

Abstract
Hepatitis C virus (HCV) is highly dependent on cellular proteins for its own propagation. In order to identify the cellular factors involved in HCV propagation, we performed protein microarray assays using the HCV core protein as a probe. Of ∼9,000 host proteins immobilized in a microarray, approximately 100 cellular proteins were identified as HCV core-interacting partners. Of these candidates, mitogen-activated protein kinase-activated protein kinase 3 (MAPKAPK3) was selected for further characterization. MAPKAPK3 is a serine/threonine protein kinase that is activated by stress and growth inducers. Binding of HCV core to MAPKAPK3 was confirmed by in vitro pulldown assay and further verified by coimmunoprecipitation assay. HCV core protein interacted with MAPKAPK3 through amino acid residues 41 to 75 of core and the N-terminal half of kinase domain of MAPKAPK3. In addition, both RNA and protein levels of MAPKAPK3 were elevated in both HCV subgenomic replicon cells and cell culture-derived HCV (HCVcc)-infected cells. Silencing of MAPKAPK3 expression resulted in decreases in both protein and HCV infectivity levels but not in the intracellular HCV RNA level. We showed that MAPKAPK3 increased HCV IRES-mediated translation and MAPKAPK3-dependent HCV IRES activity was further increased by core protein. These data suggest that HCV core may modulate MAPKAPK3 to facilitate its own propagation.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped virus with a positive-sense, single-stranded RNA genome. HCV causes both acute and persistent infections and often leads to liver cirrhosis and hepatocellular carcinoma (HCC) (1). HCV belongs to the genus Hepacivirus within the Flaviviridae family (2). The HCV genome consists of 9,600 nucleotides and encodes a 3,010-amino-acid protein from a single open reading frame. This polyprotein is processed cotranslationally and posttranslationally by viral and host cellular proteases into 10 functional proteins, including structural (core, E1, and E2) and nonstructural (p7 and NS2 to NS5B) proteins (3). The structural proteins are the components of the viral particle, whereas the nonstructural proteins are involved in the replication of the viral genome.
The HCV core is the nucleocapsid protein that packages the viral RNA genome. The amino acid sequence of the core is highly conserved among six genotypes of HCV. The HCV core can form multimers and self-assemble into nucleocapsid-like particles. The core interacts with many cellular proteins and regulates transcription, signal transduction, cell cycle, steatosis, and tumorigenesis. The interaction between core and apolipoprotein AII leads to the association of core with lipid droplets and lipid accumulation in hepatocytes (4, 5). In addition, PPAR alpha activation is essential for HCV core protein-induced hepatic steatosis and HCC in mice (6). Indeed, HCV core is known to be a multifunctional protein involved in liver pathogenesis and HCC.
Mitogen-activated protein kinase (MAPK) signaling pathways regulate a variety of cellular responses, including gene expression, proliferation, differentiation, and immune responses (7). The MAPK-activated protein kinase (MAPKAPK) family, with its members MAPKAPK2/MK2 and MAPKAPK3/MK3, is one of the downstream targets of MAPK cascades (8). The MAPKAPK3 protein belongs to the serine/threonine MAPKAPK family. MAPKAPK3 is closely related to MAPKAPK2, sharing 72% nucleotide and 75% amino acid identity (9). MAPKAPK3 was characterized as a MAP kinase-activated protein kinase located in the small-cell lung cancer tumor suppressor gene region (9). To date, the functions of MAPKAPK3 protein are poorly understood. It has been previously reported that MAPKAPK3 was activated by influenza virus infection (10). Moreover, the Tsukada group recently identified 2 single-nucleotide polymorphisms located in MAPKAPK3 that were associated with interferon (IFN) therapy in patients infected with HCV genotype 1b (11). However, the functional role of MAPKAPK3 in HCV-infected cells has not been clarified yet.
In the present study, we have used functional proteomics to identify cellular proteins interacting with HCV core protein. Protein microarray technology is a powerful approach to analyze protein-protein, protein-phospholipid, protein-nucleic acid, and protein-small molecule interactions (12, 13). Protein microarray assays are rapid, systematic, relatively inexpensive, and high-throughput screening methods that have important applications in studying biological functions and in drug discovery (14, 15). Using protein microarray analysis, we have identified approximately 100 HCV core-interacting cellular proteins. Among these core partners, MAPKAPK3 was selected for further characterization. Binding of HCV core to MAPKAPK3 was verified by both in vitro pulldown and coimmunoprecipitation assays. Silencing of MAPKAPK3 expression resulted in decreases in protein, extracellular HCV RNA, and HCV infectivity levels. These data suggest that MAPKAPK3 may be involved in HCV propagation.
MATERIALS AND METHODS
Plasmid constructions.
The cDNA of HCV core protein was amplified by PCR using the cDNA derived from genotype 1b. PCR products were inserted into the BamHI and HindIII sites of the pET21b vector (Invitrogen), HindIII and BamHI sites of the pCDNA3 vector (Invitrogen), and BamHI and XbaI sites of the pcDNA4 vector (Invitrogen), respectively. The HCV core mutants were constructed using full-length core as a template. For cloning of MAPKAPK3, total RNAs extracted from Huh7.5 cells were used for reverse transcription-PCR (RT-PCR) with the indicated primers (Table 1). PCR products were inserted into the XbaI and BamHI sites of the plasmid p3XFlag-CMV10 (Sigma-Aldrich). MAPKAPK3 deletion mutants were generated by site-directed mutagenesis (Enzynomics) using primers listed in Table 1 according to the manufacturer's instructions. Small interfering RNA (siRNA)-resistant mutant MAPKAPK3 contains four silent mutations in the siRNA binding site. To generate siRNA-resistant inactive mutant (ATP-binding-defective mutant) MAPKAPK3 (16), a substitution mutation in K73A was introduced into siRNA-resistant mutant MAPKAPK3. Myc-tagged NS3, NS4B, NS5A, and NS5B plasmids have been described elsewhere (17). Flag-tagged MAPKAPK2 was kindly provided by Jun-ichi Abe (University of Rochester).
Table 1.
List of primers used in this study
| Primer | Primer sequence | Enzyme site | Purpose |
|---|---|---|---|
| Core-F | ATCAACTAGTGATGAGCACGAATCCT | SpeI | Cloning of HCV genotype 1b core into pProEX HTa and pET21b+ vectors |
| Core-R | ATTATCTAGAGCAACCGGGCAAGTT | XbaI | |
| Core-FpC3 | GCAAGCTTATGAGCACGAATCCTAAACC | HindIII | Cloning of HCV genotype 1b core into pCDNA3 vector |
| Core-FpC3 | CGGGATCCTCAAGAGCAACCGGGCAA GTTC | BamHI | |
| Myc-core-F | GCGGATCCGAACAAAAACTCATCTCAGAAGAGGATCTGATGAGCACGAATCCTAAACC | BamHI | Cloning of HCV genotype 1b core into pCDNA4 vector |
| Core-R | CGTCTAGAAGAGCAACCGGGCAAGTTC | XbaI | |
| MK3-F | CGCTCTAGA ATGGATGGTGAAACAGCAG | XbaI | Cloning of human MAPKAPK3 into p3XFLAG-CMV-10 vector |
| MK3-R | ATGGATCC CTACTGGTTGTTGCAGCCC | BamHI | |
| MK3 resist-F | ACCACCCCTGGATCAACCAGAGTATGGTAGTGCCACAGACCC | Generation of siRNA-resistant mutant of MAPKAPK3 | |
| MK3 resist-R | GGGTCTGTGGCACTACCATACTCTGGTTGATCCAGGGGTGGT | ||
| MK3 inact-F | GACAGAAGTGTGCCCTGGCGCTCCTGTATGACAGCC | Generation of siRNA-resistant inactive (ATP-binding-defective) mutant of MAPKAPK3 | |
| MK3 inact-R | GGCTGTCATACAGGAGCGCCAGGGCACACTTCTGTC | ||
| 5′ NTR-F | TGAGTGTCGTACAGCCTCCA | qRT-PCR | |
| 5′ NTR-R | ACGCTACTCGGCTAGCAGTC | ||
| qActin-F | TGACAGCAGTCGGTTGGAGCG | qRT-PCR | |
| qActin-R | GACTTCCTGTAACAACGCATCTCATA |
Antibodies.
Antibodies were purchased from the following sources: MAPKAPK3, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and Myc antibodies were from Santa Cruz, and Flag and actin antibodies were from Sigma-Aldrich; HCV core, NS3, and NS5A antibodies have been described elsewhere (17).
Preparation for microarray probe.
