

Abstract
Here, we applied the designed ankyrin repeat protein (DARPin) technology to develop novel gp120-directed binding molecules with HIV entry-inhibiting capacity. DARPins are interesting molecules for HIV envelope inhibitor design, as their high-affinity binding differs from that of antibodies. DARPins in general prefer epitopes with a defined folded structure. We probed whether this capacity favors the selection of novel gp120-reactive molecules with specificities in epitope recognition and inhibitory activity that differ from those found among neutralizing antibodies. The preference of DARPins for defined structures was notable in our selections, since of the four gp120 modifications probed as selection targets, gp120 arrested by CD4 ligation proved the most successful. Of note, all the gp120-specific DARPin clones with HIV-neutralizing activity isolated recognized their target domains in a conformation-dependent manner. This was particularly pronounced for the V3 loop-specific DARPin 5m3_D12. In stark contrast to V3-specific antibodies, 5m3_D12 preferentially recognized the V3 loop in a specific conformation, as probed by structurally arrested V3 mimetic peptides, but bound linear V3 peptides only very weakly. Most notably, this conformation-dependent V3 recognition allowed 5m3_D12 to bypass the V1V2 shielding of several tier 2 HIV isolates and to neutralize these viruses. These data provide a proof of concept that the DARPin technology holds promise for the development of HIV entry inhibitors with a unique mechanism of action.
INTRODUCTION
The highly specific interaction of the human immunodeficiency virus (HIV) envelope spike, a trimer composed of gp120 and gp41 heterodimers, with the cellular receptor CD4 and a coreceptor (typically CCR5 or CXCR4) initiates HIV entry and is indispensable for infection (reviewed in reference 1). The potential of HIV entry-blocking agents for intervention strategies has been acknowledged for a long time, and a number of agents targeting the viral envelope proteins and the cellular receptor CD4, CCR5, or CXCR4 have been developed (2, 3). However, thus far, only two entry inhibitors, the peptidic fusion inhibitor T-20, targeting gp41 (4), and the small-molecule CCR5 inhibitor maraviroc (5), have been used clinically.
The design of appropriate inhibitors, in particular those that target the viral envelope proteins, faces challenges that are in many respects similar to those encountered by natural and vaccine-induced antibody (Ab) responses. Both neutralizing antibodies and inhibitors need to access vulnerable sites on the HIV envelope trimer. The complex quaternary structure of the trimeric envelope spike, however, efficiently shields functionally important domains. Conserved domains are positioned facing inward in the trimeric complex, only to be exposed transiently upon receptor engagement (6–8), while the outer trimer surface is protected by flexible, variable loops and extensive glycosylation (7, 9–11). These strategies act in concert to shield the envelope complex from immune recognition and inhibitor attack (8, 12). Most of the gp120 outer domain is not functionally important (7, 13) and allows the virus to rapidly mutate and evade any envelope-targeting agent (12). Antibody-based vaccines and entry inhibitors thus need to act against a wide spectrum of genetically divergent HIV strains to be effective, ideally by targeting conserved yet easily accessed sites on the viral envelope that are important for the function of the envelope trimer and cannot be altered without significant fitness costs. Rare broadly active and potent neutralizing antibodies can emerge in natural infection (14). Vaccine immunogens capable of eliciting such responses, however, still need to be developed and, likewise, the means to design and select inhibitors that target conserved and accessible domains still need to be created.
Here, we employed the designed ankyrin repeat protein (DARPin) technology (15–19) to derive HIV envelope-specific entry inhibitors. We have previously utilized the technology successfully to develop DARPins specific for CD4, which proved to be highly potent in inhibiting entry of divergent HIV strains (18). DARPins, like the natural ankyrin repeat proteins from which they are derived (20), have exceptional binding properties and recognize particularly well targets with a defined rigid surface, such as folded proteins. Although they share many properties with antibodies, most notably their high target specificity and affinity, DARPins differ from antibodies in size, structure, and preference for folded proteins as targets (19). In the current study, we sought to explore whether these distinct properties of DARPins allow selection of binding molecules that recognize epitopes on gp120 differing from those targeted by antibodies and that, upon binding to virions, interfere with HIV entry.
To this end, we performed four independent series of ribosome display selections of gp120-specific DARPins, utilizing different gp120 molecules and epitope display approaches (Fig. 1A and B). Our selections yielded a variety of gp120-directed DARPin molecules that all proved to depend to a higher degree on structural conservation of the envelope protein than gp120-specific antibodies recognizing overlapping domains. Most noteworthy, DARPin 5m3_D12 proved to recognize the V3 loop in a highly conformation-dependent manner. This clone recognized the V3 loop on wild-type (wt) gp120 protein, as well as in the context of the trimeric spike on virus particles, and bound to structurally arrested V3 loop mimetic peptides, but unlike V3 loop antibodies, it showed dramatically reduced capacity to bind to linear V3 loop peptides. Importantly, V3 loop antibodies largely fail to interact with the V3 loop on envelope trimers in the presence of active V1V2 shielding on tier 2 viruses and therefore, in most cases, have only marginal or no neutralization activity (21). On the other hand, DARPin 5m3_D12 was in part capable of bypassing the V1V2 shielding of tier 2 HIV isolates, albeit in a strain-dependent manner, enabling it to block the entry of these virus isolates.
Fig 1.

Selection strategies employed for the generation of gp120-specific DARPins. (A) Schematic view of a ribosome display DARPin selection round. (B) The four employed DARPin selection strategies and numbers of gp120-specific binders obtained. *, 3 clones in selection III and 1 clone in selection IV with borderline reactivity for gp120 had affinities too low (≥10 μM) for further analysis.
MATERIALS AND METHODS
Reagents.
Reagents were kindly provided by the following: soluble CD4 (sCD4) by W. Olson (Progenics Pharmaceuticals Inc., Tarrytown, NY); monoclonal antibodies (MAbs) IgG1b12 (22), b6 (23), and PGT128 (24) by D. Burton (Scripps Research Institute, La Jolla, CA); MAb 1-79 (25) by M. Nussenzweig (Rockefeller University, New York, NY); MAb 2G12 (26) by H. Katinger (Polymun, Vienna, Austria); MAbs 17b, 19b, 48D, and A32 (27) by J. Robinson (Tulane University); and MAb 447-52D (28) by S. Zolla Pazner and MAb F425 B4e8 (29) by M. Posner and L. Cavacini via the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. CD4M47 was synthesized as described previously (30). Linear and cyclic V3 mimetic peptides from strains JR-FL and MN were synthesized as described previously (31). The following V3 loop peptides were used (the sequences are in parentheses): linear peptides, linear (JR-FL) (NNTRKSIHIGPGRAFYTTGEIIG) and linear (MN) (GGGGYNKRKRIHIGPGRAFYTTKNIIG); structural V3 loop mimetics that were cyclized by a d-Pro–l-Pro (DPP) dipeptide that stabilizes the hairpin conformation, cyclic IY (MN) (KRIHIGPGRAFYTTDPP), cyclic IY (MNmut) (KRIHIGAGRAFYTTDPP), cyclic IY (JR-FL) (KSIHIGPGRAFYTTDPP), cyclic IF (MN) (KRIHIGPGRAFYTDPP), cyclic IF (JR-FL) (TCKSIHIGPGRAFYTCGDPP) with the Cys residues disulfide bonded, cyclic HY (JR-FL) (SIHIGPGRAFYTTDPP), cyclic HF (MN) (RIHIGPGRAFYTDPP), cyclic HF1 (JR-FL) (SIHIGPGRAFYTDPP), cyclic HF2 (JR-FL) (RCSIHIGPGRAFYTCGDPP) with the Cys residues disulfide bonded, and cyclic HF3 (JR-FL) (RKSIHIGPGRAFYTTGDPP); and a cyclic peptide cyclized by a disulfide bond between the two cysteines, cyclic SS (JR-FL) (GNCRKSIHIGPGRAFYTTGCG).
For the peptide enzyme-linked immunosorbent assay (ELISA), biotinylated peptides were used. The linear (MN) peptide was biotinylated directly, and cyclic IY (MN) had a PEG08 linker between the mimetic and biotin (31). All synthetic peptides were ≥95% pure by analytical high-performance liquid chromatography (HPLC) and gave electrospray mass spectrometry (MS) spectra consistent with the expected masses.
gp120 proteins.
