K, Banerjee A, Frey SE, Belshe RB, Ray R. 2011. A weak neutralizing antibody response to hepatitis C virus envelope glycoprotein enhances virus infection. PLoS One 6:e23699 doi:10.1371/journal.pone.0023699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ray R, Meyer K, Banerjee A, Basu A, Coates S, Abrignani S, Houghton M, Frey SE, Belshe RB. 2010. Characterization of antibodies induced by vaccination with hepatitis C virus envelope glycoproteins. J. Infect. Dis. 202:862–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ray R. 2011. Progress toward development of a hepatitis C vaccine with broad shoulders. Sci. Transl. Med. 3:94ps33 doi:10.1126/scitranslmed.3002772 [DOI] [PubMed] [Google Scholar]
- 23. Amet T, Ghabril M, Chalasani N, Byrd D, Hu N, Grantham A, Liu Z, Qin X, He JJ, Yu Q. 2012. CD59 incorporation protects hepatitis C virus against complement-mediated destruction. Hepatology 55:354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, Greve JW, Buurman WA. 2009. Activation of the complement system in human nonalcoholic fatty liver disease. Hepatology 50:1809–1817 [DOI] [PubMed] [Google Scholar]
- 25. Fukumori Y, Yoshimura K, Ohnoki S, Yamaguchi H, Akagaki Y, Inai S. 1989. A high incidence of C9 deficiency among healthy blood donors in Osaka, Japan. Int. Immunol. 1:85–89 [DOI] [PubMed] [Google Scholar]
- 26. Miura T, Goto S, Iguchi S, Shimada H, Ueno M, Nishi S, Narita I. 2011. Membranoproliferative pattern of glomerular injury associated with complement component 9 deficiency due to Arg95Stop mutation. Clin. Exp. Nephrol. 15:86–91 [DOI] [PubMed] [Google Scholar]
- 27. Morgan BP, Campbell AK, Compston DA. 1984. Terminal component of complement (C9) in cerebrospinal fluid of patients with multiple sclerosis. Lancet ii:251–254 [DOI] [PubMed] [Google Scholar]
- 28. Yang LB, Li R, Meri S, Rogers J, Shen Y. 2000. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer's disease. J. Neurosci. 20:7505–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harriman GR, Esser AF, Podack ER, Wunderlich AC, Braude AI, Lint TF, Curd JG. 1981. The role of C9 in complement-mediated killing of Neisseria. J. Immunol. 127:2386–2390 [PubMed] [Google Scholar]
- 30. Podack ER, Tschopp J. 1982. Polymerization of the ninth component of complement (C9): formation of poly(C9) with a tubular ultrastructure resembling the membrane attack complex of complement. Proc. Natl. Acad. Sci. U. S. A. 79:574–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tschopp J, Müller-Eberhard HJ, Podack ER. 1982. Formation of transmembrane tubules by spontaneous polymerization of the hydrophilic complement protein C9. Nature 298:534–538 [DOI] [PubMed] [Google Scholar]
- 32. Dankert JR, Esser AF. 1987. Bacterial killing by complement. C9-mediated killing in the absence of C5b-8. Biochem. J. 244:393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joiner KA, Goldman RC, Hammer CH, Leive L, Frank MM. 1983. Studies on the mechanism of bacterial resistance to complement-mediated killing. VI. IgG increases the bactericidal efficiency of C5b-9 for E. coli 0111B4 by acting at a step before C5 cleavage. J. Immunol. 131:2570–2575 [PubMed] [Google Scholar]
- 34. Drogari-Apiranthitou M, Kuijper EJ, Dekker N, Dankert J. 2002. Complement activation and formation of the membrane attack complex on serogroup B Neisseria meningitidis in the presence or absence of serum bactericidal activity. Infect. Immun. 70:3752–3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Homann C, Varming K, Høgåsen K, Mollnes TE, Graudal N, Thomsen AC, Garred P. 1997. Acquired C3 deficiency in patients with alcoholic cirrhosis predisposes to infection and increased mortality. Gut 40:544–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El-Serag HB, Anand B, Richardson P, Rabeneck L. 2003. Association between hepatitis C infection and other infectious diseases: a case for targeted screening? Am. J. Gastroenterol. 98:167–174 [DOI] [PubMed] [Google Scholar]
- 37. Ramos-Casals M, Muñoz S, Medina F, Jara LJ, Rosas J, Calvo-Alen J, Brito-Zerón P, Forns X, Sánchez-Tapias JM. 2009. Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC Registry). J. Rheumatol. 36:1442–1448 [DOI] [PubMed] [Google Scholar]
- 38. Keeffe EB, Iwarson S, McMahon BJ, Lindsay KL, Koff RS, Manns M, Baumgarten R, Wiese M, Fourneau M, Safary A, Clemens R, Krause DS. 1998. Safety and immunogenicity of hepatitis A vaccine in patients with chronic liver disease. Hepatology 27:881–886 [DOI] [PubMed] [Google Scholar]
- 39. Chlabicz S, Grzeszczuk A, Łapiński TW. 2002. Hepatitis B vaccine immunogenicity in patients with chronic HCV infection at one year follow-up: the effect of interferon-alpha therapy. Med. Sci. Monit. 8:CR379–CR383 [PubMed] [Google Scholar]
- 40. Buxton JA, Kim JH. 2008. Hepatitis A and hepatitis B vaccination responses in persons with chronic hepatitis C infections: a review of the evidence and current recommendations. Can. J. Infect. Dis. Med. Microbiol. 19:197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montefiori DC, Cornell RJ, Zhou JY, Zhou JT, Hirsch VM, Johnson PR. 1994. Complement control proteins, CD46, CD55, and CD59, as common surface constituents of human and simian immunodeficiency viruses and possible targets for vaccine protection. Virology 205:82–92 [DOI] [PubMed] [Google Scholar]
- 42. Saifuddin M, Ghassemi M, Patki C, Parker CJ, Spear GT. 1994. Host cell components affect the sensitivity of HIV type 1 to complement-mediated virolysis. AIDS Res. Hum. Retroviruses 10:829–837 [DOI] [PubMed] [Google Scholar]
- 43. Schmitz J, Zimmer JP, Kluxen B, Aries S, Bögel M, Gigli I, Schmitz H. 1995. Antibody-dependent complement-mediated cytotoxicity in sera from patients with HIV-1 infection is controlled by CD55 and CD59. J. Clin. Invest. 96:1520–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sjöberg AP, Trouw LA, Blom AM. 2009. Complement activation and inhibition: a delicate balance. Trends Immunol. 30:83–90 [DOI] [PubMed] [Google Scholar]