

Abstract
Evasion of immune T cell responses is crucial for persistent viruses to establish a normal carrier state. Most studies on active immune modulation mechanisms have focused on the stage of virus production in infected cells, when large numbers of viral antigens and potential immune modulators are expressed. For oncogenic viruses such as Kaposi's sarcoma-associated herpesvirus (KSHV), which is carried as a lifelong infection, usually with little harmful effect, but can cause various tumors, the immune evasion strategies can also be relevant in the context of tumorigenesis. Here we report that the virus-encoded interferon regulatory factor 3 (vIRF3) latent viral gene expressed in KSHV-related tumors functions as a potent immunevasin. Expression of vIRF3 downregulates surface major histocompatibility complex class II (MHC-II) DR expression with slow kinetics but, more importantly, can substantially inhibit recognition by KSHV-specific CD4 T cells prior to its effects on MHC-II DR downregulation in model cell systems. This property of vIRF3 is only partly due to its ability to inhibit the transcription of CIITA and, thus, MHC-II expression; CIITA-independent inhibition of MHC-II transcripts and another as yet unidentified posttranscriptional mechanism are also involved in qualitatively modulating the availability of specific peptide/MHC-II complexes at the cell surface. Consistent with these observations, the vIRF3-expressing KSHV-associated primary effusion lymphoma (PEL) lines are generally resistant to recognition by KSHV-specific CD4 T cells. Interestingly, some PEL lines exhibit small subpopulations with lower vIRF3 expression that can be recognized. These data implicate vIRF3 as a critical determinant of the MHC-II antigen presentation function in KSHV-associated PELs that is likely to be important in the pathogenesis of these tumors.
INTRODUCTION
Infection with Kaposi's sarcoma-associated herpesvirus (KSHV) results in a lifelong carriage state, usually with little harmful effect. However, the virus can cause Kaposi's sarcoma, an AIDS-associated malignancy, and has been proposed to be the etiological agent of primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (1–4). KSHV can establish latent infections in the B cell compartment and has the potential to transform cell growth (5), a property that is relevant to its mechanisms of persistence in immunocompetent hosts as well as its oncogenic potential.
Successful persistence of viral infection depends on the establishment of a balance between host immune responses and viral immune evasion. The innate immune system provides an immediate defense against infection and is very important for controlling viral infections. Remarkably, KSHV encodes four proteins named virus-encoded interferon regulatory factors (vIRFs) that are homologues of cellular IRFs. The vIRFs have been shown to inhibit IFN responses (6). With regard to adaptive immune responses, KSHV genes reported to modulate major histocompatibility complex class I (MHC-I) antigen presentation include K3 and K5 (7, 8). However, KSHV is poorly understood in terms of its interference with the MHC-II antigen presentation pathway.
Mechanisms for interfering with the MHC-II antigen presentation pathway have been reported for other herpesviruses and involve multiple points on the MHC class II antigen presentation pathway (9). For example, US2 (10), US3 (11), and pp65 (12) of human cytomegalovirus (HCMV) and glycoprotein B of human herpes simplex virus 1 (13) all target the HLA-DR molecule for diversion and/or degradation during membrane transport. More recently, Epstein-Barr virus (EBV)-encoded BZLF1 has been shown to target CD74, the invariant-chain chaperone of HLA-DR, to evade CD4 T cell recognition (14). The gp42 protein of EBV also binds to MHC-II to sterically inhibit CD4 T cell receptor interactions (15).
Using peripheral blood mononuclear cells from healthy KSHV-infected donors, Sabbah et al. successfully identified and isolated CD4 T cell responses to LANA, a protein expressed in all KSHV-infected cells, whether normal or malignant. However, most PEL cell lines were not be able to be recognized by these CD4 T cell clones, except via reexpression of the MHC class II transactivator (CIITA) in the PELs (16). A study from another group showed that vIRF3 can inhibit the PIII and PIV promoters of CIITA and can inhibit the transcription and translation of MHC-II elements (17), which provides a potential explanation for the poor recognition of PELs by the LANA-specific CD4 T cells. However, the small MHC-II downregulation observed in the latter study cannot fully explain the substantially reduced MHC-II expression observed in the PELs. There is also no direct evidence that this inhibition of transcription of CIITA by vIRF3 can impair the recognition by CD4 T cells.
In the study described here, we have carried out a more detailed analysis of the effects of vIRF3 on MHC-II antigen presentation and, importantly, have extended the work to include functional T cell recognition assays as a readout. We demonstrate for the first time that vIRF3 does indeed impair the recognition by KSHV-specific CD4 T cells in B cells and PEL lines and that the ability of PEL cells to be recognized by LANA-specific CD4 T cells correlated with vIRF3 expression at the single-cell level. Using quantitative reverse transcription-PCR (RT-PCR), we demonstrate that vIRF3 can inhibit the transcription of CIITA and MHC-II elements, through which vIRF3 can substantially downregulate MHC-II expression, but with slow kinetics that lag behind an effect on functional presentation of antigen to CD4 T cells. The data reveal additional mechanisms of impairment of MHC-II antigen presentation by vIRF3 which are partly mediated through a CTIIA-independent inhibition of the transcription of MHC-II elements.
MATERIALS AND METHODS
Plasmids and transfection.
The vIRF3 gene was subcloned into the EcoRI/XhoI sites of the pCDNA3-IRES-nls-GFP plasmid vector and was verified by restriction digestion and sequence analysis. The pCDNA3-cyto-EBNA1 plasmid was kindly provided by Graham Taylor (18). The Mel JuSol (MJS) melanoma-derived cell line (19) was maintained in RPMI 1640 medium (Gibco BRL) supplemented with 10% fetal calf serum (FCS). Transient transfection of MJS cells with plasmid DNA was routinely performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Targets for the T cell recognition assay clone were generated by cotransfection of HLA-DRB51 MJS cells (14) with the cyto-EBNA1, empty-IRES-GFP, or vIRF3-IRES-GFP expression plasmid.