Codon-optimized sequences of strain JR-FL gp120 wild type, gp120D368R, gp120I420R, and the loop deletion mutants (32, 33) were custom synthesized (GeneArt, Germany), fused to a C-terminal AviTag (sequence, LNDIFEAQKIEWHE; Avidity, USA) and cloned into the expression vector CMV/R (34). Recombinant gp120 was produced by transient transfection of HEK 293T Freestyle suspension cells maintained in serum-free medium as described by the manufacturer (Invitrogen). gp120 was purified from culture supernatants using Galanthus nivalis lectin resin (Vector Laboratories) as described previously (35). The retrieved gp120 was monobiotinylated using BirA as described by the manufacturer (Roche), followed by Superdex200 size exclusion chromatography (GE Healthcare) to derive monomeric gp120. To obtain deglycosylated gp120, 500 μg of gp120 was subjected to 0.15 U α(1,2,3,6)-mannosidase (from jack bean; Europa Bioproducts) for 30 h at 37°C in the presence of protease inhibitors (Complete Mini EDTA-free Protease Inhibitor Cocktail Tablets; Roche Applied Science, Switzerland). The gp120ΔV1V2 construct was created as previously described (21). The ΔV1 and ΔV3 gp120 loop deletions were created with the following linker sequences for deleted loops (HXB2 numbering): ΔV1 linker, C131KDVNAGEIKNC157; ΔV3 linker, C296TGAGHC331.
DARPin selection by ribosome display.
Selection with the N2C and N3C DARPin libraries was performed essentially as described previously (36–38). During ribosome display, a library of DARPins is translated in vitro without stop codons, so that a ternary complex of ribosome, mRNA, and the still attached, newly translated DARPin is formed. Thereby, the genotype is coupled to the phenotype, allowing the enrichment of DARPins that specifically bind to the respective target (Fig. 1A). After each selection round, RNA of bound DARPin species is amplified by reverse transcription (RT)-PCR. The obtained cDNA constitutes a sublibrary of the starting library. Selection in the absence of the target protein is performed in parallel to monitor the extent of background (off-target) cDNA amplification. Comparisons of the quantities of amplified RT-PCR products from target and background selection indicate when enrichment of target-specific DARPins occurs (38). Commonly, several consecutive rounds (>3) of selection are necessary to derive sublibraries that are highly enriched for target-specific DARPins. During this process, a balance between maintaining diversity in the library (in order not to lose specific binders with low affinity too early) and stringency of panning (to deselect nonspecific binders) has to be achieved (37). Stringency can be modulated by the intensity of the washing steps, tuning of RT-PCR cycles, or depletion of low-affinity binders by addition of excess, nonimmobilized target (off-rate selection [see below]). Prior to the experiments described here, we performed a series of analyses to probe which selection conditions would prove best for our different targets (data not shown).
In summary, we found that in order to achieve selection of gp120-specific binders, adding the target in solution rather than immobilized on a surface was key, as previously described for other targets (38). Biotinylated gp120 was bound to streptavidin-coupled magnetic microbeads (MyOne-T1 Dynabeads; Invitrogen), and panning was performed in microtubes. Early rounds of selection (rounds 1 to 3) were kept at low stringency with typically 3 to 5 short washing steps (5 min), while the intensity of washing was increased during subsequent rounds of selection (typically 8 to 10 washing steps of 10 min each). The most important adaptation to the standard ribosome display protocol previously described (18, 36, 37) was the omission of heparin in the panning buffer. Heparin is usually added during panning to prevent nonspecific RNA binding to the protein or surfaces. Since gp120 has been reported to have multiple heparin binding sites (39) and in vitro efficiently interacts with heparin (40), addition of heparin during the selection process could block otherwise accessible sites on gp120. Indeed, we found that addition of heparin completely obliterated gp120-specific DARPin selection (data not shown). Thus, while omission of heparin can lead, to some extent, to false-positive binder enrichment, as judged by RT-PCR, selection of DARPins in the absence of heparin proved the only possible approach to select gp120-specific DARPins (data not shown).
The number of selection rounds performed in each selection is shown in Fig. 1B. Sublibraries for which a target-specific enrichment by RT-PCR was evident were screened for DARPin molecules that specifically bind to gp120 by ELISA (see below). In an alternate screening approach, N3C sublibraries from selection II that yielded 1st- and 2nd-generation binders based on ELISA of gp120 were reprobed and tested directly for inhibition of HIV by pseudovirus inhibition screening against strains JR-FL, JR-FLΔV1V2, and NL4-3; simian-human immunodeficiency virus (SHIV) SF162 P3; and murine leukemia virus (MuLV) on TZM-bl cells (see below).
Detection of DARPin and MAb binding to target protein and peptides by ELISA.
Twenty nanomolar biotinylated gp120 or peptide was immobilized to white high-binding microplates (Costar) that had been coated with 60 nM Neutravidin (Pierce) either overnight at 4°C or for 1 h at 37°C, followed by three wash steps with TBST (Tris-buffered saline containing 0.1% Tween 20), pH 7.5.
gp120 proteins were either probed unliganded or triggered with 50 nM sCD4 or CD4M47. Serial dilutions of either purified DARPins or crude extract from DARPin-producing Escherichia coli were added in TBSTB (TBST with 0.5% bovine serum albumin), pH 7.5. Unbound material was washed off in TBST, and bound DARPins were detected via their His tags using a mouse anti-polyhistidine-alkaline phosphatase antibody (clone HIS-1; Sigma-Aldrich) and Tropix CDP-star chemiluminescent substrate (Applied Biosystems). MAbs were detected with polyclonal anti-human IgG (Fc specific)-alkaline phosphatase antibody produced in goat (Sigma-Aldrich). Emission in relative light units (RLU) was detected on a Dynex Technologies Luminometer. For competition ELISA, gp120 was pretreated with 33 nM the respective MAb or sCD4, and the subsequent ELISA steps were performed as described above. The percentages of binding in the presence of competitor were determined at saturating DARPin concentrations. For peptide competition ELISA, the DARPin 5m3_D12 or MAbs 447-52D and F425-4e8 were pretreated with the indicated concentrations of peptide, and the subsequent ELISA steps were performed as described above.
Affinity maturation of DARPin binders by diversification by error-prone PCR and off-rate selection.
The sublibraries of interest were randomized by amplification via error-prone PCR (41), using a dNTP-Mutagenesis Kit (Jena Biosciences) including 50 μM each of the nucleotide analogs 8-oxo-2′-dGTP (8-oxo-dGTP) and 6-(2-deoxy-β-d-ribofuranosyl)-3,4-dihydro-8H-pyrimido-[4,5-c][1,2]oxazin-7-one-triphosphate (dPTP) in the RT-PCR of ribosome display. For the subsequent panning round, a one-to-one mixture of error-prone and original PCR products was used as a template for RNA transcription.
To select clones with improved affinity, DARPin-ribosome complexes were first allowed to bind to bead-immobilized targets before a 100- to 300-fold molar excess of nonbiotinylated gp120 was added as a competitor for 1 to 2 h, followed by 10 to 15 10-min washing steps. DARPin binders with a fast off rate are captured by the nonbiotinylated competitor target in solution and are lost during subsequent washing steps. In contrast, high-affinity binders with a low off rate remain bound to the bead-coupled target protein, and their mRNA will eventually be preferentially eluted and amplified. This off-rate selection was followed by a ribosome display round in the absence of competitor and subsequent washing 10 times for 5 min each time to reenrich for specific binders, since the proportion of specific binders is lowered in resulting sublibraries by the off-rate selection step (42). Afterward, individual clones from the sublibraries were screened by binding ELISA as described above.
Molecular phylogenetic analysis by the maximum-likelihood method.
Phylogenetic analyses were conducted in MEGA5 (43) using the maximum-likelihood method based on the JTT matrix-based model (44). The trees with the highest log likelihood are shown.
DARPin purification and analysis.
DARPins were produced in the E. coli XL10 Gold strain with the pQE30 expression plasmid with isopropyl β-d-1-thiogalactopyranoside (IPTG) induction and purified using Ni-nitrilotriacetic acid (NTA) affinity chromatography as described previously (45). The DARPin proteins obtained were checked by size exclusion chromatography coupled to multiangle light scattering (SEC-MALS) for the oligomerization state. DARPins that exhibited a tendency to form dimers or higher-order aggregates were excluded from further analysis. For direct neutralization screening, small-scale DARPin preparations were purified from 400 μl bacterial lysate in 96-well plates using Ni-NTA-coated magnetic beads (His Mag Sepharose Ni; GE Healthcare).