HCV core protein expressed in Escherichia coli was purified using Invitrogen Ni-nitrilotriacetic acid (Ni-NTA) agarose beads according to the manufacturer's instructions. Proteins were renatured as we reported previously (18), and aliquots were stored at −70°C.
Protein array screening.
Protein microarray screening was performed using version 5.0 of the Invitrogen Protoarray. The array was first incubated with blocking buffer (50 mM HEPES [pH 7.5], 25% glycerol, 0.08% Triton X-100, 200 mM NaCl, 20 mM reduced glutathione, and 0.1 mM dithiothreitol [DTT]) for 1 h at 4°C with gentle shaking, and then 6 μg of purified-HCV core protein diluted in 120 μl of probing buffer (phosphate-buffered saline [PBS] containing 0.1% Tween 20) was added to the array. Following incubation at 4°C for 1.5 h, the array was washed five times in ice-cold buffer and then treated with Anti-V5-Alexa Fluor 647 antibody (Invitrogen) for 1.5 h at 4°C. The array was dried, and the images were analyzed using a PerkinElmer Scanarray Ex-pressHT system and Invitrogen Prospector version 5.2 software. Significant interactions were identified based on a Z-score cutoff value of 3.0.
Cell culture.
All cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin in 5% CO2 at 37°C. Both IFN-cured and HCV subgenomic replicon cells were grown as previously reported (17).
Immunoprecipitation.
HEK293T cells were cotransfected with 4 μg of Myc-tagged core and 2 μg of Flag-tagged MAPKAPK3 or MAPKAPK2. Total amounts of DNA were adjusted by adding an empty vector. At 48 h after transfection, cells were harvested and an immunoprecipitation assay was performed as we reported previously (17).
In vitro pulldown assay.
Approximately 1 μg of purified core protein was incubated with 30 μl of Ni-NTA agarose beads for 1 h at 4°C with gentle shaking. The beads were then washed three times in buffer (50 mM Na2HPO4 [pH 8.0], 100 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1% protease cocktail inhibitor) and were incubated with cell lysate expressing Flag-tagged MAPKAPK3 for 2 h at 4°C. The sample was washed five times in lysis buffer, and then bound protein was detected by immunoblot analysis using anti-Flag monoclonal antibodies.
Immunoblot analysis.
Equal amounts of proteins were subjected to SDS-PAGE and electrotransferred to a nitrocellulose membrane. The membrane was blocked in TBS-Tween (20 mM Tris-HCl [pH 7.6], 500 mM NaCl, and 0.25% Tween 20) containing 5% nonfat dry milk for 1 h and then incubated overnight at 4°C with the indicated antibodies in TBS-Tween containing 1% nonfat dry milk. Following three washes in TBS-Tween, the membrane was incubated with either horseradish peroxidase-conjugated goat anti-rabbit antibody or goat anti-mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) in TBS-Tween for 1 h at room temperature. Proteins were detected using an ECL kit (Amersham Biosciences).
Luciferase reporter gene assay.
Huh7.5 cells were cotransfected with pRL-HL plasmid together with β-galactosidase and the indicated plasmids. At 48 h after transfection, cells were harvested, and then luciferase assays were performed as described previously (17).
RNA interference.
siRNAs targeting MAPKAPK3 (sense, 5′-GGAUCAACCAAUCGAUGGU-3′; antisense, 5′-ACCAUCGAUUGGUUGAUCC-3′), MAPKAPK2 (sense, 5′-ACGAGCAGAUCAAGAUAAA-3′; antisense, 5′-UUUAUCUUGAUCUGCUCGU-3′), and the universal negative control were purchased from Bioneer. siRNA targeting the 5′ nontranslated region (NTR) of Jc1 (5′-CCUCAAAGAAAAACCAAACUU-3′) was used as a positive control (17). siRNA transfection was performed using a Lipofectamine RNAiMax reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Focus-forming assay.
Huh7.5 cells were seeded at 2 × 104 cells in 4-well chamber culture slides (Millipore). Following 24 h of incubation, cells were inoculated with serial dilutions of cell culture media harvested from cell culture-derived HCV (HCVcc)-infected cells. At 2 days after inoculation, indirect immunofluorescence was performed for the presence of intracellular core antigen to determine the number of focus-forming units (FFU)/ml as we reported previously (17).
Quantification of MAPKAPK and HCV RNA.
Total RNAs were isolated from HCVcc-infected cells, cell culture media, or replicon cells using the RiboEx LS reagent (Geneall Biotechnology) and reverse transcribed using the ReverTra Ace kit (Toyobo). Quantitative real-time PCR (qRT-PCR) experiments were done using an iQ5 multicolor Real-time PCR Detection system (Bio-Rad Laboratories, Hercules, CA) as we reported previously (17).
MTT assay.
Huh7.5 cells were seeded at 4 × 104 cells in a 24-well plate and transfected with the indicated siRNAs. At 4 days after transfection, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (Sigma) was added to the cells, and the cultures were incubated at 37°C for 2 h. Cell viability was determined as we reported previously (17).
Immunofluorescence assay.
Huh7.5 cells cultured on glass slides were fixed with 4% paraformaldehyde in PBS at room temperature for 12 min. After two washes with PBS, fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min and were blocked with 0.5% bovine serum albumin (BSA) for 2 h. The cells were incubated overnight with a mouse anti-MAPKAPK3 antibody and a rabbit anticore antibody, respectively. After three washes with PBS, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG or tetramethylrhodamine isothiocyanate (TRITC)-conjugated donkey anti-rabbit IgG for 2 h at room temperature. After three washes with PBS, cells were analyzed using the Zeiss LSM 700 laser confocal microscopy system (Carl Zeiss, Inc., Thornwood, NY).
RESULTS
Identification of cellular proteins interacting with HCV core using protein arrays.
To identify cellular proteins interacting with HCV core protein, core protein was purified (Fig. 1A) as described in Materials and Methods. Purified core protein was verified using anticore, anti-V5, and anti-His antibodies (Fig. 1B) and then applied on the spots of the human protein microarray kit (Invitrogen), containing approximately 9,000 human proteins. Statistically significant candidate interactors were determined by measuring the mean spot intensity with a Z-score of 3 and more as a cutoff value (Fig. 1C). Analysis of the scanned array data showed that approximately 100 cellular proteins were identified as HCV core interactors (Table 2). MAPKAPK3 was identified as one of the candidate hits, and both positive- and negative-control hits are shown in Fig. 1D. Ontological data revealed that one-half of the putative HCV core interactors were both protein and nucleic acid binding proteins involved in gene regulation, cell cycle, proliferation, and development (Fig. 2).
Fig 1.

Probing of the human protein microarray with HCV core protein. (A, B) HCV core protein was expressed in E. coli and purified by a single-step Ni-NTA agarose affinity chromatography. Purified proteins of various concentrations were separated by SDS-PAGE and either stained with Coomassie brilliant blue (A) or immunoblotted with the indicated antibodies (B). BSA was run in parallel for reference. (C) Frequency histogram for the core-probed protein microarray showing the range of Z-scores obtained. Protein interactors with a Z-score greater than three (Z ≥ 3) were considered significant. Scores were calculated using Invitrogen Protoarray version 5.2 software. (D) Identification of MAPKAPK3 in protein microarray (left panel). Both positive and negative controls are shown (right panel).
Table 2.