Retrovirus and transduction.
Retroviral constructs were engineered by cloning vIRF3 or HLA-B81 as a control into the pLZRS retroviral vector. Immediately downstream from the inserted gene was an internal ribosome entry site (IRES) and the marker gene, either truncated nerve growth factor (ΔNGFR) or green fluorescent protein (GFP). Vesicular stomatitis virus-pseudotyped retrovirus particles were produced in GP2-293 cells cotransfected with the pVSV-G envelope vector. Virus in the culture supernatant at 72 h was concentrated by ultracentrifugation (15 ml) and used to infect 5 × 105 target cells overnight. Lymphoblastoid cell lines (LCLs) were established from healthy donor B cells using the B95.8 strain of EBV. Both LCLs and PELs were maintained in RPMI 1640 medium (Gibco BRL) supplemented with 10% FCS. For the retrovirus that coexpresses NGFR, the transduced LCL or PEL cells were magnetically sorted using magnetically activated cell sorting NGFR-specific beads as directed by the manufacturer (Miltenyi Biotech).
Stable transfection and establishment of pvIRF3-tet cell lines.
A derivative of the doxycycline (DOX)-dependent expression vector pRTS-1 (20) was kindly provided by J. Mautner, Munich, Germany; vIRF3 or the reverse vIRF3 sequence as a control was introduced into the vector by standard DNA cloning procedures to create vectors pRTS-CD2-vIRF3 and pRTS-CD2-control, respectively. The pRTS-CD2-vIRF3 or pRTS-CD2-control vector was introduced into an EBV-negative derivative of the Akata BL line, Akata-A3, by electroporation of 10 μg plasmid DNA into 107 cells in Opti-MEM medium (Invitrogen) at 280 V and 960 μF using a Bio-Rad electroporation apparatus. Transfected cells were cultured in RPMI 1640 medium supplemented with 10% FCS. After 24 h, the transfected cell population was enriched by staining with OX34 antibody to rat CD2 and positively selected by magnetic cell sorting with anti-mouse IgG2a/b microbeads and LS columns (Miltenyi Biotech) according to the manufacturer's guidelines. Cells were thereafter expanded and maintained in RPMI 1640 medium supplemented with 10% FCS. Expression of vIRF3 was induced by addition of titrated doses of DOX, and the induced cells were positively selected by magnetic cell sorting with anti-NGFR microbeads and LS columns (Miltenyi Biotech). The purity of the sorted cells was checked by flow cytometry.
Flow cytometry analysis of cell surface MHC molecules.
Cell surface expression of MHC-II DR or NGFR on viable cells was determined by staining with phycoerythrin (PE)-conjugated anti-DR (Biolegend) or fluorescein isothiocyanate (FITC)-conjugated anti-NGFR (Biolegend). Cell surface expression of MHC-II DQ on viable cells was determined by staining with anti-DQ (Biolegend), followed by allophycocyanin (APC)-conjugated anti-mouse IgG (Biolegend). Stained cells were analyzed on a BD Accuri C6 flow cytometer, and the data were processed using FlowJo software (Tree Star).
Western blotting.
Goat antibodies to calregulin, mouse anti-DRα, mouse anti-DRβ, mouse anti-DMα, rat anti-LANA-1, and rabbit anti-CIITA were purchased from Santa Cruz Biotechnology. Mouse anti-KSHV-encoded vIRF3 was purchased from Novus Biologicals. Total cell lysates were denatured in reducing sample buffer (final concentrations, 2% sodium dodecyl sulfate [SDS], 72.5 mM Tris-HCl, pH 6.8, 10% glycerol, 0.2 M sodium 2-mercaptoethane-sulfonate, 0.002% bromophenol blue) and then sonicated and heated to 100°C for 5 min. Solubilized proteins equivalent to 105 cells/20-μl sample were separated by SDS-polyacrylamide gel electrophoresis (PAGE) on 4 to 12% acrylamide gradient bis-Tris NuPage minigels with MOPS (morpholinepropanesulfonic acid) running buffer (Invitrogen). Following electroblotting to polyvinylidene difluoride membranes, immunoblotting with specific primary antibodies followed by detection with appropriate alkaline phosphatase-conjugated secondary antibodies and a CDP-Star chemiluminescence detection kit (Tropix; Applied Biosystems) was performed as previously described (21).
T cell function assays.
T cells were grown in 10% FCS in RPMI 1640 medium supplemented with 30% supernatant from the interleukin-2 (IL-2)-producing MLA-144 cell line and 50 U/ml recombinant IL-2. The effector CD4+ T cell clones SNP (specific for amino acids 474 to 493 of EBV EBNA1 and restricted through DRB51), LAP/LRS (specific for amino acids 191 to 210 of KSHV LANA1 and restricted through DQ6), and LAP (specific for amino acids 191 to 205 of KSHV LANA1 and restricted through DP1) were generated as described elsewhere (16, 18). The effector CD8+ T cell clone CLG (specific for amino acids 426 to 434 of LMP2 and restricted through MHC-I A0201) was described elsewhere (22). The capacity of CD4+ and CD8+ T cell clones to recognize target LCLs, MJS cells, or BCBL-1 cells was measured by a gamma interferon (IFN-γ)-specific enzyme-linked immunosorbent assay (ELISA). Briefly, 104 effector T cells were incubated for 18 h at 37°C in V-bottom microtest plate wells with 105 target cells, before assaying the supernatants for IFN-γ release by ELISA (Endogen) in accordance with the manufacturer's recommended protocol. Antigen-sensitized targets were either incubated with peptides for 1 h or cultured for 18 h with LANA protein preparations (16) in AIM-V medium (Invitrogen).