Virus preparation.
Env-pseudotyped viruses were prepared by cotransfection of HEK 293-T cells with plasmids carrying the respective Env genes and the luciferase reporter HIV vector pNLluc-AM as described previously (46). The following envelope genes were used: NL4-3 (47); JR-FL (48); SF162-LS (49); NAB1pre-cl39x, NAB2pre-cl_3, NAB3pre-cl_43, NAB4pre-cl_1, NAB5pre-cl_1, NAB10pre-cl_2, and NAB12pre-cl_7 (50); ZA110.C.10.14; and Envs with V1V2 deleted (21). ZA110.C.10.14 (21) was isolated in the frame of the Zurich Primary HIV Infection Study (ZPHI), Division of Infectious Diseases and Hospital Epidemiology, supported by the University of Zurich's Clinical Research Priority Program (CRPP).
The panel of reference subtype B Env clones comprising the envelope proteins 6535.3, AC10.0.29, CAAN5342.A2, PVO.4, QH0692.42, REJO4541.67, RHPA4259.7, SC422661.8, THRO4156.18, TRO.11, and WITO4160.33 (51) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Neutralization assay using Env-pseudotyped virus.
The neutralization activities of DARPins and MAbs were evaluated on TZM-bl cells as described previously (46). The virus input was chosen to yield virus infectivity corresponding to 5,000 to 20,000 RLU in the absence of inhibitors. The antibody concentrations or reciprocal plasma titers causing 50% reduction in viral infectivity (50% inhibitory concentration [IC50] or 50% neutralization titer [NT50]) were calculated by fitting pooled data from two or three independent experiments to sigmoid dose-response curves (variable slope) using Prism software (GraphPad Software). If 50% inhibition was not achieved at the highest or lowest drug or plasma concentration, a “greater than or less than” value was recorded.
For competition inhibition assays with V3 peptides, MAb and DARPin inhibitor concentrations that lead to a 95% reduction in virus entry signal were chosen as fixed doses. The respective inhibitor at the fixed dose and increasing serial dilutions of the different V3 peptides were preincubated for 1 h, and then virus was added and the entire mixture was incubated for an additional hour before TZM-bl cells were added. Infection was measured after 48 h as described previously (46). The percent inhibition of the DARPin and MAb in the presence of competing peptide was calculated in relation to control wells containing the corresponding concentration of the peptide in the absence of inhibitor.
RESULTS
Selection of gp120-specific DARPin molecules.
The high structural flexibility of the surface unit gp120 within the viral envelope trimer and its extensive glycosylation are considered major barriers to the selection of neutralizing antibodies, both in natural infection and upon vaccination (8, 10, 52). In selection I (Fig. 1B), we sought to determine whether DARPins can overcome these barriers and efficiently bind to this flexible, highly glycosylated target. DARPin DNA libraries encoding either two (N2C) or three (N3C) internal randomized ankyrin repeats between an N- and C-terminal capping repeat (15) were subjected to ribosome display selections and panned against recombinant gp120 from strain JR-FL (Fig. 1B). Enrichment of specific binders in both the N2C and the N3C libraries was observed based on RT-PCR products after four rounds of ribosome display (data not shown). A total of 285 individual clones from each library were expressed in E. coli, crude extracts were screened for gp120 binding by ELISA, and reactive clones were sequenced. None of the N2C clones and only a single clone from the N3C library proved to encode a DARPin that specifically bound to JR-FL gp120 (Fig. 1B), indicating that DARPins have a limited capacity to recognize the wild-type, conformationally flexible gp120 envelope glycoprotein.
DARPin panning on gp120 target proteins with decreased flexibility and degree of glycosylation.
This low frequency in selecting gp120-specific DARPin binders prompted us to probe three alternative selection strategies to determine whether modified gp120 proteins with reduced structural flexibility or decreased camouflage by glycosylation proved to be better selection targets. In a first approach, we limited the structural flexibility of gp120 by ligation with the CD4-mimetic miniprotein CD4M47, which arrests gp120 in the CD4-bound conformation (30) (Fig. 1B, selection II). In a further strategy, we probed whether removal of the V1V2 domain, the largest and most flexible of the gp120 variable loop domains (53, 54), improves the efficiency of gp120-specific DARPin selection (Fig. 1B, selection III, gp120ΔV1V2). Additionally, we explored whether deglycosylated gp120 allows selection of gp120-specific DARPins at higher frequency (Fig. 1B, selection IV), as sites vulnerable to neutralizing antibody attack are known to be efficiently shielded by the Env protein's heavy glycosylation (9–11).
After three to five rounds of ribosome display, 95 clones from each sublibrary were screened by ELISA for reactivity with the respective target protein used during panning. gp120 conformationally arrested by ligation with the CD4 mimetic proved the most effective target, as already after three rounds of selection we derived two specific binders from the N2C library and nine specific binders from the N3C library of selection II. In selection III (target, gp120ΔV1V2) and selection IV (target, deglycosylated gp120), the derived N2C and N3C sublibraries yielded no gp120-specific clones. Although a few N2C clones (3 clones in selection III and 1 clone in selection IV) with low reactivity with gp120 were detected, they had too low affinity (≥10 μM) for gp120 to allow further follow-up analysis (data not shown).
Affinity maturation of gp120-specific DARPins.
Our primary screen of sublibraries from the four independent selection strategies yielded in total 12 gp120-specific clones (referred to as 1st-generation binders) (Fig. 1B and 2), which all originated from selections I and II. Of note, specific binding of these 1st-generation binders in ELISA to the respective gp120 target protein used in the selection was detected only at high nanomolar to micromolar concentrations, indicative of relatively low affinities of these DARPins for gp120 (Fig. 3 and data not shown). In order to select for gp120-specific DARPins with improved affinities, the sublibraries of all four selection strategies were subjected to additional selection rounds with the aim of diversifying specific binders and specifically selecting for high-affinity binders using off-rate selection. Clones obtained after this affinity maturation step are referred to as 2nd-generation binders (Fig. 1B and 2). Overall, the affinity maturation and off-rate selection step proved successful. Selection I yielded four improved derivatives of the N3C clone 4a3_H3, selected in the primary screen, and four additional N2C binders, whereas selection II yielded 5 further N3C clones upon affinity maturation.
Fig 2.

Phylogenetic analysis of gp120-specific DARPin clones derived by different selection approaches. The sequence relationships of clones obtained from four different selection strategies are shown as phylogenetic trees derived using the maximum-likelihood method based on the JTT matrix-based model. The data are based on protein sequence alignments of N2C and N3C binders from each respective selection. First-generation binders are shown in black and 2nd-generation binders in blue. Binders shown with inverse colored boxes were chosen to be analyzed in detail. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site.
Fig 3.

Analysis of DARPin binding specificity to gp120. Shown is an overview of the binding strengths of individual purified DARPin clones obtained from different selection strategies to gp120 and derivatives as assessed by ELISA, with the half-maximal binding concentration for each of the gp120-specific DARPin binders to recombinant gp120JR-FL wt (green), in complex with CD4 (blue) or CD4M47 (red), or gp120JR-FL with the V1V2 loop deleted (orange). No bar is shown when no binding to the respective construct was detected up to a concentration of 4 μM. The error bars indicate the standard errors of the mean (SEM).
For selections III and IV, which yielded only particularly weak binders in the 1st generation, we retrieved several 2nd-generation binders with improved reactivity for the respective target (13 N2C binders from selection III and 10 N2C binders from selection IV) (Fig. 1B).
Characterization of gp120-specific 1st- and 2nd-generation DARPins.
Sequence analysis of gp120-specific DARPins obtained in the 1st and 2nd generations of selection revealed that individual clones derived from the same N2C or N3C sublibrary were in most cases highly related (Fig. 2; see Fig. S1 in the supplemental material). Based on the sequence analysis and binding efficacy in the initial screen, distinct clones from all selections were chosen for detailed binding and inhibitory-activity analysis. To this end, DARPin preparations were purified and checked by SEC-MALS for the oligomeric state. DARPins that had a tendency to form dimers or higher-order aggregates were excluded from further analysis (data not shown). The 10 N2C DARPin clones from selection IV differed in only a few amino acids (Fig. 2). Unfortunately, all selection IV clones needed to be excluded from follow-up, as they formed higher-order oligomers. For all other clones from selections I, II, and III chosen for follow-up, the binding properties of purified DARPins to (i) full-length wt gp120 of strain JR-FL, (ii) JR-FL gp120 liganded to CD4M47, (iii) JR-FL gp120 liganded to soluble CD4, and (iv) JR-FL gp120 with the V1V2 loop deleted were determined. Figure 3 depicts gp120 binding data for representative clones of each selection series. In general, the derived DARPin clones showed gp120 binding patterns that were in line with their respective selection strategies.