Candidate core interactors identified by protein microarray screening
| Gene function and name | Gene product | NCBI accession no. | Z-score |
|---|---|---|---|
| Kinase | |||
| MAPKAPK3 | MAP kinase-activated protein kinase 3 | BC001662 | 3.89 |
| AURKA | Aurora kinase A, transcript variant 2 | NM_003600 | 3.27 |
| VRK1 | Vaccinia-related kinase 1 | NM_003384 | 4.89 |
| CSNK2A2 | Casein kinase 2, alpha prime polypeptide | NM_001896 | 4.47 |
| IPPK | Inositol 1,3,4,5,6-pentakisphosphate 2-kinase | NM_022755 | 3.37 |
| STK6 | Serine/threonine protein kinase 6 | BC001280 | 3.23 |
| MAPKAPK5 | Mitogen-activated protein kinase-activated protein kinase 5, transcript variant 1 | NM_003668 | 3.13 |
| RABEPK | Rab9 effector protein with kelch motifs | BC047023 | 3.09 |
| DNA binding | |||
| PPARG | Peroxisome proliferator-activated receptor gamma | BC006811 | 3.51 |
| DUX3 | Double homeobox 3 | NM_012148 | 6.83 |
| NR4A1 | Nuclear receptor subfamily 4, group A, member 1, transcript variant 1 | NM_002135 | 4.65 |
| C17orf85 | Uncharacterized protein | NM_018553 | 4.00 |
| PIN4 | Protein (peptidylprolyl cis/trans isomerase) NIMA-interacting, 4 | NM_006223 | 4.00 |
| DRAP1 | DR1-associated protein 1 (negative cofactor 2 alpha) | NM_006442 | 3.82 |
| ZNF239 | Zinc finger protein 239 | BC026030 | 3.64 |
| RAD51AP1 | RAD51-associated protein 1 | BC016330 | 3.47 |
| CNOT8 | CCR4-NOT transcription complex, subunit 8 | NM_004779 | 3.26 |
| HMGN3 | High-mobility group nucleosomal binding domain 3, transcript variant 2 | NM_138730 | 3.36 |
| TERF1 | Telomeric repeat binding factor (NIMA-interacting) 1 | BC029378 | 3.28 |
| KLF3 | Kruppel-like factor 3 (basic) | BC051687 | 3.24 |
| MSL3L1 | Male-specific lethal 3-like 1 (Drosophila), transcript variant 2 | NM_078630 | 3.21 |
| EBAG9 | Estrogen receptor binding site associated, antigen, transcript variant 1 | NM_004215 | 3.12 |
| FOS | V-fos FBJ murine osteosarcoma viral oncogene homolog | NM_005252 | 3.14 |
| HMGB2 | High-mobility group box 2 | NM_002129 | 3.04 |
| RNA binding | |||
| ZRANB2 | Zinc finger, RAN-binding domain containing 2, transcript variant 1 | NM_203350 | 6.18 |
| SART3 | Squamous cell carcinoma antigen recognized by T cells 3 | BC041638 | 3.70 |
| RPL23A | Ribosomal protein L23a | NM_000984 | 5.28 |
| EIF1AY | Eukaryotic translation initiation factor 1A, Y linked | BC005248 | 5.34 |
| SRP19 | Signal recognition particle, 19 kDa | BC010947 | 3.93 |
| RPL22 | 60S ribosomal protein L22 | NM_000983 | 3.40 |
| SFRS9 | Splicing factor, arginine/serine-rich 9 | NM_003769 | 3.31 |
| SFRS5 | Splicing factor, arginine/serine-rich 5 | BC018823 | 3.31 |
| RPL29 | 60S ribosomal protein L29 | NM_000992 | 3.00 |
| RPL30 | Ribosomal protein L30 | NM_000989 | 3.14 |
| DDX55 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 55 | BC030020 | 3.10 |
| Protein binding | |||
| EHHADH | Peroxisomal bifunctional enzyme | NM_001966 | 3.34 |
| CTTN | Cortactin, transcript variant 2 | NM_138565 | 5.30 |
| CCNG1 | Cyclin G, transcript variant 1 | NM_004060 | 5.14 |
| WDR5 | WD repeat domain 5, transcript variant 1 | NM_017588 | 4.56 |
| RPS28 | 40S ribosomal protein S28 | NM_001031 | 4.06 |
| GCC1 | GRIP and coiled-coil domain-containing protein 1 | BC008902 | 3.88 |
| EPB41L4A | Band 4.1-like protein 4A | NM_022140 | 3.95 |
| SPC24 | SPC24, NDC80 kinetochore complex component, homolog (Saccharomyces cerevisiae) | NM_182513 | 3.32 |
| PDAP1 | PDGFA-associated protein 1 | NM_014891 | 3.35 |
| HDGFRP3 | Hepatoma-derived growth factor, related protein 3 | NM_016073 | 3.58 |
| DYNC1I1 | Dynein, cytoplasmic 1, intermediate chain 1 | BC037854 | 3.46 |
| PSMD4 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 4, transcript variant 2 | NM_153822 | 3.46 |
| FGF12 | Fibroblast growth factor 12 | BC022524 | 3.53 |
| UBD | Ubiquitin D | NM_006398 | 3.42 |
| NOC2L | Nucleolar complex associated 2 homolog (S. cerevisiae) | BC003555 | 3.39 |
| SUDS3 | Sin3 histone deacetylase corepressor complex component SDS3 | BC093990 | 3.05 |
| DNAJC8 | DnaJ homolog subfamily C member 8 | NM_014280 | 3.24 |
| CCNB3 | Cyclin B3, transcript variant 2 | NM_033671 | 3.15 |
| MAP2 | Microtubule-associated protein 2, transcript variant 2 | NM_031845 | 3.21 |
| INTS3 | Integrator complex subunit 3 | NM_023015 | 3.00 |
| OTUD6B | OTU domain containing 6B | BC029760 | 3.04 |
| SPANXN4 | Sperm protein associated with the nucleus on the X chromosome N4 | NM_001009613 | 3.07 |
| SERTAD1 | SERTA domain containing 1 | BC002670 | 3.81 |
| Calcium/metal ion binding | |||
| CALU | Calumenin | NM_001219 | 3.55 |
| PPP2R3B | Protein phosphatase 2 (formerly 2A), regulatory subunit B″, beta, transcript variant 2 | NM_199326 | 5.63 |
| ELOF1 | Elongation factor 1 homolog (S. cerevisiae) | NM_032377 | 4.47 |
| TRIM44 | Tripartite motif-containing 44 | NM_017583 | 4.44 |
| ASXL1 | Additional sex combs like 1 (Drosophila) | BC064984 | 4.01 |
| POLB | Polymerase (DNA directed), beta | NM_002690 | 3.30 |
| PCLO | Piccolo (presynaptic cytomatrix protein) | BC001304 | 3.10 |
| JMJD2D | Jumonji domain containing 2D | NM_018039 | 3.13 |
| GTPase/ATP/GTP binding | |||
| HRAS | V-Ha-ras Harvey rat sarcoma viral oncogene homolog | BC006499 | 3.68 |
| UBE2S | Ubiquitin-conjugating enzyme E2S | BC004236 | 5.17 |
| SUPT16H | FACT complex subunit SPT16 | BC021561 | 4.31 |
| TTLL7 | Tubulin tyrosine ligase-like family, member 7 | BC048970 | 3.38 |
| TBC1D22B | TBC1 domain family member 22B | NM_017772 | 3.21 |
| Other | |||
| RPL39L | Ribosomal protein L39-like | NM_052969 | 4.39 |
| C20orf198 | Chromosome 20 open reading frame 198 | NM_139016 | 3.47 |
| PRR15 | Proline rich 15 | NM_175887 | 3.32 |
| SPRR4 | Small proline-rich protein 4 | NM_173080 | 3.02 |
| GLIPR2 | Golgi body-associated plant pathogenesis-related protein 1 | NM_022343 | 4.00 |
| NOL7 | Nucleolar protein 7, 27 kDa | NM_016167 | 3.64 |
| IFRD2 | Interferon-related developmental regulator 2 | BC001327 | 3.03 |
| CLIP4 | CAP-GLY domain containing linker protein family, member 4 | NM_024692 | 3.01 |
| DEXI | Dexamethasone-induced transcript | NM_014015 | 5.15 |
| Unknown | |||
| C11orf51 | Chromosome 11 open reading frame 51 | NM_014042 | 4.79 |
| CCDC28A | Coiled-coil domain-containing protein 28A | BC000758 | 4.56 |
| HSPC111 | Hypothetical protein HSPC111 | BC040106 | 3.79 |
| C8orf33 | Chromosome 8 open reading frame 33 | NM_023080 | 3.86 |
| VCY | Variable charge, Y-linked 1B | BC056508 | 3.87 |
| C9orf9 | Chromosome 9 open reading frame 9 | NM_018956 | 4.00 |
| LOC158812 | Spermatid nuclear transition protein 4 | XM_088679 | 3.80 |
| C12orf41 | Chromosome 12 open reading frame 41 | BC013900 | 3.86 |
| CCDC72 | Coiled-coil domain containing 72 | NM_015933 | 3.84 |
| C7orf49 | Chromosome 7 open reading frame 49 | NM_024033 | 3.70 |
| FAM126B | Family with sequence similarity 126, member B | NM_173822 | 3.56 |
| C13orf27 | Chromosome 13 open reading frame 27 | NM_138779 | 3.47 |
| C11orf63 | Chromosome 11 open reading frame 63, transcript variant 2 | NM_199124 | 3.35 |
| FAM116B | Family with sequence similarity 116, member B | NM_001001794 | 3.27 |
| C1orf63 | Chromosome 1 open reading frame 63 | NM_020317 | 3.35 |
| C1orf35 | Chromosome 1 open reading frame 35 | NM_024319 | 3.12 |
| FAM71C | Family with sequence similarity 71, member C | NM_153364 | 3.00 |
| AMMECR1L | AMME chromosomal region gene 1-like | NM_031445 | 3.21 |
| LRRC48 | Leucine-rich repeat containing 48 | BC050419 | 3.11 |
| SLAIN2 | SLAIN motif family, member 2 | BC031691 | 3.03 |
| C9orf71 | Chromosome 9 open reading frame 71 | NM_153237 | 3.08 |
| C12orf45 | Chromosome 12 open reading frame 45 | NM_152318 | 3.07 |
Fig 2.