QRT-PCR assay.
Total RNA was isolated from cultured cell lines using a Qiagen RNeasy kit and treated with DNase I (Turbo DNA-free kit; Ambion). Quantitative RT-PCR (QRT-PCR) assays for CIITA, DRA, DRB, DQA, DPA, DMA, DOA, CD74, HLA-A, and PGK were performed with TaqMan gene expression assays (Applied Biosystems), duplexed with GAPDH (glyceraldehyde-3-phosphate dehydrogenase) assays for normalization.
RESULTS
vIRF3 expression level is related to MHC-II antigen presentation in PELs.
In a previous paper (16), we speculated that the poor recognition of PELs by MHC-II-matched LANA-specific CD4 T cells was due to the expression of vIRF3. On closer examination of the VG-1 PEL line (23), we observed normal levels of MHC class II cell surface receptor (both MHC-II DR and MHC-II DQ) expression in a small subpopulation of cells (high DR DQ population) (Fig. 1A). Flow cytometry cell sorting of MHC-II DR-stained VG-1 cells enabled the isolation of two populations: one with high DR expression and the other with low DR expression (Fig. 1B). These two populations of VG-1 cells were assayed for recognition by MHC-II-matched LANA-specific CD4 T cells restricted through DQ6 (LAP/LRS peptide specific) or DP1 (LAP peptide specific) (16). Recognition was measured from the release of IFN-γ from the effector T cells. Little or no recognition of VG-1 cells with low DR expression was observed, but there was clear recognition of VG-1 cells with high DR expression that was in some cases comparable to the recognition of control B cells pulsed with synthetic target peptide (Fig. 1C). Aliquots of cells from replicate experiments were analyzed by immunoblotting, and band intensities were quantified. The mean expression of vIRF3 expression in VG-1 cells with high DR expression compared with that in VG-1 cells with low DR expression was 51.5% ± 6.3% (Fig. 1D). Expression of LANA showed less of a reduction, with a mean expression of 75.5% ± 3.4% in VG-1 cells with high DR expression compared with that for VG-1 cells with low DR expression. Similar results were obtained with another PEL cell line, BC-1 (data not shown).
Fig 1.

vIRF3 expression level is related to MHC-II antigen presentation in PELs. (A) To measure surface MHC-II DR and DQ expression, VG-1 cells were stained with anti-DQ, followed by APC-conjugated anti-mouse IgG and then PE-conjugated anti-DR. The cells were analyzed by flow cytometry and displayed as a dot plot of APC-conjugated DQ versus PE-conjugated DR fluorescence. (B) To purify the high-DR-expressing VG-1 cells stained with PE-conjugated anti-DR, cells were analyzed on a Mo-Flow cell sorter and the weakest DR-expressing cells and the strongest DR-expressing cells were gated (left, showing a dot plot of side scatter [SS] versus PE fluorescence) and sorted. (Right) Analysis of cell populations sorted for high DR expression (dashed line) and low DR expression (solid line). (C) Sensitivity of sorted VG-1 cells with high DR and low DR expression (black bars, 1 × 105 cells; gray bars, 2 × 104 cells) to LAP/LRS or LAP CD4+ effector T cells (both specific for LANA-derived peptide). Target and effector cells were cocultured for 18 h before assaying for T cell recognition by IFN-γ-specific ELISA. The DQ6- and DP1-positive LCLs pulsed with peptides (pep) served as positive-control targets. Error bars for IFN-γ release in the histograms indicate the standard deviations of triplicate cultures. (D) Sorted VG-1 cells with high DR and low DR expression were analyzed by SDS-PAGE and immunoblotting with antibodies specific for vIRF3, LANA, or calregulin as a loading control. The blot is from one representative experiment. The histogram shows the mean ± standard deviation of 3 independent replicate experiments quantified by densitometry, where the densities of the vIRF3 or LANA bands were normalized relative to the density of their own calregulin loading control.
These data extend previously published results to provide strong support for the idea that vIRF3 expression level is indeed related to MHC-II antigen presentation in PELs.
vIRF3 can modulate MHC-II antigen presentation in MJS cells.
A potential mechanistic explanation for the inverse correlation between vIRF3 and MHC-II antigen presentation in PELs might be found in the recent demonstration that vIRF3 inhibits the promoters of CIITA, leading to inhibition of the transcription and translation of MHC-II elements (17). To investigate this possibility in more detail, we tested the effect of vIRF3 on MHC-II antigen presentation using CD4 T cell effectors in a functional assay previously developed in our Birmingham laboratories (14). MJS melanoma cells expressing MHC-II DRB51 were transfected with a cyto-EBNA1 expression plasmid together with an empty IRES-GFP vector or with a vIRF3-IRES-GFP vector expressing vIRF3. The cultures were then cocultured with MHC-II DRB51-restricted CD4 T cells specific for the SNP EBNA1-derived peptide, and T cell recognition was measured from the release of IFN-γ from the effector cells. Mock-transfected cells were not recognized, but good recognition was seen with target cells cotransfected with cyto-EBNA1 and the control IRES-GFP vector (Fig. 2A). A substantial 50% reduction in recognition of cyto-EBNA1 was observed when the target cells were cotransfected with vIRF3 (Fig. 2A).
Fig 2.