(i) Selection I.
Clones derived from the N2C and N3C sublibraries of selection I were in both cases closely related (Fig. 2). While both N2C and N3C DARPins bound wt gp120 efficiently, this binding was abolished in the presence of sCD4 but remained comparable to that of wt gp120 when gp120 was complexed with CD4M47 (Fig. 3). Of note, the N2C clone 6a2_A1 bound gp120 with V1V2 deleted efficiently, while the N3C clone H3_1B3 failed to do so, indicating that these clones recognize distinct epitopes.
(ii) Selection II.
Selection II (CD4M47-liganded gp120) yielded clones with substantially divergent sequences and binding reactivities within the N3C sublibrary (Fig. 2). CD4 ligation proved to be a decisive determinant for DARPin binding to gp120 among selection II clones (Fig. 3). None of the probed 1st-generation clones bound unliganded wt gp120. All N3C clones bound to CD4M47-liganded gp120 but not to CD4M47 alone (Fig. 2 and data not shown). In contrast, the N2C binder 3m2_A12 bound only to sCD4-liganded gp120 but none of the other probed targets, even though this binder was selected against CD4M47-liganded gp120. The 2nd-generation clone 5m3_D12 was the only selection II DARPin that recognized non-CD4-triggered wt gp120, albeit with markedly lower efficacy than when triggered with either sCD4 or CD4M47. In addition, 5m3_D12 bound gp120 with V1V2 deleted with high affinity.
(iii) Selection III.
Despite relatively high sequence diversity among the 13 N2C clones obtained from selection III, the probed clones displayed largely overlapping binding patterns (Fig. 3 and data not shown). Two N2C clones, AKNF1_10 and AKNF1_14, were selected for further analysis. In accordance with their selection against gp120 with V1V2 deleted, this envelope mutant was recognized with highest efficiency, but the clones also proved to recognize wt gp120. CD4 ligation again had different effects on the binding capacities of these clones. While CD4M47 ligation had no influence, sCD4 abolished the capacity of the DARPins to interact with gp120 (Fig. 3).
Defining binding domains of gp120-specific DARPins.
In order to map the binding sites recognized by the gp120-specific DARPins in more detail, we studied the capacities of representative members of each selection to bind to a panel of recombinant JR-FL gp120 proteins that included (i) full-length wt protein; (ii) the CD4 binding site (CD4bs) mutant gp120D368R, known to obliterate CD4 and CD4bs antibody binding (55–57); and (iii) the coreceptor binding site (CoRbs) mutant gp120I420R, known to eliminate binding of antibodies recognizing the CD4-induced (CD4i) coreceptor binding site (27, 58). The panel also included mutant gp120 proteins lacking (i) the V1 loop (gp120ΔV1), (ii) the entire V1V2 domain (gp120ΔV1V2), and (iii) the V3 loop (gp120ΔV3) (Fig. 4). The functionality of all gp120 proteins used in these studies was verified by assessing the binding of gp120-specific antibodies and CD4 (Fig. 4A). The observed binding patterns of the MAbs were in accordance with the respective epitopes of the antibodies, with CD4bs-specific MAb b12 and CD4IgG2 lacking the capacity to bind the gp120D368R mutant protein, the V3-loop-specific MAbs failing to bind gp120ΔV3, and the CD4i MAbs 17b and 48d showing enhanced capacity to bind gp120ΔV1V2 in the non-CD4-triggered conformation. Likewise, binding of CD4i MAbs to gp120I420R was obliterated and was strongly reduced for gp120ΔV3, in line with the known contribution of the V3 loop and bridging sheet to the binding domains of these antibodies (27, 58, 59).
Fig 4.

DARPin mapping of reactivity with mutant gp120 proteins. Shown is binding of gp120-specific MAbs (A) and DARPins (B) to the indicated JR-FL-derived gp120 proteins by ELISA. The ELISA binding signal of DARPins and MAbs to wt gp120 at saturating concentration was set to 100%, and mutant gp120 binding is depicted relative to this value. For clones that bind gp120 only in a liganded form, the CD4 ligand was used in each ELISA. Since the CD4 ligands do not bind gp120D368R, the construct could not be employed in the liganded form. The heat maps indicate 100% wt-like binding in green and no binding in red, with a linear gradient through yellow at 50%. n/a, not applicable; n/d, not done.
With the exception of DARPins from selection II, which were selected against CD4-triggered gp120 and which require this conformation for effective binding, all the DARPins bound efficiently to the CD4bs mutant gp120D368R (Fig. 4B). Most strikingly, only the selection II DARPin 5m3_D12 bound the CoRbs mutant gp120I420R, indicating that the structural arrest conferred by this mutant is not tolerated by most of the selected DARPins. The V1 loop deletion, in contrast, was tolerated by all groups. Deletion of the entire V1V2 domain had different effects. The selection I DARPin H3_1_B3 failed to bind gp120ΔV1V2. Binding of the three selection II DARPins 3m3_A8, 3m3_B9, and 3m3_F12 was markedly reduced in the absence of the V1V2 domain, while the remaining DARPins bound equally well in the presence and absence of the V1V2 region. With a single exception (3m2_A12), deletion of the V3 loop led to complete or nearly complete loss of DARPin binding. Thus, both the gp120I420R mutation and V3 loop deletion conferred structural rearrangements in monomeric gp120 that affected recognition by DARPins but were largely tolerated by gp120-specific Abs.
Inhibition of HIV entry by gp120-specific DARPins.
We next evaluated the efficacy of the DARPins to inhibit entry of envelope-pseudotyped HIV into TZM-bl cells (46, 60), probing a panel of subtype B tier 1 (including highly neutralization-sensitive viruses with V1V2 deleted) and tier 2 viruses (Fig. 5). We had previously determined the inhibitory capacities of MAbs b12 and 2G12 against the same virus panel (21, 46), and these data are shown in comparison. The majority of DARPin clones showed a moderate to potent inhibition of the highly neutralization-sensitive viruses lacking the V1V2 domain and the tier 1 virus isolates NL4-3 and SF162 but lacked activity against tier 2 viruses. Notably, only one clone, 5m3_D12, showed some breadth against tier 2 viruses. The same DARPin also blocked in vitro infection of activated macaque peripheral blood mononuclear cells (PBMCs) with SHIV162P3 (data not shown). None of the selected DARPin clones affected the entry of virions carrying the unrelated retroviral MuLV envelope, confirming that the observed inhibition by DARPins is indeed HIV specific. Interestingly, DARPins H3_1B3, 3m3_B9, and 3m3_F12 blocked wt NL4-3 and wt SF162 but lost neutralizing activity in the absence of the V1V2 loop. The latter finding is in line with our observation that these clones had a strongly reduced capacity to bind gp120ΔV1V2, indicating that they bind to domains on gp120 encompassing the V1V2 region. Similarly, 3m3_A8 showed decreased activity against SF162, but not NL4-3, in the absence of the V1V2 domain. DARPins AKNF1_10 and AKNF1_14, selected against JR-FL gp120 with V1V2 deleted, neutralized tier 1 isolates and viruses with V1V2 deleted with high potency (in the low nanomolar to subnanomolar range) but failed to block wild-type tier 2 isolates with one exception (6535.3 neutralization by AKNF1_14). Thus, as can be expected from the panning against gp120 with V1V2 deleted, DARPins AKNF1_10 and AKNF1_14 are limited in their action by V1V2 shielding. They can only access their binding domains when the envelope trimer has adopted an open, tier 1-like conformation for which V1V2 shielding is less efficient or when the V1V2 shielding is artificially removed, but not when the trimer is in the closed conformation adopted by tier 2 viruses. Despite the fact that all DARPins in selections I and II were derived from selection against wt JR-FL gp120, only DARPin 5m3_D12 blocked entry of wt JR-FL efficiently. Since DARPin 5m3_D12 also showed the highest reactivity against tier 2 viruses probed in our panel, we focused for the remaining analysis on this clone.
Fig 5.