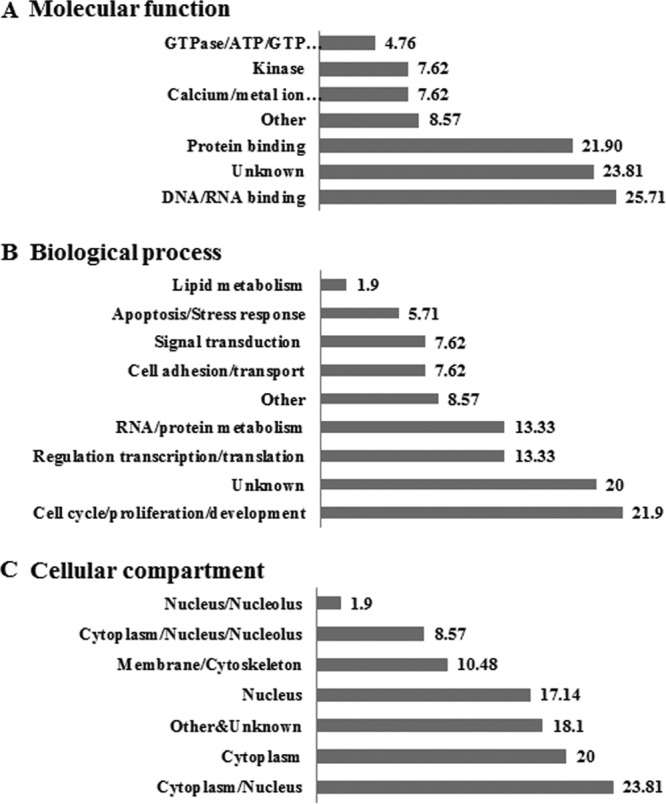
Ontological analysis of the HCV core interactors. Gene ontologies were determined for each of the core interactors, and the results for each of the three standard ontological categories were plotted as percentages.
MAPKAPK3 interacts with HCV core derived from both genotype 1b and genotype 2a.
Because MAPK was involved in proinflammatory cytokine regulation in influenza virus-infected cells (10), we selected MAPKAPK3 to explore a possible involvement in HCV-infected cells. To verify the protein array data, we performed an in vitro binding assay using His-tagged core protein purified from E. coli and cell lysates expressing Flag-tagged MAPKAPK3. Figure 3A shows that MAPKAPK3 selectively interacted with HCV core protein. Coimmunoprecipitation data further confirmed that MAPKAPK3 specifically interacted with core derived from both genotypes 1b and 2a (Fig. 3B). Next, we investigated whether HCV core protein interacted with endogenous MAPKAPK3 in Huh7.5 cells electroporated with Jc1 RNA. Cell lysates harvested at 4 days after electroporation were immunoprecipitated with either an anti-MAPKAPK3 antibody or control IgG, and bound proteins were analyzed by immunoblotting with an anticore antibody. Indeed, HCV core interacts with endogenous MAPKAPK3 protein (Fig. 3C). These data suggest that MAPKAPK3 may colocalize with core in HCV-infected cells. To investigate this possibility, Huh7.5 cells were either mock infected or infected with Jc1, followed by an immunofluorescence assay. As expected, MAPKAPK3 was widely expressed in both the nucleus and the cytoplasm in mock-infected cells (Fig. 3D). It was noteworthy that MAPKAPK3 accumulated mainly in the cytoplasm in HCV-infected cells. Figure 3D shows that both MAPKAPK3 and HCV core were colocalized in the cytoplasm as indicated by the yellow fluorescence. Since MAPKAPK3 is closely related to MAPKAPK2, sharing 72% nucleotide and 75% amino acid identity (9), we asked whether core could interact with MAPKAPK2. For this purpose, HEK293T cells cotransfected with Myc-tagged core and Flag-tagged MAPKAPK3/MAPKAPK2 were immunoprecipitated with an anti-Myc antibody and bound protein was immunoblotted with an anti-Flag antibody. Figure 3E shows that MAPKAPK3, but not MAPKAPK2, interacted with HCV core. In fact, MAPKAPK2 was not identified as a candidate for core interactor in the protein microarray (Fig. 1D). Collectively, these data suggest that MAPKAPK3 specifically interacts with HCV core both in vitro and in vivo.
Fig 3.

MAPKAPK3 interacts with HCV core protein. (A) HEK293T cells were transiently transfected with either vector or Flag-tagged MAPKAPK3. Total cell lysates harvested at 36 h after transfection were incubated with His-tagged core protein purified from E. coli. Bound proteins were precipitated by Ni-NTA beads and detected by immunoblotting (IB) with an anti-Flag monoclonal antibody (top). Core protein was verified with an anticore antibody (bottom). (B) HEK293T cells were cotransfected with Flag-tagged MAPKAPK3 and a Myc-tagged core expression plasmid derived from either genotype 1b or 2a. At 48 h after transfection, cell lysates were immunoprecipitated (IP) with an anti-Myc monoclonal antibody, and bound proteins were then detected by IB analysis using an anti-Flag monoclonal antibody (top panel). Protein expressions of Flag-tagged MAPKAPK3 and Myc-tagged core were verified by IB with an anti-Flag monoclonal antibody and anticore polyclonal antibody, respectively (lower panels). The asterisk indicates IgG heavy chain. (C) HCV core protein interacts with the endogenous MAPKAPK3 in HCV-infected cells. Huh7.5 cells were electroporated with 10 μg of Jc1 RNA. Cell lysates harvested at 4 days after electroporation were immunoprecipitated (IP) with either an anti-MAPKAPK3 antibody or control IgG. Bound protein was immunoblotted with either MAPKAPK3 antibody (upper panel, lanes 3 and 4) or anticore antibody (lower panel, lanes 3 and 4). The asterisk indicates a heavy chain. MK3, MAPKAPK3. (D) Huh7.5 cells were either mock infected or infected with Jc1 for 4 h. At 48 h postinfection, cells were fixed in 4% paraformaldehyde, and immunofluorescence staining was performed by using an anti-MAPKAPK3 monoclonal antibody and fluorescein isothiocyanate-conjugated goat anti-mouse IgG to detect MAPKAPK3 (green) and a rabbit anticore antibody and TRITC-conjugated donkey anti-rabbit IgG to detect core (red). Dual staining showed colocalization of MAPKAPK3 and core as yellow fluorescence in the merged image. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to label nuclei (blue). (E) HEK293T cells were cotransfected with Myc-tagged core and Flag-tagged MAPKAPK3 or Flag-tagged MAPKAPK2. At 48 h after transfection, cell lysates were IP with an anti-Myc monoclonal antibody, and bound proteins were then detected by IB analysis using an anti-Flag monoclonal antibody (top panel). Protein expression of Myc-tagged core was verified by IB with anticore polyclonal antibody (bottom panel). The asterisk indicates IgG heavy chain.
HCV core interacts with MAPKAPK3 through the N-terminal half of the kinase domain in MAPKAPK3 and aa 41 to 75 residues of core.
To determine the region in core responsible for MAPKAPK3 binding, the interactions between MAPKAPK3 and various deletion mutants of core (Fig. 4A) were determined by a transfection-based coimmunoprecipitation assay. As shown in Fig. 4B, MAPKAPK3 interacted with a mutant encompassing amino acids (aa) M41 to 173 but not with the mutant encompassing aa M76 to 173 of core. This result indicated that the region spanning aa 41 to 75 of core was responsible for binding with MAPKAPK3. Next, we determined the region in MAPKAPK3 for core binding. We constructed various domain mutants of MAPKAPK3 (Fig. 4C), and the binding domain was determined as described above. Figure 4D demonstrated that core interacted with a mutant harboring an intact kinase domain but not with a mutant harboring the C-terminal half of the kinase domain in MAPKAPK3, indicating that core interacted with MAPKAPK3 through the region encompassing the N-terminal half of the kinase domain.
Fig 4.