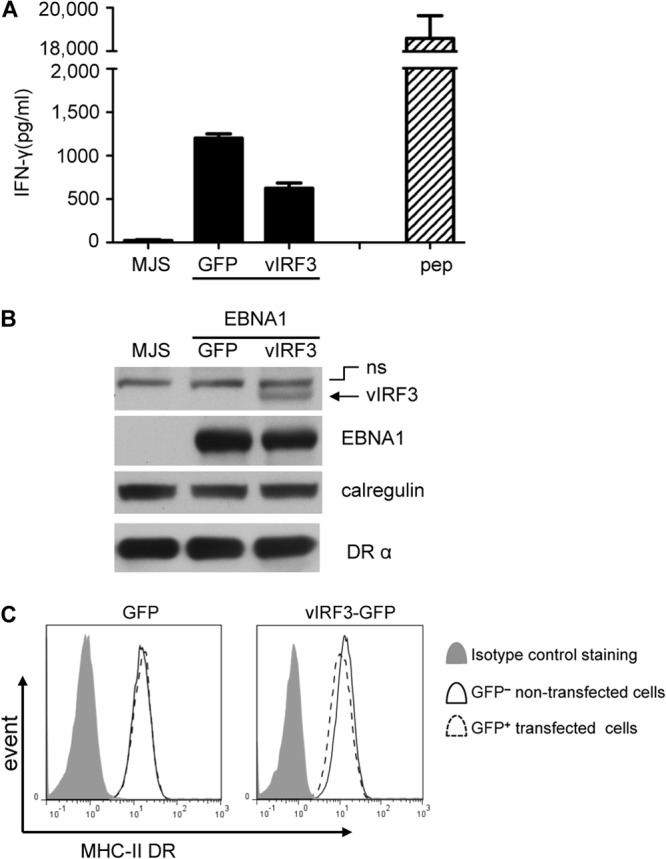
vIRF3 can modulate MHC-II antigen presentation in MJS cells. DRB51-expressing MJS cells were cotransfected with the pCDNA-cyto-EBNA1 expression vector together with IRES-GFP (GFP) or vIRF3-GFP (vIRF3) expression plasmids. (A) At 24 h posttransfection, the cells were used as targets for SNP CD4+ effector T cells (specific for an EBNA1-derived peptide) for a further 18 h before assaying for T cell recognition by IFN-γ-specific ELISA. The DRB51-expressing MJS cells pulsed with peptides served as controls. Error bars for the IFN-γ release in the histograms indicate the standard deviations of triplicate cultures. (B) Total cell lysates of target cells were analyzed by immunoblotting using antibodies specific for EBNA1, vIRF3, DRα, or calregulin as a loading control. A nonspecific (ns) band on the blot is indicated. (C) Viable transfected target cells were stained with PE-conjugated anti-DR and analyzed by flow cytometry. Histograms show the surface MHC-II DR expression on GFP− cells and GFP+ cells.
Replicate aliquots of cells analyzed by immunoblotting confirmed expression of vIRF3 in the cultures transfected with the vIRF3-GFP plasmid, in which about 10% of the cells became GFP positive (GFP+), and the levels of EBNA1 were similar between the cells cotransfected with cyto-EBNA1 and IRES-GFP or vIRF3-GFP (Fig. 2B). Two-color flow cytometry analysis of viable cells stained for surface MHC-II DR suggested that vIRF3 caused a small but reproducible reduction (about 15%) in the level of cell surface DR molecules on GFP+ cells compared to the expression on untransfected GFP-negative (GFP−) cells in the same cultures (Fig. 2C). In four independent experiments, the mean expression of surface MHC-II DR in the GFP+ cells compared to untransfected GFP− cells was 85.8% ± 7.5% (P = 0.0089).
vIRF3 can inhibit CIITA transcription and reduce surface MHC-II DR expression, but with slow kinetics.
The key finding from the transient-transfection experiments whose results are shown in Fig. 2 was that a substantial reduction of CD4 T cell recognition was observed with only a very small reduction of cell surface MHC-II DR expression. As vIRF3 is a latent gene that is constitutively expressed in KSHV-positive PELs (24, 25), it is physiologically relevant to also check the phenotype of vIRF3 at later time points. To this end, we generated a bicistronic retrovirus, pLZRS-vIRF3-IRES-NGFR, that coexpresses vIRF3 with a truncated inactive NGFR, which was used as a marker to identify and purify the transduced population. The MJS cells were transduced with control, LZRS-B81-IRES-NGFR, or LZRS-vIRF3-IRES-NGFR retroviruses, and surface MHC-II expression was monitored through two-color flow cytometry staining of surface MHC-II DR and NGFR as a surrogate marker of vIRF3 expression (Fig. 3A). At day 2, the relative fluorescence intensity of surface DR staining on the NGFR-positive (NGFR+) (vIRF3-positive [vIRF3+]) cells was about 60% compared to that of untransduced NGFGR-negative (NGFR−) (vIRF3-negative [vIRF3−]) cells in the same culture (Fig. 3A, left). However, by day 5, the surface DR expression was substantially reduced to about 13% by vIRF3 (Fig. 3A, middle), and at day 7, the surface DR expression was reduced to about 7% on NGFR+ (vIRF3+) cells compared to the expression on untransduced NGFR− (vIRF3−) cells (Fig. 3A right, and B).
Fig 3.