HIV entry inhibition by gp120-specific DARPins. DARPin inhibition of a panel of 21 Env-pseudotyped viruses of tier 1 (wt and with V1V2 deleted) and tier 2 and of MuLV as a control was probed in the TZM-bl assay. The IC50s are depicted. Virus envelopes with V1V2 deleted are shown in red, wt tier 1 envelopes in blue, wt tier 2 envelopes in black, and the MuLV control in purple. For comparison, inhibition data for the MAbs IgG1-b12 and 2G12 from Rusert et al. (21, 46) are depicted. DARPins were tested to a maximal concentration of 5 μM and MAbs to 666 nM.
DARPin 5m3_D12 recognizes the V3 loop.
To obtain further information on the epitope recognized by 5m3_D12, we performed competition binding experiments using a panel of gp120-directed MAbs specific for the gp120 core, C terminus, CD4bs, CoRbs, and V3 loop and the glycan-dependent MAbs 2G12 and PGT128 (Fig. 6). 5m3_D12 binding to gp120 was competed off only by MAbs directed to the V3 tip (447-52D and 1-79). The fact that none of the other DARPins was affected by these MAbs and the failure of 5m3_D12 to bind to gp120 with the V3 loop deleted (Fig. 4) provided strong evidence that 5m3_D12 binds the V3 loop directly. MAb PGT128, which recognizes a glycan-dependent motif in the V3 stem region, had, in contrast to the tip-specific MAbs, only a marginal influence on 5m3_D12 binding. Of note, PGT128 had a more substantial effect on all other DARPins in our panel, suggesting that PGT128 binding induces conformational rearrangements that afflict a variety of domains within gp120. In line with the CoRbs mutant analysis and the selection against a CD4-liganded target, the CoRbs epitope-directed MAb 17b (and to a lesser extent MAb 48d) affected the binding of all DARPins from selection II. Of note, 5m3_D12, which preferentially binds to CD4- and CD4-M47-triggered gp120, failed to bind to gp120 in the presence of MAbs b12 and b6 but recognized gp120 in the presence of VRC01, suggesting that the latter MAb induces conformational changes required for DARPin recognition similarly to CD4 (Fig. 6).
Fig 6.
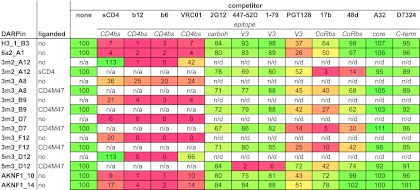
DARPin mapping of reactivity with gp120 in the presence of gp120-specific MAbs. Binding of DARPins to wt JR-FL gp120 (CD4 triggered or not, as indicated) in ELISA in the presence of the depicted gp120-specific MAbs was monitored. “None” indicates binding to gp120 in the absence of competitor MAb and was set to 100%. The binding signal in the presence of the competing MAbs is expressed relative to this value. For clones that bind gp120 only when liganded, binding in the presence of the CD4 ligand was set to 100%. Ligands were not employed if they targeted the same epitope as the competitor used. The heat map indicates 100% binding in the absence of competitor in green and complete inhibition (0% binding) of binding in red, with a linear gradient through yellow at 50%. n/a, not applicable; n/d, not done.
DARPin 5m3_D12 recognizes the V3 loop in a conformation-dependent manner.
To determine whether 5m3_D12 is indeed specific for the V3 loop, we performed direct binding studies and competition binding experiments using both linear peptides and a panel of V3 loop mimetics based on structures in the Protein Data Bank (PDB) of V3 loop peptides bound to the MAbs F425-B4e8 (29), 2219 (61), 537-10D (62), and 447-52D (63) (Fig. 7A). The peptides in each complex adopt β-hairpin conformations, but differences arise in the orientations of side chains on each face of the hairpin (the register of the hairpin). In the complex with 2219, the I307 and F317 side chains point to the same side of the hairpin and comprise a cross-strand hydrogen-bonding (HB) pair, whereas in the complex with F425-B4e8, I307 and Y318 form the HB pair, and with 537-10D the bond is between H308 and F317 (Fig. 7A). In designing the mimetics, the V3 loop sequences were transplanted onto a d-Pro–l-Pro template in order to stabilize the backbone hairpin conformation and fix the hairpin register (64). The pair of residues directly attached to the template should orient their side chains onto the same face of the hairpin and occupy an HB position. In this way, the four cyclic peptide mimetics referred to here as HF, IY, IF, and HY (Fig. 7A) were designed and should structurally mimic the V3 peptides complexed with the respective MAbs. The IY mimetic was reported previously (31), whereas the other mimetics studied here were produced and characterized in the same way. The nuclear magnetic resonance (NMR) structures of the mimetics were determined in aqueous solution, which confirmed the expected hairpin structures (Fig. 7A; see Fig. S2 and S3 and Tables S1 to S5 in the supplemental material). All V3 peptides comprised the conserved GPG motif at the tip of the loop, as well as parts of the loop stem.
Fig 7.

(A) DARPin 5m3_D12 interacts preferentially with structural mimetics of the V3 loop. (Top row) Backbone ribbon representations of linear V3 peptides bound to the MAbs 537-10D, F425-B4e8, 2219, and 447-52D (from PDB files 3GHE, 2QSC, 2B0S, and 2ESX). The C(β) atoms of selected side chains are shown as balls, and the residues shown are color coded. Cross-strand hydrogen-bonded residues are indicated by light-blue dotted lines. (Middle row) The designed backbone cyclic V3 loop mimetics with the HF, IY, IF, and HY registers. (Bottom row) Representative solution NMR structures determined for each mimetic, which confirm for each a stable β-hairpin backbone conformation and the predicted hairpin registers. The d-Pro–l-Pro template is shown for each at the bottom of the structure in orange. (B) Binding of DARPin 5m3_D12 to immobilized recombinant JR-FL gp120 liganded with the CD4-mimetic CD4M47 was studied by ELISA in the presence of increasing concentrations of linear V3 peptides and V3 mimetics. The data are shown relative to 5m3_D12 binding without competitor peptides. (C) The same analysis as in panel B is shown for MAbs 447-52D and F425-4e8.
We performed competition binding studies of 5m3_D12 binding to plate-immobilized JR-FL gp120 triggered by CD4M47 in the presence and absence of linear V3 loop peptides and V3 mimetics from strains JR-FL and MN (Fig. 7B). Interestingly, 5m3_D12 showed a very clear preference for structurally arrested mimetics of the IY register, as they competed effectively with gp120 for DARPin binding whereas the linear peptides based on the same sequences did not. The mimetic IY (MNmut), which contains a proline-to-alanine substitution in the conserved GPG motif, showed reduced competition (Fig. 7B). In contrast to IY mimetics, mimetics with HY and IF registers did not compete off 5m3_D12 binding to gp120. The mimetics with an HF register and the cyclic SS, a V3 mimetic cyclized by a disulfide bond, showed weak to moderate competition.
Overall, the competition experiments highlighted a strong dependence of 5m3_D12 on a specific V3 conformation, which is in sharp contrast to V3 loop-directed antibodies (31). Interestingly, the MAbs 447-52D (the epitope model for the HY mimetic) and F425-B4e8 (the epitope model for the IY mimetics) reacted equally well with the linear V3 peptide and their modeled epitope mimetic but showed reduced efficacy in binding mimetics with a different register (Fig. 7C). We verified these findings by performing direct V3 peptide binding experiments using plate-immobilized biotinylated peptides. DARPin 5m3_D12, but none of the DARPins from other groups (Fig. 8 and data not shown), bound to the mimetic with an IY register based on the V3 sequence of the MN virus strain. Binding to the linear V3 peptide from the same strain by the DARPin was approximately 100-fold weaker. In contrast, the V3-specific MAbs 1-79 (25), 19b (65), and 447-52D bound both the linear and structurally arrested peptides at comparable levels, with 1-79 even displaying a slight preference for the linear peptide (Fig. 8).
Fig 8.

DARPin 5m3_D12 interacts preferentially with structural mimetics of the V3 loop and inhibits entry of tier 2 virus JR-FL. Shown are direct binding (left) to plate-immobilized linear MN V3 peptide [linear (MN)] and cyclic IY MN V3 mimetic [cyclic IY (MN)] in ELISA and entry inhibition in TZM-bl cells (right) of pseudoviruses JR-FL and JR-FLΔV1V2 by DARPin 5m3_D12, MAb 1-79, MAb 19b, and MAb 447-52D.