HCV core protein interacts with MAPKAPK3 through amino acid residues 41 to 75 of core and the N-terminal half of the kinase domain of MAPKAPK3. (A) Schematic illustration of both wild-type and mutants of HCV core expression plasmid. aa, amino acids. (B) MAPKAPK3 interacts with amino acid residues 41 to 75 of core. HEK293T cells were cotransfected with Flag-tagged MAPKAPK3 and Myc-tagged core expression plasmids. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-Myc monoclonal antibody, and bound proteins were immunoblotted with an anti-Flag monoclonal antibody. Protein expressions of Myc-tagged core and Flag-tagged MAPKAPK3 were verified by immunoblotting with an anti-Myc or anti-Flag monoclonal antibody using the same cell lysates. (C) Schematic illustration of both wild-type and mutants of MAPKAPK3 expression plasmid. (D) HCV core interacts with the N-terminal region of the kinase domain of MAPKAPK3. HEK293T cells were cotransfected with the indicated combinations of expression plasmids. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-Myc monoclonal antibody, and bound proteins were immunoblotted with an anti-Flag monoclonal antibody (right panel). Arrows denote bound proteins. The asterisks indicate IgG heavy and light chains. Protein expressions of Myc-tagged core and Flag-tagged MAPKAPK3 were verified by immunoblotting with rabbit anticore antibody or anti-Flag monoclonal antibody using the same cell lysates (left panel).
HCV increases MAPKAPK3 expression level.
To investigate whether the MAPKAPK3 expression level was regulated by HCV, we determined both mRNA and protein levels of MAPKAPK3 in replicon cells. As shown in Fig. 5A, the MAPKAPK3 mRNA level was significantly increased in replicon cells compared with IFN-cured cells. Figure 5B demonstrates that the MAPKAPK3 protein level was pronouncedly elevated in HCV replicon cells. We further demonstrated that both mRNA and protein levels of MAPKAPK3 were significantly increased in both Jc1-infected cells (Fig. 5C and D) and JFH1-infected cells (Fig. 5E and F) compared with mock-infected cells. These data suggested that HCV nonstructural protein was responsible for upregulation of MAPKAPK3 expression. For this purpose, Huh7 cells were transiently transfected with either vector or Myc-tagged core, NS3, NS4B, NS5A, or NS5B, individually. At 72 h after transfection, total cell lysates were immunoblotted with the indicated antibodies. As shown in Fig. 5G, NS5A, but not other HCV proteins, increased the MAPKAPK3 protein level in a dose-dependent manner. Collectively, these data clearly demonstrate that HCV increases the MAPKAPK3 expression level.
Fig 5.
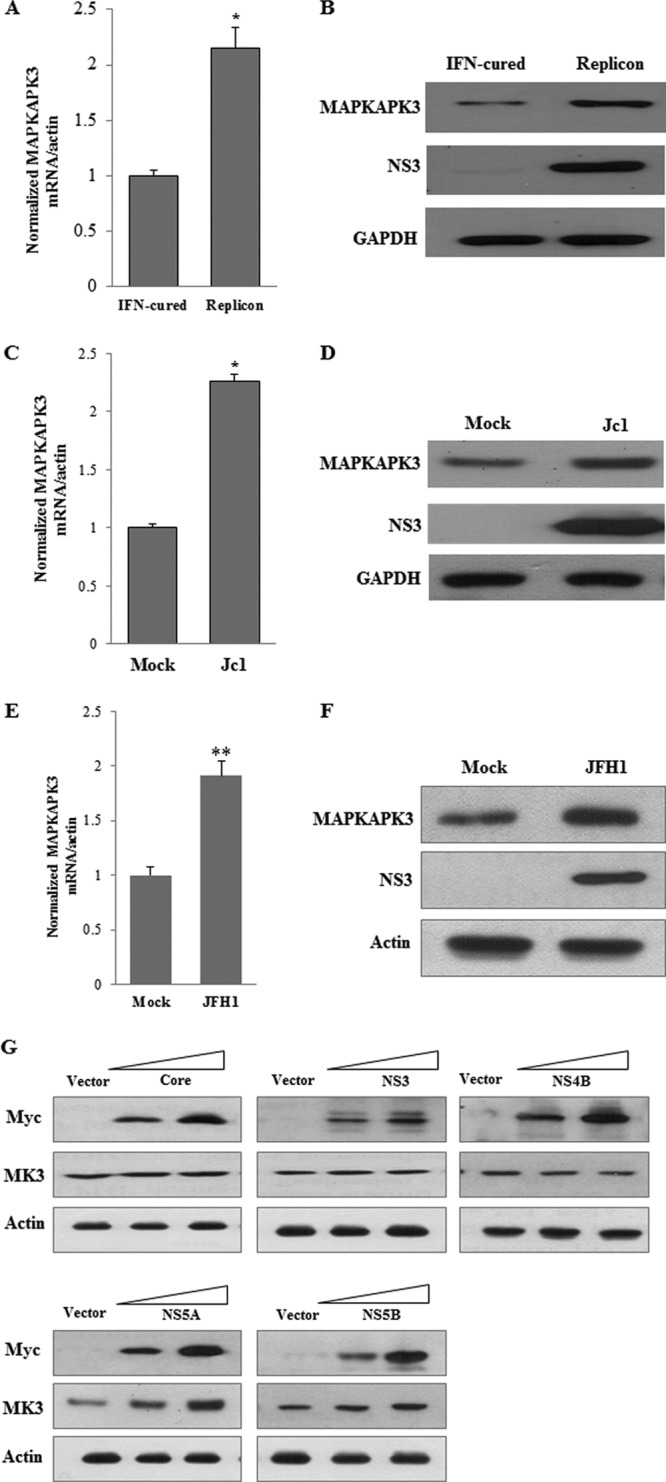
HCV increases MAPKAPK3 expression level. (A) MAPKAPK3 mRNAs isolated from either IFN-cured or subgenomic replicon cells were quantified by qPCR. The asterisk indicates a significant difference (*, P < 0.05) from the value for the mock control. Error bars indicate standard deviations. (B) Total cell lysates harvested from either IFN-cured or subgenomic replicon cells were immunoblotted with the indicated antibodies. (C) Huh7.5 cells were either mock infected or infected with HCV Jc1 for 4 h. At 96 h postinfection, MAPKAPK3 mRNAs were analyzed by qPCR. Data from two independent experiments were quantified. (D) Total cell lysates harvested at 96 h postinfection were immunoblotted with the indicated antibodies. (E) Huh7.5 cells were either mock infected or infected with HCV JFH1 for 4 h. At 96 h postinfection, MAPKAPK3 mRNAs were analyzed by qPCR. Data from two independent experiments were quantified. The asterisks indicate significant differences (**, P < 0.01). Error bars indicate standard deviations. (F) Total cell lysates harvested at 96 h postinfection were immunoblotted with the indicated antibodies. (G) Huh7 cells were transiently transfected with either vector or increasing amounts of Myc-tagged core or NS3, NS4B, NS5A, or NS5B expression plasmid, individually. At 72 h after transfection, total cell lysates were immunoblotted with the indicated antibodies.
Knockdown of MAPKAPK3 suppresses HCV protein expression level in HCV subgenomic replicon cells.
To investigate the effect of MAPKAPK3 on HCV replication, HCV subgenomic replicon cells were transfected with siRNA targeting MAPKAPK3 or the indicated control siRNA constructs. Figure 6A demonstrated that silencing of MAPKAPK3 dramatically impaired both NS3 and NS5A protein levels in replicon cells. Of note, the suppressive activity of MAPKAPK3 siRNA was as efficient as that of positive siRNA (Fig. 6A, lane 2 versus lane 3). Knockdown of MAPKAPK3 mRNA was confirmed by qPCR (Fig. 6B). Interestingly, the intracellular HCV RNA level was not altered by knockdown of MAPKAPK3 (Fig. 6C). These data suggest that MAPKAPK3 may be involved in the translational step of the HCV life cycle.
Fig 6.