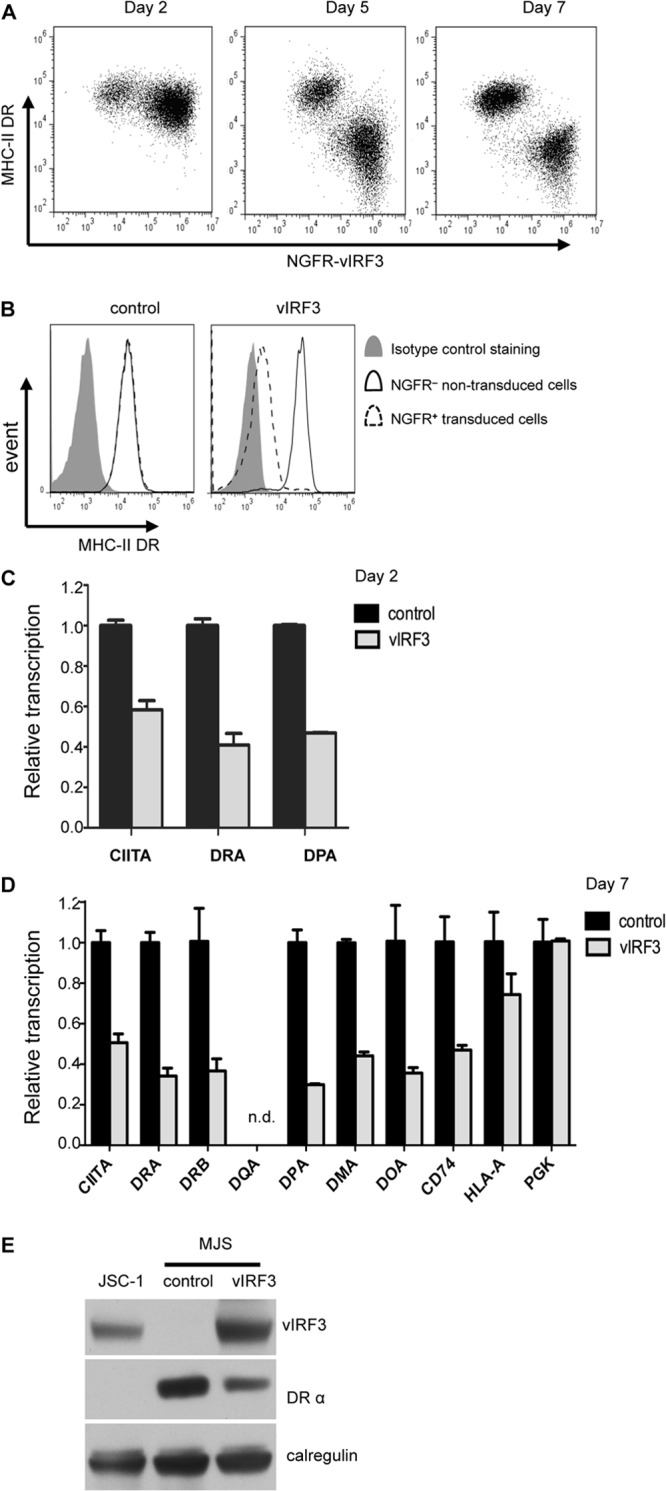
vIRF3 inhibits CIITA transcription and substantially downregulates surface MHC-II DR expression with slow kinetics. MJS cells were transduced with retrovirus pLZRS-vIRF3-IRES-NGFR or a control vector, pLZRS-B81-IRES-NGFR. (A) Transduced MJS cells were harvested at the indicated time points, stained with PE-conjugated anti-DR plus FITC-conjugated anti-NGFR, and analyzed by flow cytometry to determine the levels of DR expression on transduced (NGFR+) cells and nontransduced (NGFR−) cells in the same cultures. (B) Histograms show the surface MHC-II DR expression on NGFR− cells and NGFR+ cells at day 7 after retrovirus transduction. (C) At day 2 after transduction, the transduced cells from control and vIRF3-transduced cultures were purified with anti-NGFR magnetic beads. Relative levels of CIITA, DRA, and DPA transcripts in total RNA isolated from transduced and sorted cells normalized to the levels of the GAPDH transcripts measured were measured by QRT-PCR assay. The error bars represent the standard deviations of triplicate reactions. (D) At day 7 after transduction, mRNA from sorted cells was assayed by QRT-PCR for an extended panel of transcripts: CIITA, DRA, DRB, DQA, DPA, DMA, DOA, CD74, HLA-A, and PGK. The error bars represent the standard deviations of triplicate reactions. (E) Total cell lysates of the purified NGFR+ populations from control and vIRF3-transduced cultures were analyzed by SDS-PAGE alongside lysates from JSC-1 cells. The samples were immunoblotted with antibodies specific for vIRF3, HLA-DRα, or calregulin as a loading control.
To enable analysis of MHC-II locus transcripts and protein, the control retrovirus- and vIRF3 retrovirus-transduced cells in these experiments were purified by NGFR sorting at each time point after transduction. QRT-PCR assays with total mRNA isolated from these samples showed that vIRF3 can effectively inhibit the transcription of CIITA and MHC-II elements DR and DP as early as 2 days after transduction (Fig. 3C and D). More extensive transcriptional analyses on the day 7 samples showed that the transcripts of CIITA and all MHC-II elements tested (including DRA, DRB, DPA, DMA, DOA, and CD74) were significantly downregulated following vIRF3 expression in MJS cells (P < 0.01) (Fig. 3D).
Immunoblot analysis of cell lysates at day 7 confirmed the expression of vIRF3 and a corresponding reduction in total cellular DRα protein expression in the cell lysates from vIRF3-transduced cells compared to control retrovirus-transduced cells (Fig. 3E).
Together, these data show that vIRF3 can substantially reduce total and surface MHC-II DR protein expression but that it does so with much slower kinetics than the more immediate inhibition of CIITA expression and subsequent impairment of MHC-II element transcription.
Ectopic expression of CIITA can partly abolish surface MHC-II DR downregulation by vIRF3.
To investigate whether the delayed substantial reduction of surface MHC-II DR protein expression in MJS cells was due to the inhibition of CIITA transcription, we generated an MJS cell line with ectopic overexpression of CIITA driven by a retrovirus promoter. It was confirmed by immunoblotting that CIITA was overexpressed in the CIITA-expressing MJS cell line, which resulted in a small increase of the already high levels of MHC-II DR expression in MJS cells that was evident by flow cytometry (Fig. 4A). In the next set of experiments, MJS cells and CIITA-expressing MJS cells were transduced with control/GFP or vIRF3/GFP bicistronic retroviruses to examine whether constitutive overexpression of CTIIA might affect the ability of vIRF3 to downregulate surface MHC-II DR expression. Expression of vIRF3 was confirmed by immunoblotting of cell lysates (Fig. 4C). Flow cytometry revealed that when maximal inhibition of MHC-II DR expression by vIRF3 was achieved (i.e., by day 9 in Fig. 4B), the level of surface DR on vIRF3/GFP+-expressing control MJS cells was reduced to about 11% of the expression in GFP−, non-vIRF3-expressing MJS cells, whereas in CIITA-expressing MJS cells, the expression of vIRF3 reduced the level of surface DR expression to 28% of the level of expression in non-vIRF3-expressing cells. Reproducibly, the constitutive overexpression of CTIIA in MJS cells resulted in about 3 times more surface DR in vIRF3-expressing cells.