Most interestingly, DARPin 5m3_D12 is able to neutralize not only JR-FL with V1V2 deleted, but also wt JR-FL-pseudotyped virus, albeit with 2-log-unit lower potency (Fig. 8). This contrasts with the V3-directed MAbs 1-79, 19b, and 447-52D, which are more than 5 log units less potent against wt JR-FL than JR-FL with V1V2 deleted (Fig. 8). Since the V3 loop is potently shielded against antibody attack by the V1V2 domain, most V3 loop antibodies defined to date cannot, or can only with low efficacy, neutralize HIV (21). In the absence of V1V2 shielding, however, V3 loop antibodies display remarkable potency and cross-reactivity (21).
DARPin 5m3_D12 is thus able to partially circumvent V1V2 shielding. Intriguingly, we observed a notable capacity of DARPin 5m3_D12 to block divergent strains of wt HIV (Fig. 5). However, while our data suggest that 5m3_D12 is able to partially bypass the V1V2 shielding on genetically divergent isolates, the activity of 5m3_D12 against viruses lacking the V1V2 domain was still markedly enhanced, indicating that access of 5m3_D12 is also to some extent restricted by the V1V2 shield.
We next compared the neutralization breadth of 5m3_D12 to those of V3-specific MAbs 447-52D (28) and 1-79 against subtype B viruses (Fig. 9). Both MAbs recognize an epitope in the V3 tip that is expected to at least partially overlap the 5m3_D12 epitope (Fig. 6). DARPin 5m3_D12 neutralized a subset of strains with considerable potency (Fig. 9, red symbols) but showed no efficacy against the residual strains. Intriguingly, the majority of the strains that were sensitive to 5m3_D12 were also neutralized by MAb 1-79, but not MAb 447-52D, indicating distinct differences also among V3 loop antibodies in bypassing V1V2 shielding.
Fig 9.

Subtype B tier 2 inhibition by DARPin 5m3_D12 and V3-specific MAbs. The neutralization breadth against subtype B tier 2 viruses of DARPin 5m3_D12 and V3-specific monoclonal antibodies 1-79 and 447-52D was probed in TZM-bl pseudotype virus inhibition assays, and the IC50s are shown. Isolates inhibited by DARPin 5m3_D12 are shown in red.
To obtain insights into the binding properties of 5m3_D12 in the context of the native envelope spike during virus entry, we next performed competition inhibition experiments using V3 peptides and mimetics. Neutralization activity of DARPin 5m3_D12 against SF162 and JR-FL-pseudotyped virus with V1V2 deleted was diminished by addition of V3 IY mimetics in a dose-dependent fashion, whereas the corresponding linear peptides and the mimetic with an HF3 register showed no effect (Fig. 10). This contrasted with the pattern observed for MAb 1-79, where the neutralization activity was more strongly affected by addition of linear V3 peptides than V3 mimetics of the IY register. A V3 mimetic with an HF register had an intermediate effect. In summary, these experiments underlined the fact that DARPin 5m3_D12 recognizes the V3 loop in a structurally constrained manner.
Fig 10.

IY V3 loop mimetics efficiently compete with the native V3 loop on intact viral spikes for binding to DARPin 5m3_D12. Inhibitory activities in the TZM-bl assay of DARPin 5m3_D12 and MAb 1-79 against pseudoviruses JR-FLΔV1V2 and SF162 were assessed in the presence and absence of competing linear V3 peptides and cyclic V3 mimetics.
DISCUSSION
HIV has evolved an array of schemes, the foremost being heavy glycosylation and conformational masking, to shield vulnerable sites on the envelope trimer from immune recognition and attack by neutralizing antibodies to preserve the functionality of the entry complex (8, 10). Engineered entry inhibitors by and large suffer from the same limitations as antibodies in accessing relevant domains on the envelope trimer. In the present study, we investigated the potential of DARPin technology in selecting novel gp120-reactive entry inhibitors. Both antibodies and the smaller DARPins have the ability to recognize their targets with high affinity and specificity while differing entirely in structure (19). We reasoned that the distinct binding properties of DARPins may allow the selection of novel gp120-reactive molecules with specificities in epitope recognition and inhibitory activity that differ from those found among neutralizing antibodies.
Antibodies and the N2C (15-kDa) and N3C (18-kDa) types of DARPins employed in the present study cover binding footprints of comparable sizes. DARPins recognize the target by using their surface of α-helices and their row of β-hairpins, resulting in a groove-like binding surface, and thus, they prefer to bind to the surface of a globular protein domain, or at least structurally well-defined loops (16–18, 66–68). Antibodies can do this, too, but they can also bind to an unstructured terminal peptide or long loop, which adapts to a pocket or groove between the antibody variable domains. Conversely, the complementary determining regions (CDRs) of the antibody, especially a long CDR-H3, can bind within a pocket of the target or at the side of a domain (69). The preference of DARPins for recognizing structural components could be advantageous in selecting HIV envelope-directed inhibitors, particularly as recent findings of potent broadly active neutralizing antibodies revealed a high prevalence of antibodies recognizing conformational inter- and intraprotomer loop-spanning binding domains (70, 71). DARPins also have many favorable biophysical properties, such as exceptional stability and high-yield prokaryotic production, which renders this type of molecule a promising candidate for use as a topical microbicide. Furthermore, the construction of bispecific binders is very straightforward, making biepitopic targeting and thus very high avidities achievable (72).
In the present study, we selected HIV entry-blocking clones from DARPin libraries via ribosome display, in which gp120 was presented as a panning target in four modifications aiming to investigate the influence of gp120 conformational flexibility and glycosylation on the efficacy of DARPin selection. We found that DARPins have a relatively limited capacity to recognize the wt, conformationally flexible gp120 envelope glycoprotein. Of the four gp120 modifications probed, only gp120 liganded with the CD4M47 miniprotein (and thereby structurally arrested) yielded a broader range of DARPin binders recognizing different epitopes. Common to all selected gp120-reactive DARPins was a relatively strong dependence on specific gp120 conformations. This was particularly evident in the mutant gp120 mapping we performed. The CoRbs mutant gp120I420R and the V3 loop deletion variant were, with one exception each, not recognized by the selected DARPins, irrespective of which selection they came from, while gp120-specific antibodies recognized the same mutant proteins unless their epitope was known to be directly affected by the inferred mutations (Fig. 4).
Preferences regarding specific gp120 conformations were also apparent among DARPins selected against CD4M47-liganded gp120. Only the V3 loop-specific N3C clone 5m3_D12 was able to bind to nonliganded gp120. All other DARPins derived from this selection series require CD4 ligation (by either sCD4 or the CD4 miniprotein CD4M47) in order to bind to gp120.
The V3-specific DARPin 5m3_D12 was the only DARPin we selected with a notable activity to block subtype B tier 2 virus infection. Even though this activity was limited to a relatively small number of isolates, it is noteworthy, considering that V3 loop-specific antibodies commonly fail to inhibit these types of isolates, as they cannot bypass V1V2 shielding (21). Thus, although 5m3_D12 is to some extent restricted by V1V2 shielding, it interacts with the V3 loops of certain subtype B tier 2 isolates in a way that allows bypassing of the V1V2 shield and effective inhibition of entry. Investigations of the breadth of 5m3_D12 against viruses from other subtypes are under way.
In stark contrast to the interaction of V3 loop antibodies with their epitope on V3, 5m3_D12 depends on a specific conformation of the V3 loop domain in order to recognize the loop and block infection. The preference of DARPin 5m3_D12 for a mimetic structure with the IY register (31) suggests that the IY β-hairpin structure is close to the predominant conformation the gp120 V3 loop adopts after CD4 binding. Both epitope binding and the neutralization activity of 5m3_D12 crucially relied on this V3 loop conformation. Overall, our findings highlight that the preference of DARPins to recognize specific structures may be utilized in further selection strategies to our advantage. Mimetics as employed here have promise as targets for selection, as they could allow us to more efficiently steer DARPin selection toward a specific site.
In summary, our study highlights the potential of the DARPin technology in retrieving HIV envelope-reactive binders with unique properties that harbor entry-inhibitory capacity. In particular, the conformation dependence of the DARPin-target interaction may prove advantageous for selecting potent entry inhibitors with novel specificities, including alternatives to quaternary antibodies (73, 74), i.e., targeting epitopes only present on the fully assembled native envelope spike. Once identified, HIV-specific DARPin binders with inhibitory activity open multiple avenues for improving their potency. Besides affinity maturation, multivalent constructs (of DARPins with one or more specificities) to cross-link subunits within a trimer or neighboring trimers have promise in boosting efficacy (75).