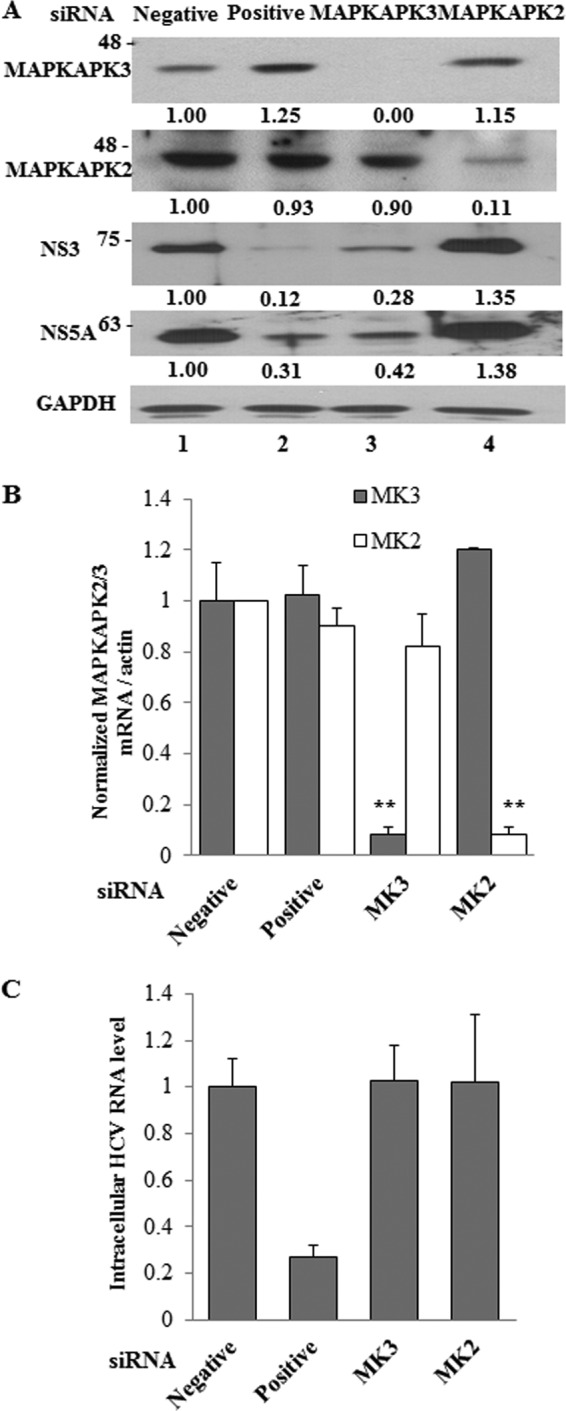
Knockdown of MAPKAPK3 impairs HCV protein expression level in HCV subgenomic replicon cells. (A) Huh7 cells harboring HCV subgenomic replicon were transfected with the indicated siRNA constructs. Total cell lysates harvested at 72 h after transfection were immunoblotted with the indicated antibodies. The band intensity was quantified using ImageJ software. (B, C) Total RNAs were extracted from cells at 72 h after siRNA transfection, and either MAPKAP2/3 mRNAs (B) or intracellular HCV RNAs (C) were quantified by qPCR. Negative, universal negative-control siRNA; positive, HCV-specific siRNA; MK3, MAPKAPK3-specific siRNA; MK2, MAPKAPK2-specific siRNA. The asterisks indicate significant difference (**, P < 0.01) from the value for the negative control. Experiments were carried out in duplicate. Error bars indicate standard deviations.
Knockdown of MAPKAPK3 suppresses HCV propagation.
To further investigate the effect of MAPKAPK3 on HCV propagation, Huh7.5 cells transfected with siRNA targeting MAPKAPK3 or the indicated control siRNA constructs were infected with Jc1. At 48 h postinfection, both protein and RNA levels were determined. Silencing of MAPKAPK3 significantly suppressed both NS3 and core protein levels (Fig. 7A). It was noteworthy that the suppressive activity of MAPKAPK3 siRNA was as efficient as that of positive siRNA (Fig. 7A, lane 2 versus lane 3). Moreover, MAPKAPK3 but not MAPKAPK2 specifically impaired HCV protein expression. As described for replicon cells above, the intracellular HCV RNA level was not affected by knockdown of MAPKAPK3 in Jc1-infected cells (Fig. 7B). Silencing of MAPKAPK3 expression led to strong inhibition of the extracellular HCV RNA level (Fig. 7C). We further showed that knockdown of MAPKAPK3 significantly inhibited both HCV protein and extracellular HCV RNA levels in JFH1-infected cells (Fig. 7D and E). To confirm this result, naïve Huh7.5 cells were infected with Jc1 harvested from Fig. 7C and HCV infectivity was determined. As shown in Fig. 7F, HCV infectivity was significantly suppressed in MAPKAPK3 knockdown cells. Because transfection of siRNAs induced no cytotoxicity (Fig. 7G), the silencing effect was specific to MAPKAPK3 in HCV-infected cells. To rule out the off-target effect of MAPKAPK3 siRNA, we generated a siRNA-resistant MAPKAPK3 mutant and a siRNA-resistant ATP-binding-defective MAPKAPK3 mutant (16). As shown in Fig. 7H, exogenous expression of a siRNA-resistant MAPKAPK3 mutant, but neither wild-type MAPKAPK3 nor the ATP-binding-defective MAPKAPK3 mutant, rescued HCV protein expression (lane 4). Collectively, these data suggest that MAPKAPK3 is required for HCV propagation.
Fig 7.

Knockdown of MAPKAPK3 suppresses HCV propagation. (A) Huh7.5 cells were transfected with the indicated siRNAs. At 2 days after siRNA transfection, cells were infected with Jc1 for 4 h, and then cell lysates harvested at 48 h postinfection were immunoblotted with the indicated antibodies. The band intensity was quantified using ImageJ software. (B) Intracellular HCV RNAs were isolated at 48 h postinfection and were analyzed by qPCR. (C) Extracellular RNAs isolated from these cells were quantified by qPCR. (D) Huh7.5 cells were transfected with the indicated siRNAs. At 2 days after siRNA transfection, cells were infected with JFH1 for 4 h, and then cell lysates harvested at 48 h postinfection were immunoblotted with the indicated antibodies. (E) Total RNAs were extracted from Huh7.5 cells at 48 h postinfection. Both intracellular and extracellular HCV RNAs were quantified by qPCR. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01). Experiments were carried out in triplicate. Error bars indicate standard deviations. MK3, MAPKAPK3; MK2, MAPKAPK2. (F) Culture supernatants harvested from experiment depicted in panel C were used to infect naïve Huh7.5 cells. Virus titers were determined by focus-forming assays. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from the value for the negative control. (G) Huh7.5 cells were transfected with 50 nM the indicated siRNAs. At 4 days after siRNA transfection, cell viability was determined by MTT assay. Negative, universal negative-control siRNA; positive, HCV-specific positive-control siRNA; MK3, MAPKAPK3; MK2, MAPKAPK2. (H) Huh7.5 cells were transfected with the indicated siRNAs. At 48 h after siRNA transfection, cells were infected with Jc1 for 4 h and then further transfected with the indicated combinations of expression plasmids. At 48 h after plasmid transfection, cell lysates were immunoblotted using the indicated antibodies. MAPKAPK3-WT, Flag-tagged wild-type MAPKAPK3; MAPKAPK3-SR, Flag-tagged siRNA-resistant mutant MAPKAPK3; MAPKAPK3-KA, Flag-tagged siRNA-resistant ATP-binding-defective mutant MAPKAPK3. Experiments were carried out in duplicate.
Protein interaction between core and MAPKAPK3 is required for HCV IRES-mediated translation.
Translation of the HCV RNA is mediated mainly by an internal ribosome entry site (IRES) that is located within the 5′ untranslated region (UTR) of the viral RNA (19–21). Because silencing of MAPKAPK3 had no effect on intracellular HCV RNA level, we further investigated the effect of MAPKAPK3 on the translational step of the HCV life cycle. For this purpose, we took advantage of the pRL-HL construct (Fig. 8A) (22). Indeed, overexpression of MAPKAPK3 significantly increased HCV IRES activity (Fig. 8B, left panel). We also showed that MAPKAPK3-dependent HCV IRES-mediated translation was increased by core protein in a dose-dependent manner. To verify the effect of MAPKAPK3 on HCV IRES-mediated translation, we performed a reporter assay using an ATP-binding-defective MAPKAPK3 mutant (MK3-KA). As shown in Fig. 8B (left panel), the inactive MAPKAPK3 mutant was unable to increase HCV IRES-mediated translation. These data indicate that MAPKAPK3 is involved in HCV IRES-mediated translation. It has been previously reported that HCV core stimulated HCV IRES activity (23, 24). Indeed, overexpression of core protein increased HCV IRES activity 1.5-fold compared to vector control, and core-mediated IRES activity was further elevated by MAPKAPK3 in a dose-dependent manner (Fig. 8B, right panel). We further showed that an ATP-binding-defective mutant of MAPKAPK3 was unable to bind to the HCV core protein (Fig. 8C), indicating that protein interaction was important for HCV protein expression. Next, we investigated the functional relevance of protein interplay between core and MAPKAPK3 in HCV IRES-mediated translation. Huh7.5 cells were transfected with either negative siRNA or MAPKAPK3 siRNA, and then cells were further transfected with either empty vector or core plasmid in the absence or presence of siRNA-resistant mutant MAPKAPK3 together with reporter plasmids. As shown in Fig. 8D, core protein alone increased HCV IRES activity in a dose-dependent manner (left panel). Knockdown of MAPKAPK3 impaired HCV IRES-mediated translational activity. However, expression of a siRNA-resistant MAPKAPK3 mutant rescued IRES activity, and this activity was enhanced by core protein in a dose-dependent manner (Fig. 8D, left panel). Therefore, the effect of core on HCV IRES activity was MAPKAPK3 dependent. We further assessed the functional significance of protein interaction between core and MAPKAPK3 on HCV IRES-mediated translation using a binding-defective mutant of core. For this purpose, Huh7.5 cells were cotransfected with wild-type MAPKAPK3 and full-length core together with reporter plasmids. Cells were further transfected with various mutant constructs of core plasmid. As shown in Fig. 8D (right panel, lane 2), MAPKAPK3-dependent HCV IRES activity was significantly increased by full-length core protein. In competition assays, all core mutants except M76-173 efficiently abrogated the protein interaction between wild-type core and MAPKAPK3 and thus impaired the HCV IRES activity (Fig. 8D, right panel). Since M76-173 is a binding-defective mutant core (Fig. 4B), these data indicate that the protein interplay between core and MAPKAPK3 plays a crucial role in HCV propagation.