Fig 4.

Ectopic expression of CIITA partly abolishes vIRF3-mediated downregulation of surface MHC-II DR. (A) MJS cells transduced with a control IRES-NGFR retrovirus or with a CIITA-IRES-NGFR retrovirus. (Left) Cell lysates prepared from MJS and CIITA-expressing MJS cells were analyzed by immunoblotting using antibodies specific for CIITA, the DRα chain, or calregulin as a loading control; (right) transduced cells were stained with PE-conjugated anti-DR and then analyzed by flow cytometry. Solid-line histogram, MHC-II DR expression on MJS cells; dashed-line histogram, expression on CIITA-expressing MJS cells; shaded histogram, isotype control staining. (B) MJS and CIITA-expressing MJS cells transduced with retrovirus pLZRS-vIRF3-IRES-GFP were harvested at the indicated times over 9 days for staining with PE-conjugated anti-DR and analysis by flow cytometry. Histograms show the surface MHC-II DR expression on GFP− cells and GFP+ cells at each time point. The numbers at the top of the histogram boxes are the relative fluorescent intensity compare with that of the nontransfected GFP− population, which was equal to 100, and that of the isotype control, which was equal to 0. (C) MJS and CIITA-expressing MJS cells were transduced with retrovirus pLZRS-vIRF3-IRES-GFP, and expression of vIRF3 at 9 days postinfection was confirmed by immunoblotting of cell lysates alongside lysates from JSC-1 cells. Con, control.
These data are consistent with the reduction of surface MHC-II DR expression and modulation of MHC-II antigen presentation by vIRF3 being partially due to the inhibition of the transcription of CIITA and the fact that the delayed effect on protein expression is due to the known long half-life of MHC-II molecules. However, it is important to note that the reduction of surface MHC-II DR expression cannot be fully reversed by the ectopic overexpression of CIITA.
vIRF3 inhibits CIITA transcription in a B cell line.
All the work described above was carried out in the MJS cell model, but in the context of PELs, it would be physiologically more relevant to examine the behavior of vIRF3 in B cells. To this end, we generated from the EBV-negative Akata-A3 Burkitt lymphoma line transfectants with a DOX-inducible vIRF3-expressing vector, pRTS-CD2-vIRF3. The vIRF3 gene and an NGFR-IRES-GFP sequence were regulated by a bidirectional DOX-regulated promoter which allows the purification of vIRF3-expressing cells following DOX induction. By titrating the concentration of DOX, at 2 ng/ml we achieved expression levels of vIRF3 that were similar to those in PELs (Fig. 5A). As we know from the previous experiments in MJS cells that vIRF3 affects MHC-II with slow kinetics, we harvested the B cells at 7 to 11 days postinduction. Two-color flow cytometry analysis of viable cells stained for surface MHC-II DR suggested that vIRF3 reduced the level of cell surface DR molecules only slightly but reproducibly by about 15% (Fig. 5B). Immunoblot analysis of purified GFP/NGFR+ cells confirmed the expression of vIRF3 in the cells containing the vIRF3 vector, but the levels of DRα, DRβ, DMα, and the MHC-I heavy chain were not noticeably altered by vIRF3 (Fig. 5C). Analysis of RNA by QRT-PCR showed that the transcripts of CIITA and, with the exception of MHC-II DMA, all of the MHC-II elements tested were substantially reduced (P < 0.01) following vIRF3 expression compared with the levels in control Akata-A3 cells (Fig. 5D).
Fig 5.

vIRF3 inhibits CIITA transcription in a B cell line. (A) Akata-A3 cells transfected with pRTS-CD2-vIRF3 were induced with the indicated concentrations of DOX and sorted with NGFR-specific magnetic beads. The lysates of purified NGFR+ cells were analyzed by SDS-PAGE alongside lysates from uninduced vIRF3-expressing Akata-A3 cells and JSC-1 cells. The samples were immunoblotted with antibodies specific for vIRF3 or calregulin as a loading control. (B) Akata-A3 cells transfected with pRTS-CD2-vIRF3 or pRTS-CD2-control were induced with 2 ng/ml of DOX, and viable cells were stained with PE-conjugated anti-DR for analysis by flow cytometry. Histograms show the surface MHC-II DR expression on GFP− cells and GFP+ cells in both cultures. (C) Akata-A3 cells transfected with pRTS-CD2-vIRF3 or pRTS-CD2-control were induced with 2 ng/ml of DOX and sorted with NGFR magnetic beads. The lysates of purified NGFR+ cells were analyzed by SDS-PAGE and immunoblotting with antibodies specific for vIRF3, DRα, DRβ, DMα, MHC-I (HC10), or calregulin as a loading control. (D) Relative levels of CIITA, DRA, DRB, DQA, DPA, DMA, DOA, CD74, and HLA-A transcripts in total RNA isolated from transduced and sorted cells normalized to the levels of the GAPDH transcripts measured were measured by QRT-PCR assay. The error bars represent the standard deviations of triplicate reactions.
Therefore, physiological levels of vIRF3 do effectively inhibit CIITA and MHC-II element transcription in B cells but do not necessarily impact the levels of MHC-II protein molecules in these cells in the time scale of this experiment.
vIRF3 can modulate MHC-II antigen presentation in LCLs.