With the increasing understanding of the architecture of the viral spike (21, 76, 77), the possibility to generate stable soluble trimers that closely resemble the native spike (78, 79), and the means to generate structurally arrested peptide mimetics of gp120 microdomains (31), a number of tools have become available which have promise to tailor future DARPin selections to specific domains of interest. As discussed above, based on our current data, this holds particular promise to improve envelope-specific DARPin identification and to harness the distinctive binding properties of DARPins for HIV inhibitor development.
Supplementary Material
ACKNOWLEDGMENTS
Funding was provided by Swiss National Science Foundation (SNF) grant 310000-120739 to A.T. and National Institutes of Health (NIH) grant R01AI084133 to M.R. and A.T.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 13 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00152-13.
REFERENCES
- 1. Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb. Perspect. Med. 2:a006866 doi:10.1101/cshperspect.a006866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. U. S. A. 100:11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaertner H, Cerini F, Escola JM, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I, Nedellec R, Salkowitz J, Gorochov G, Mosier D, Hartley O. 2008. Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc. Natl. Acad. Sci. U. S. A. 105:17706–17711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4:1302–1307 [DOI] [PubMed] [Google Scholar]
- 5. Walker DK, Abel S, Comby P, Muirhead GJ, Nedderman AN, Smith DA. 2005. Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab. Dispos. 33:587–595 [DOI] [PubMed] [Google Scholar]
- 6. Schief WR, Ban YE, Stamatatos L. 2009. Challenges for structure-based HIV vaccine design. Curr. Opin. HIV AIDS 4:431–440 [DOI] [PubMed] [Google Scholar]
- 7. Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711 [DOI] [PubMed] [Google Scholar]
- 8. Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682 [DOI] [PubMed] [Google Scholar]
- 9. Starcich BR, Hahn BH, Shaw GM, McNeely PD, Modrow S, Wolf H, Parks ES, Parks WP, Josephs SF, Gallo RC, Wong-Staal F. 1986. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45:637–648 [DOI] [PubMed] [Google Scholar]
- 10. Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312 [DOI] [PubMed] [Google Scholar]
- 11. Go EP, Irungu J, Zhang Y, Dalpathado DS, Liao HX, Sutherland LL, Alam SM, Haynes BF, Desaire H. 2008. Glycosylation site-specific analysis of HIV envelope proteins (JR-FL and CON-S) reveals major differences in glycosylation site occupancy, glycoform profiles, and antigenic epitopes' accessibility. J. Proteome Res. 7:1660–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parren PW, Moore JP, Burton DR, Sattentau QJ. 1999. The neutralizing antibody response to HIV-1: viral evasion and escape from humoral immunity. AIDS 13(Suppl. A):S137–S162 [PubMed] [Google Scholar]
- 13. Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burton DR, Ahmed R, Barouch DH, Butera ST, Crotty S, Godzik A, Kaufmann DE, McElrath MJ, Nussenzweig MC, Pulendran B, Scanlan CN, Schief WR, Silvestri G, Streeck H, Walker BD, Walker LM, Ward AB, Wilson IA, Wyatt R. 2012. A blueprint for HIV vaccine discovery. Cell Host Microbe 12:396–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. 2003. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 332:489–503 [DOI] [PubMed] [Google Scholar]
- 16. Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grütter MG, Plückthun A. 2004. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22:575–582 [DOI] [PubMed] [Google Scholar]
- 17. Zahnd C, Wyler E, Schwenk JM, Steiner D, Lawrence MC, McKern NM, Pecorari F, Ward CW, Joos TO, Plückthun A. 2007. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 369:1015–1028 [DOI] [PubMed] [Google Scholar]
- 18. Schweizer A, Rusert P, Berlinger L, Ruprecht CR, Mann A, Corthesy S, Turville SG, Aravantinou M, Fischer M, Robbiani M, Amstutz P, Trkola A. 2008. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog. 4:e1000109 doi:10.1371/journal.ppat.1000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boersma YL, Plückthun A. 2011. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr. Opin. Biotechnol. 22:849–857 [DOI] [PubMed] [Google Scholar]
- 20. Bork P. 1993. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17:363–374 [DOI] [PubMed] [Google Scholar]
- 21. Rusert P, Krarup A, Magnus C, Brandenberg OF, Weber J, Ehlert AK, Regoes RR, Günthard HF, Trkola A. 2011. Interaction of the gp120 V1V2 loop with a neighboring gp120 unit shields the HIV envelope trimer against cross-neutralizing antibodies. J. Exp. Med. 208:1419–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barbas CF, III, Bjorling E, Chiodi F, Dunlop N, Cababa D, Jones TM, Zebedee SL, Persson MA, Nara PL, Norrby E, Burton DR. 1992. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proc. Natl. Acad. Sci. U. S. A. 89:9339–9343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parren PW, Gauduin MC, Koup RA, Poignard P, Fisicaro P, Burton DR, Sattentau QJ. 1997. Relevance of the antibody response against human immunodeficiency virus type 1 envelope to vaccine design. Immunol. Lett. 57:105–112 [DOI] [PubMed] [Google Scholar]
- 24. Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, Pietzsch J, Ott RG, Anthony RM, Zebroski H, Hurley A, Phogat A, Chakrabarti B, Li Y, Connors M, Pereyra F, Walker BD, Wardemann H, Ho D, Wyatt RT, Mascola JR, Ravetch JV, Nussenzweig MC. 2009. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458:636–640 [DOI] [PubMed] [Google Scholar]
- 26. Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, Katinger H. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol. 70:1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thali M, Moore JP, Furman C, Charles M, Ho DD, Robinson J, Sodroski J. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67:3978–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gorny MK, Conley AJ, Karwowska S, Buchbinder A, Xu JY, Emini EA, Koenig S, Zolla-Pazner S. 1992. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J. Virol. 66:7538–7542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pantophlet R, Aguilar-Sino RO, Wrin T, Cavacini LA, Burton DR. 2007. Analysis of the neutralization breadth of the anti-V3 antibody F425-B4e8 and re-assessment of its epitope fine specificity by scanning mutagenesis. Virology 364:441–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stricher F, Huang CC, Descours A, Duquesnoy S, Combes O, Decker JM, Kwon YD, Lusso P, Shaw GM, Vita C, Kwong PD, Martin L. 2008. Combinatorial optimization of a CD4-mimetic miniprotein and cocrystal structures with HIV-1 gp120 envelope glycoprotein. J. Mol. Biol. 382:510–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riedel T, Ghasparian A, Moehle K, Rusert P, Trkola A, Robinson JA. 2011. Synthetic virus-like particles and conformationally constrained peptidomimetics in vaccine design. Chembiochem 12:2829–2836 [DOI] [PubMed] [Google Scholar]
- 32. Haas J, Park EC, Seed B. 1996. Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr. Biol. 6:315–324 [DOI] [PubMed] [Google Scholar]
- 33. Andre S, Seed B, Eberle J, Schraut W, Bultmann A, Haas J. 1998. Increased immune response elicited by DNA vaccination with a synthetic gp120 sequence with optimized codon usage. J. Virol. 72:1497–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barouch DH, Yang ZY, Kong WP, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, Letvin NL, Nabel GJ. 2005. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J. Virol. 79:8828–8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Selvarajah S, Puffer B, Pantophlet R, Law M, Doms RW, Burton DR. 2005. Comparing antigenicity and immunogenicity of engineered gp120. J. Virol. 79:12148–12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zahnd C, Amstutz P, Plückthun A. 2007. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4:269–279 [DOI] [PubMed] [Google Scholar]
- 37. Dreier B, Plückthun A. 2011. Ribosome display: a technology for selecting and evolving proteins from large libraries. Methods Mol. Biol. 687:283–306 [DOI] [PubMed] [Google Scholar]
- 38. Dreier B, Plückthun A. 2012. Rapid selection of high-affinity binders using ribosome display. Methods Mol. Biol. 805:261–286 [DOI] [PubMed] [Google Scholar]
- 39. Moulard M, Lortat-Jacob H, Mondor I, Roca G, Wyatt R, Sodroski J, Zhao L, Olson W, Kwong PD, Sattentau QJ. 2000. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 74:1948–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vives RR, Imberty A, Sattentau QJ, Lortat-Jacob H. 2005. Heparan sulfate targets the HIV-1 envelope glycoprotein gp120 coreceptor binding site. J. Biol. Chem. 280:21353–21357 [DOI] [PubMed] [Google Scholar]