Fig 8.

MAPKAPK3 increases HCV IRES translation. (A) Schematic diagram of pRL-HL plasmid. (B) (Left panels) Huh7.5 cells were cotransfected with vector, Flag-tagged wild-type MAPKAPK3, or Flag-tagged mutant MAPKAPK3-KA individually in the absence or presence of increasing amounts of Myc-tagged core together with pRL-HL and β-galactosidase expression plasmids. Luciferase activities were determined at 48 h after transfection and then normalized to β-galactosidase. HCV IRES activity was expressed as the FLuc/RLuc ratio (left, upper panel). Protein expressions of MAPKAPK3, MAPKAPK3-KA, core, and actin were verified using the same cell lysates with the indicated antibodies (left, lower panel). The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01). Experiments were carried out in duplicate. Error bars indicate standard deviations. (Right panels) Huh7.5 cells were cotransfected with either vector or Myc-tagged core in the absence or presence of increasing amounts of Flag-tagged wild-type MAPKAPK3 together with pRL-HL and β-glactosidase expression plasmids. Luciferase activities were determined at 48 h after transfection and then normalized to β-galactosidase. HCV IRES activity was expressed as the FLuc/RLuc ratio (right, upper panel). The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01) from the value for the vector control. Experiments were carried out in duplicate. Error bars indicate standard deviations. Protein expressions of core, MAPKAPK3-WT, MAPKAPK3-KA, and actin were verified using the same cell lysates with the indicated antibodies (right, lower panels). (C) HEK293T cells were cotransfected with the indicated combinations of expression plasmids. At 48 h after transfection, cell lysates were immunoprecipitated with an anti-Myc monoclonal antibody, and bound proteins were then detected by immunoblot analysis using an anti-Flag monoclonal antibody. Protein expressions of Flag-MAPKAPK3, Flag-MAPKAPK3-SR, Flag-MAPKAPK3-KA, Myc-core, and actin in the same cell lysates were verified by immunoblot analysis using an anti-Flag monoclonal antibody, anticore polyclonal antibody, and anti-actin antibody, respectively. Asterisks indicate IgG heavy and light chains. (D) (Left panels) Huh7.5 cells were transfected with 50 nM the indicated siRNAs. At 24 h after transfection, cells were further transfected with either vector (white bars) or two different amounts of Myc-tagged core (black bars) in the absence or presence of siRNA-resistant mutant MAPKAPK3 (Flag-MAPKAPK3-SR) together with pRL-HL and β-galactosidase expression plasmids. Luciferase activities were determined at 48 h after transfection and normalized to β-galactosidase. HCV IRES activity was expressed as the ratio of FLuc to RLuc (upper panel). Experiments were carried out in duplicate. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01). Error bars indicate standard deviations. Protein expressions of MAPKAPK3, MAPKAPK3-SR, core, and actin were verified using the same cell lysates with the indicated antibodies (lower panels). (Right panels) Huh7.5 cells were cotransfected with Flag-tagged MAPKAPK3-WT and pcDNA3 core together with pRL-HL and β-galactosidase expression plasmids. At 24 h after transfection, cells were further transfected with either vector or Myc-tagged core mutants as indicated. Luciferase activities were determined at 36 h after transfection and normalized to β-galactosidase. HCV IRES activity was expressed as the ratio of FLuc to RLuc (right, upper panel). Experiments were carried out in duplicate. The asterisks indicate significant differences (*, P < 0.05; **, P < 0.01). Error bars indicate standard deviations. ns, P value was not significant. Protein expressions of MAPKAPK3-WT, pCDNA3-core, Myc-tagged core mutants, and actin were verified using the same cell lysates with the indicated antibodies (right, lower panels).
DISCUSSION
The HCV core protein modulates various cellular signal transduction pathways by interacting with a variety of cellular factors and is involved in HCV-induced pathogenesis (25–28). To identify the cellular factors involved in HCV propagation, we employed a protein array screening using HCV core protein as a probe. Of 9,000 human proteins, approximately 100 cellular proteins were identified as the novel HCV core interactors. Among them, more than 25% were RNA/DNA binding proteins, while ∼21% were proteins involved in cell cycle, proliferation, and development. Since MAPKAPK3 has been implicated in influenza virus propagation (10), we explored the possible involvement of MAPKAPK3 in HCV propagation. MAPKAPK3 is a member of the MAPKAP kinase family. It is activated by three members of the MAPK family, i.e., extracellular signal-regulated kinase (ERK), p38, and Jun N-terminal kinase (JNK) (16).
Protein interaction between MAPKAPK3 and HCV core was confirmed by both in vitro binding assay and coimmunoprecipitation assays. MAPKAPK3 specifically interacted with HCV core derived from genotypes 1b and 2a. We further verified that both MAPKAPK3 and HCV core proteins were colocalized in the cytoplasm of Jc1-infected cells. Overexpression of MAPKAPK3 significantly increased HCV IRES-mediated translational activity, and this activity was synergistically enhanced by core protein. These data suggest that protein interplay between core and MAPKAPK3 may facilitate the cellular milieu to increase HCV propagation.
Here we demonstrated that MAPKAPK3 supported the translation of viral proteins but not transcription of viral RNA. We showed that siRNA-mediated knockdown of MAPKAPK3 significantly suppressed the HCV protein expression level, whereas intracellular HCV RNA levels were unchanged in HCV replicon cells. Likewise, knockdown of MAPKAPK3 significantly reduced HCV protein and extracellular HCV RNA levels, but not the intracellular HCV RNA level, in both Jc1- and JFH1-infected cells. This was further verified by the significant decrease of HCV infectivity in MAPKAPK3 knockdown cells. It has been previously reported that MAPKAPK3 was activated by influenza virus (10). Likewise, HCV also increased the MAPKAPK3 expression level. Since NS5A but not other HCV proteins elevated the MAPKAPK3 protein level, it is possible that NS5A may be involved in this process. Indeed, stress-induced protein kinase p38 MAPK and JNK have been demonstrated in the HCV subgenomic replicon cells and in NS5A-expressing Huh7 cells (29). Hence, the upregulation of MAPKAPK3 in cells overexpressing HCV NS5A protein might result from the stress induced by the upstream transducer of MAPK signaling cascades. However, the detailed mechanism of how HCV upregulates expression of MAPKAPK3 mRNA and protein levels needs further investigation.
MAPKAPK3 has substrates overlapping or identical to those of MAPKAPK2 both in vitro and in vivo, including HSP25, HSP27, CREB, E47, and SRF (30–33). Major functions of both MAPKAPK2 and MAPKAPK3 are regulations of cell cycle, cytokines, and chromatin and actin remodeling (34). Nevertheless, MAPKAPK2 was not identified as a core interactor in our protein array screening data. Indeed, there was no direct protein interaction between core and MAPKAPK2. Moreover, knockdown of MAPKAPK2 showed no effect on HCV propagation. These data showed that MAPKAPK3 but not MAPKAPK2 was specifically involved in HCV propagation.