The results in Fig. 5 led us to examine what might be the effect of vIRF3 on functional T cell recognition in B cells. To this end, we generated EBV-transformed B LCLs that stably express vIRF3 through transduction with the pLZRS-vIRF3-IRES-NGFR retrovirus, followed by two rounds of sorting for NGFR-expressing cells. Expression of vIRF3 in the vIRF3-expressing LCLs was confirmed by immunoblotting, although it was clear that the levels were lower than those of vIRF3 expression in PELs (Fig. 6A). Also, there was no obvious difference in total cellular and surface MHC-II DR expression between the control LCLs and vIRF3-expressing LCLs (Fig. 6A).
Fig 6.
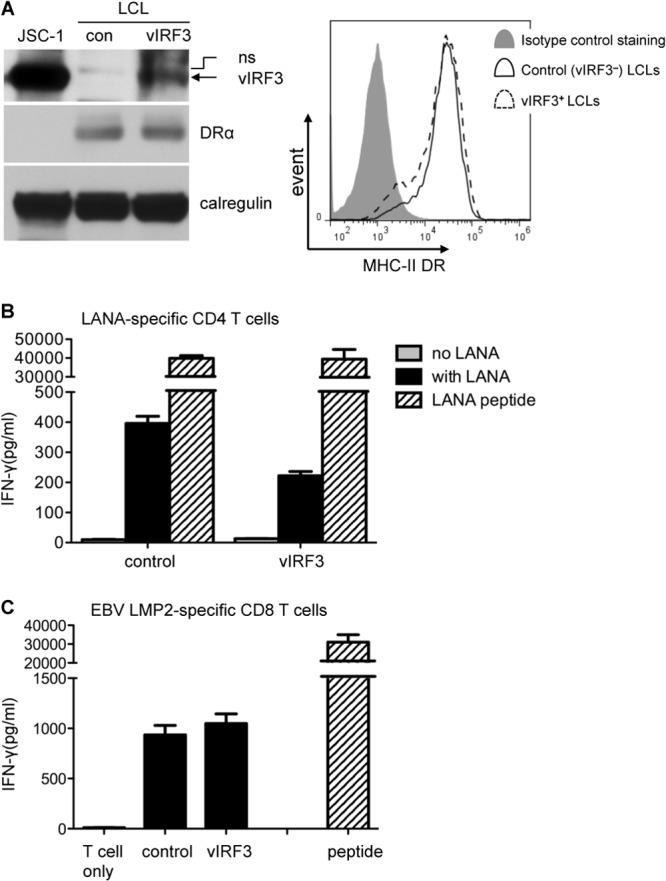
vIRF3 can modulate MHC-II antigen presentation in LCLs. (A) HLA-DQ6- and HLA-A2-typing LCLs were transduced with pLZRS-vIRF3-IRES-NGFR or with control pLZRS-B81-IRES-NGFR retrovirus and sorted with NGFR magnetic beads. The lysates of the purified NGFR+ cells from both transductions, along with those of the KSHV+ control PEL line JSC-1, were analyzed by SDS-PAGE and immunoblotting with antibodies specific for vIRF3, DRα, or calregulin as a loading control. Histograms show the surface MHC-II DR expression on LCL control cells and vIRF3-expressing LCL cells. (B) Control LCL cells and vIRF3-expressing LCL cells were pulsed with LANA protein or a control protein overnight and were then cocultured with LAP/LRS CD4+ effector T cells (DQ6 restricted and specific for LANA-derived peptide) for a further 18 h before assaying for T cell recognition by IFN-γ-specific ELISA. The LCL cells pulsed with peptides served as a control. Error bars for IFN-γ release in the histograms indicate the standard deviations of triplicate cultures. (C) The control LCL cells and vIRF3-expressing LCL cells were cocultured with CLG CD8+ effector T cells (A2 restricted and specific for EBV LMP2-derived peptide) and assayed for IFN-γ by ELISA.
To investigate the effect of vIRF3 on MHC-II antigen presentation, control LCLs and vIRF3-expressing LCLs were fed LANA protein and then cocultured with MHC-II-matched-allele (DQ6)-restricted LAP/LRS LANA-specific CD4 T cells. T cell recognition was measured from the release of IFN-γ from the effector cells, and peptide-pulsed target cells served as controls. There was no recognition of either type of LCL pulsed with control protein. Good recognition was seen with both LCLs when fed with LANA protein, but there was an approximately 50% reduction of T cell recognition in the LANA-fed vIRF3-expressing LCLs compared with that in control LCLs (Fig. 6B). Furthermore, this reduction was not observed when the LCLs were pulsed with specific LANA peptides. In additional control experiments, the recognition by HLA-A0201-restricted CD8 T cells specific for the GLC peptide of EBV LMP2 did not differ between the control and vIRF3-expressing LCLs (Fig. 6C).
vIRF3 modulates MHC-II antigen presentation in BCBL-1 cells independently of CIITA inhibition.
In our previous study, we observed that one unique PEL line, BCBL-1, can be recognized by LANA-specific CD4 T cells and correlated with the lowest level of vIRF3 expression (16). To investigate whether overexpression of vIRF3 in this PEL can modulate MHC-II antigen presentation, we transduced BCBL-1 cells with control pLZRS-B81-IRES-NGFR retrovirus and pLZRS-vIRF3-IRES-NGFR retrovirus. At 7 to 10 days postinduction, these cells were analyzed in various assays. Two-color flow cytometry analysis of viable cells stained for surface MHC-II DR showed that vIRF3 reduced the level of cell surface MHC-II DR expression by about 50% on NGFR/vIRF3+ cells compared to the level of expression on NGFR/vIRF3− cells in the same cultures (Fig. 7A). Immunoblot analysis of pure populations of vIRF3-expressing BCBL-1 cells confirmed the overexpression of vIRF3 in the vIRF3-expressing BCBL-1 cells compared with the level of expression in control BCBL-1 cells and a concomitant small level of reduction of total MHC-II DR expression following vIRF3 expression (Fig. 7B).