- 41. Zaccolo M, Williams DM, Brown DM, Gherardi E. 1996. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255:589–603 [DOI] [PubMed] [Google Scholar]
- 42. Zahnd C, Sarkar CA, Plückthun A. 2010. Computational analysis of off-rate selection experiments to optimize affinity maturation by directed evolution. Protein Eng. Des. Sel. 23:175–184 [DOI] [PubMed] [Google Scholar]
- 43. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282 [DOI] [PubMed] [Google Scholar]
- 45. Kohl A, Binz HK, Forrer P, Stumpp MT, Plückthun A, Grütter MG. 2003. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. U. S. A. 100:1700–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rusert P, Mann A, Huber M, von Wyl V, Günthard HF, Trkola A. 2009. Divergent effects of cell environment on HIV entry inhibitor activity. AIDS 23:1319–1327 [DOI] [PubMed] [Google Scholar]
- 47. Adachi A, Koenig S, Gendelman HE, Daugherty D, Gattoni-Celli S, Fauci AS, Martin MA. 1987. Productive, persistent infection of human colorectal cell lines with human immunodeficiency virus. J. Virol. 61:209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819–822 [DOI] [PubMed] [Google Scholar]
- 49. Cheng-Mayer C, Weiss C, Seto D, Levy JA. 1989. Isolates of human immunodeficiency virus type 1 from the brain may constitute a special group of the AIDS virus. Proc. Natl. Acad. Sci. U. S. A. 86:8575–8579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trkola A, Kuster H, Rusert P, von Wyl V, Leemann C, Weber R, Stiegler G, Katinger H, Joos B, Günthard HF. 2008. In vivo efficacy of human immunodeficiency virus neutralizing antibodies: estimates for protective titers. J. Virol. 82:1591–1599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pejchal R, Wilson IA. 2010. Structure-based vaccine design in HIV: blind men and the elephant? Curr. Pharm. Des. 16:3744–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kwon YD, Finzi A, Wu X, Dogo-Isonagie C, Lee LK, Moore LR, Schmidt SD, Stuckey J, Yang Y, Zhou T, Zhu J, Vicic DA, Debnath AK, Shapiro L, Bewley CA, Mascola JR, Sodroski JG, Kwong PD. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Gils MJ, Bunnik EM, Boeser-Nunnink BD, Burger JA, Terlouw-Klein M, Verwer N, Schuitemaker H. 2011. Longer V1V2 region with increased number of potential N-linked glycosylation sites in the HIV-1 envelope glycoprotein protects against HIV-specific neutralizing antibodies. J. Virol. 85:6986–6995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Olshevsky U, Helseth E, Furman C, Li J, Haseltine W, Sodroski J. 1990. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J. Virol. 64:5701–5707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pantophlet R, Ollmann Saphire E, Poignard P, Parren PW, Wilson IA, Burton DR. 2003. Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J. Virol. 77:642–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Y, Migueles SA, Welcher B, Svehla K, Phogat A, Louder MK, Wu X, Shaw GM, Connors M, Wyatt RT, Mascola JR. 2007. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat. Med. 13:1032–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xiang SH, Doka N, Choudhary RK, Sodroski J, Robinson JE. 2002. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retroviruses 18:1207–1217 [DOI] [PubMed] [Google Scholar]
- 59. Li Y, Svehla K, Louder MK, Wycuff D, Phogat S, Tang M, Migueles SA, Wu X, Phogat A, Shaw GM, Connors M, Hoxie J, Mascola JR, Wyatt R. 2009. Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J. Virol. 83:1045–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stanfield RL, Gorny MK, Zolla-Pazner S, Wilson IA. 2006. Crystal structures of human immunodeficiency virus type 1 (HIV-1) neutralizing antibody 2219 in complex with three different V3 peptides reveal a new binding mode for HIV-1 cross-reactivity. J. Virol. 80:6093–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Burke V, Williams C, Sukumaran M, Kim SS, Li H, Wang XH, Gorny MK, Zolla-Pazner S, Kong XP. 2009. Structural basis of the cross-reactivity of genetically related human anti-HIV-1 mAbs: implications for design of V3-based immunogens. Structure 17:1538–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rosen O, Sharon M, Quadt-Akabayov SR, Anglister J. 2006. Molecular switch for alternative conformations of the HIV-1 V3 region: implications for phenotype conversion. Proc. Natl. Acad. Sci. U. S. A. 103:13950–13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robinson JA. 2008. Beta-hairpin peptidomimetics: design, structures and biological activities. Acc. Chem. Res. 41:1278–1288 [DOI] [PubMed] [Google Scholar]
- 65. Scott CF, Jr, Silver S, Profy AT, Putney SD, Langlois A, Weinhold K, Robinson JE. 1990. Human monoclonal antibody that recognizes the V3 region of human immunodeficiency virus gp120 and neutralizes the human T-lymphotropic virus type IIIMN strain. Proc. Natl. Acad. Sci. U. S. A. 87:8597–8601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Amstutz P, Binz HK, Parizek P, Stumpp MT, Kohl A, Grütter MG, Forrer P, Plückthun A. 2005. Intracellular kinase inhibitors selected from combinatorial libraries of designed ankyrin repeat proteins. J. Biol. Chem. 280:24715–24722 [DOI] [PubMed] [Google Scholar]
- 67. Eggel A, Buschor P, Baumann MJ, Amstutz P, Stadler BM, Vogel M. 2011. Inhibition of ongoing allergic reactions using a novel anti-IgE DARPin-Fc fusion protein. Allergy 66:961–968 [DOI] [PubMed] [Google Scholar]
- 68. Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. 2007. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 5:e7 doi:10.1371/journal.pbio.0050007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. North B, Lehmann A, Dunbrack RL., Jr 2011. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406:228–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, O'Dell S, Patel N, Shahzad-ul Hussan S, Yang Y, Zhang B, Zhou T, Zhu J, Boyington JC, Chuang GY, Diwanji D, Georgiev I, Kwon YD, Lee D, Louder MK, Moquin S, Schmidt SD, Yang ZY, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Burton DR, Koff WC, Walker LM, Phogat S, Wyatt R, Orwenyo J, Wang LX, Arthos J, Bewley CA, Mascola JR, Nabel GJ, Schief WR, Ward AB, Wilson IA, Kwong PD. 2011. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480:336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong CH, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA. 2011. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science 334:1097–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Boersma YL, Chao G, Steiner D, Wittrup KD, Plückthun A. 2011. Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J. Biol. Chem. 286:41273–41285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Moore PL, Gray ES, Sheward D, Madiga M, Ranchobe N, Lai Z, Honnen WJ, Nonyane M, Tumba N, Hermanus T, Sibeko S, Mlisana K, Abdool Karim SS, Williamson C, Pinter A, Morris L. 2011. Potent and broad neutralization of HIV-1 subtype C by plasma antibodies targeting a quaternary epitope including residues in the V2 loop. J. Virol. 85:3128–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krachmarov C, Lai Z, Honnen WJ, Salomon A, Gorny MK, Zolla-Pazner S, Robinson J, Pinter A. 2011. Characterization of structural features and diversity of variable-region determinants of related quaternary epitopes recognized by human and rhesus macaque monoclonal antibodies possessing unusually potent neutralizing activities. J. Virol. 85:10730–10740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dreier B, Honegger A, Hess C, Nagy-Davidescu G, Mittl PR, Grütter MG, Belousova N, Mikheeva G, Krasnykh V, Plückthun A. 2013. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc. Natl. Acad. Sci. U. S. A. 110:E869–E877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mao Y, Wang L, Gu C, Herschhorn A, Xiang SH, Haim H, Yang X, Sodroski J. 2012. Subunit organization of the membrane-bound HIV-1 envelope glycoprotein trimer. Nat. Struct. Mol. Biol. 19:893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tran EE, Borgnia MJ, Kuybeda O, Schauder DM, Bartesaghi A, Frank GA, Sapiro G, Milne JL, Subramaniam S. 2012. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 8:e1002797 doi:10.1371/journal.ppat.1002797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, Kang YK, Depetris R, Marozsan AJ, Sanders RW, Klasse PJ, Milne JL, Wilson IA, Olson WC, Moore JP, Subramaniam S. 2011. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. U. S. A. 108:11440–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sellhorn G, Kraft Z, Caldwell Z, Ellingson K, Mineart C, Seaman MS, Montefiori DC, Lagerquist E, Stamatatos L. 2012. Engineering, expression, purification, and characterization of stable clade A/B recombinant soluble heterotrimeric gp140 proteins. J. Virol. 86:128–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.