Finally, we investigated which step of the HCV life cycle was affected by MAPKAPK3. We used a bicistronic reporter construct containing Renilla luciferase and firefly luciferase genes separated by a functional HCV IRES (22). The dual-luciferase assay showed that MAPKAPK3 was involved in HCV IRES translation but not in transcription of viral RNA (Fig. 8). This result further supported that MAPKAPK3 modulated the HCV protein level but not the intracellular HCV RNA level. Since HCV IRES-mediated translation was specifically modulated by HCV core protein in previous studies (23, 24) and knockdown of MAPKAPK3 significantly decreased HCV IRES activity in the presence of core protein in the current study, these data indicated that modulation of HCV IRES-mediated translation by core protein occurred through MAPKAPK3 protein. Moreover, overexpression of mutant core containing the necessary region to bind to MAPKAPK3 competed away the protein interaction between wild-type core and MAPKAPK3, and hence HCV IRES activity was impaired. These data indicate that direct binding of core to MAPKAPK3 is required for increasing the ability of MAPKAPK3 to stimulate HCV IRES activity. We can speculate that both NS5A and core are collaboratively involved in the modulation of MAPKAPK3 in HCV-infected cells. NS5A increases the MAPKAPK3 expression level, and core utilizes MAPKAPK3 to stimulate HCV IRES activity. Collectively, our data suggest that HCV may hijack cellular MAPKAPK3 for its own propagation and MAPKAPK3 may thus be a potential therapeutic target for HCV infections.
ACKNOWLEDGMENTS
This work was supported by the Basic Science Research Program (2012026351) and Biotechnology Development (2008-2004100) from the Ministry of Education, Science and Technology, South Korea, and by the National R&D Program (1020290) for Cancer Control, Ministry for Health and Welfare, South Korea.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 13 March 2013
REFERENCES
- 1. Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y, Choo QL, Houghton M, Kuo G. 1990. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A. 87:6547–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giannini C, Brechot C. 2003. Hepatitis C virus biology. Cell Death Differ. 10(Suppl 1):S27–S38 [DOI] [PubMed] [Google Scholar]
- 3. Lindenbach BD, Rice CM. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933–938 [DOI] [PubMed] [Google Scholar]
- 4. Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, Eder G, Schaff Z, Chapman MJ, Miyamura T, Brechot C. 1997. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. U. S. A. 175:740–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi ST, Polyak SJ, Hong T, Taylor DR, Gretch DR, Lai MM. 2002. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology 292:198–210 [DOI] [PubMed] [Google Scholar]
- 6. Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. 2008. PPAR alpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 118:683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dong C, Davis RJ, Flavell RA. 2002. MAP kinases in the immune response. Annu. Rev. Immunol. 20:55–72 [DOI] [PubMed] [Google Scholar]
- 8. Gaestel M. 2006. MAPKAP kinases-MKs—two's company, three's a crowd. Nat. Rev. Mol. Cell Biol. 7:120–130 [DOI] [PubMed] [Google Scholar]
- 9. Sithanandam G, Latif F, Duh FM, Bernal R, Smola U, Li H, Kuzmin I, Wixler V, Geil L, Shrestha S. 1996. 3pK, a new mitogen-activated protein kinase-activated protein kinase located in the small cell lung cancer tumor suppressor gene region. Mol. Cell. Biol. 16:868–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luig C, Köther K, Dudek SE, Gaestel M, Hiscott J, Wixler V, Ludwig S. 2010. MAP kinase-activated protein kinases 2 and 3 are required for influenza A virus propagation and act via inhibition of PKR. FASEB J. 24:4068–4077 [DOI] [PubMed] [Google Scholar]
- 11. Tsukada H, Ochi H, Maekawa T, Abe H, Fujimoto Y, Tsuge M, Takahashi H, Kumada H, Kamatani N, Nakamura Y, Chayama K. 2009. A polymorphism in MAPKAPK3 affects response to interferon therapy for chronic hepatitis C. Gastroenterology 136:1796–1805.e6 doi:10.1053/j.gastro.2009.01.061 [DOI] [PubMed] [Google Scholar]
- 12. Satoh J, Nanri Y, Yamamura T. 2006. Rapid identification of 14-3-3-binding proteins by protein microarray analysis. J. Neurosci. Methods 152:278–288 [DOI] [PubMed] [Google Scholar]
- 13. Tripathi LP, Kataoka C, Taguwa S, Moriishi K, Mori Y, Matsuura Y, Mizuguchi K. 2010. Network based analysis of hepatitis C virus core and NS4B protein interactions. Mol. Biosyst. 6:2539–2553 [DOI] [PubMed] [Google Scholar]
- 14. Fenner BJ, Scannell M, Prehn JH. 2010. Expanding the substantial interactome of NEMO using protein microarrays. PLoS One 5:e8799 doi:10.1371/journal.pone.0008799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hall DA, Ptacek J, Snyder M. 2007. Protein microarray technology. Mech. Ageing Dev. 128:161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ludwig S, Engel K, Hoffmeyer A, Sithanandam G, Neufeld B, Palm D, Gaestel M, Rapp UR. 1996. 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol. Cell. Biol. 16:6687–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lim YS, Tran HT, Yim SA, Hwang SB. 2011. Peptidyl-prolyl isomerase Pin1 is a cellular factor required for hepatitis C virus propagation. J. Virol. 85:8777–8788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hwang SB, Lai MMC. 1993. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J. Virol. 67:7659–7662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji H, Fraser CS, Yu Y, Leary J, Doudna JA. 2004. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc. Natl. Acad. Sci. U. S. A. 101:16990–16995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. 2001. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 291:1959–1962 [DOI] [PubMed] [Google Scholar]
- 21. Wang L, Jeng KS, Lai MM. 2011. Poly(C)-binding protein 2 interacts with sequences required for viral replication in the hepatitis C virus (HCV) 5′ untranslated region and directs HCV RNA replication through circularizing the viral genome. J. Virol. 85:7954–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Honda M, Kaneko S, Matsushita E, Kobayashi K, Abell GA, Lemon SM. 2000. Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology 118:152–162 [DOI] [PubMed] [Google Scholar]
- 23. Boni S, Lavergne JP, Boulant S, Cahour A. 2005. Hepatitis C virus core protein acts as a trans-modulating factor on internal translation initiation of the viral RNA. J. Biol. Chem. 280:17737–17748 [DOI] [PubMed] [Google Scholar]
- 24. Lourenço S, Costa F, Débarges B, Andrieu T, Cahour A. 2008. Hepatitis C virus internal ribosome entry site-mediated translation is stimulated by cis-acting RNA elements and trans-acting viral factors. FEBS J. 275:4179–4197 [DOI] [PubMed] [Google Scholar]
- 25. Cheng PL, Chang MH, Chao CH, Lee YH. 2004. Hepatitis C viral proteins interact with Smad3 and differentially regulate TGF-beta/Smad3-mediated transcriptional activation. Oncogene 23:7821–7838 [DOI] [PubMed] [Google Scholar]
- 26. Joo M, Hahn YS, Kwon M, Sadikot RT, Blackwell TS, Christman JW. 2005. Hepatitis C virus core protein suppresses NF-kappaB activation and cyclooxygenase-2 expression by direct interaction with IkappaB kinase beta. J. Virol. 79:7648–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kasprzak A, Adamek A. 2007. Role of hepatitis C virus proteins (C, NS3, NS5A) in hepatic oncogenesis. Hepatol. Res. 38:1–26 [DOI] [PubMed] [Google Scholar]
- 28. Lin W, Kim SS, Yeung E, Kamegaya Y, Blackard JT, Kim KA, Holtzman MJ, Chung RT. 2006. Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain. J. Virol. 80:9226–9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qadri I, Iwahashi M, Capasso JM, Hopken MW, Flores S, Schaack J, Simon FR. 2004. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: role of JNK, p38 MAPK and AP-1. Biochem. J. 378:919–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clifton AD, Young PR, Cohen PA. 1996. Comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. 392:209–214 [DOI] [PubMed] [Google Scholar]
- 31. Engel K, Ahlers A, Brach MA, Herrmann F, Gaestel M. 1995. MAPKAP kinase 2 is activated by heat shock and TNF-alpha: in vivo phosphorylation of small heat shock protein results from stimulation of the MAP kinase cascade. J. Cell. Biochem. 57:321–330 [DOI] [PubMed] [Google Scholar]
- 32. Heidenreich O, Neininger A, Schratt G, Zinck R, Cahill MA, Engel K, Kotlyarov A, Kraft R, Kostka S, Gaestel M, Nordheim A. 1999. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J. Biol. Chem. 274:14434–14443 [DOI] [PubMed] [Google Scholar]
- 33. Sutherland C, Alterio J, Campbell DG, Le-Bourdelles B, Mallet J, Haavik J, Cohen P. 1993. Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase an MAP-kinase-activated kinases 1 and 2. Eur. J. Biochem. 217:715–722 [DOI] [PubMed] [Google Scholar]
- 34. Ronkina N, Kotlyarov A, Gaestel M. 2008. MK2 and MK3—a pair of isoenzymes? Front. Biosci. 13:5511–5521 [DOI] [PubMed] [Google Scholar]