Fig 7.
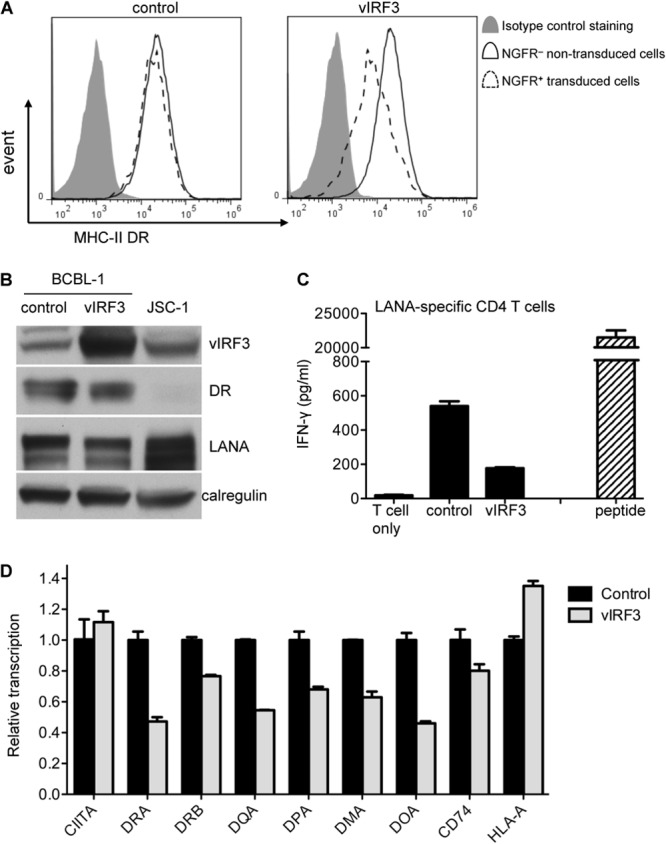
vIRF3 modulates MHC-II antigen presentation in the BCBL-1 PEL line independently of inhibition of CIITA. BCBL-1 cells were transduced with pLZRS-vIRF3-IRES-NGFR or control pLZRS-B81-IRES-NGFR retrovirus. (A) The transduced cells were stained with PE-conjugated anti-DR plus FITC-conjugated anti-NGFR and analyzed by flow cytometry for surface MHC-II DR expression on NGFR− cells and NGFR+ cells. (B) The transduced cells were sorted with NGFR-specific magnetic beads. The lysates of purified NGFR+ cells from both transductions, along with those of the control JSC-1 PEL line, were analyzed by SDS-PAGE and immunoblotting with antibodies specific for vIRF3, DRα, LANA, or calregulin as a loading control. (C) The BCBL-1 control and vIRF3-expressing BCBL-1 cells were cocultured with LAP/LRS CD4+ effector T cells (DQ6 restricted and specific for LANA-derived peptide) for 18 h before assaying for T cell recognition by IFN-γ-specific ELISA. A DQ6-positive LCL pulsed with target peptides served as a control. Error bars for IFN-γ release in the histograms indicate the standard deviations of triplicate cultures. (D) Relative levels of CIITA, DRA, DRB, DQA, DPA, DMA, DOA, CD74, and HLA-A transcripts in total RNA isolated from transduced and sorted cells normalized to the levels of the GAPDH transcripts measured were measured by QRT-PCR assay. The error bars represent the standard deviations of triplicate reactions.
To investigate whether this downregulation of MHC-II DR expression is sufficient to modulate MHC-II antigen presentation, BCBL-1 control and vIRF3-expressing BCBL-1 cells were cocultured with MHC-II-matched-allele (DQ6)-restricted LAP/LRS LANA-specific CD4 T cells, and T cell recognition was measured from the release of IFN-γ from the effector cells. The result showed that the recognition by CD4 T cells was substantially reduced with vIRF3-expressing BCBL-1 cells compared with that achieved with control BCBL-1 cells (Fig. 7C).
Finally, the effect of vIRF3 overexpression on the transcription of CIITA and MHC-II elements was investigated through QRT-PCR assays carried out on the total RNA isolated from the NGFR-sorted population. As shown in Fig. 7D, while the transcription of most of the MHC-II elements was reduced following the overexpression of vIRF3 in BCBL-1 cells, there was no reduction in the transcription of CIITA by vIRF3.
Therefore, in a PEL background, vIRF3 can reduce the surface MHC-II DR expression and inhibit MHC-II antigen presentation through a CIITA-independent mechanism.
DISCUSSION
Consistent with a recent report that vIRF3 has the potential to inhibit the transcription of CIITA and the subsequent expression of MHC-II genes (17), we have now shown, using antigen-specific recognition by CD4 T cells, that vIRF3 can modulate the MHC-II antigen presentation function in melanoma cells, B cells, and PEL cells. These functional assays revealed a level of complexity that was not evident from the previous biochemical observations. Thus, while we confirmed that vIRF3 can indeed downregulate MHC-II DR expression, the kinetics of this effect were slow and were preceded by a substantial impairment in presentation of antigen to CD4 effector T cells. Furthermore, while CIITA might contribute to the downregulation of MHC-II molecules under some circumstances, our combined data from different cell lines and particularly PEL lines show that the modulation of MHC-II antigen presentation by vIRF3 may be mediated predominantly through CIITA-independent mechanisms.
CIITA is a master regulator of the expression of MHC-II elements, including HLA-DR, HLA-DQ, HLA-DP, HLA-DM, HLA-DO, and invariant chain/CD74. It is a transcriptional coactivator that lacks an intrinsic DNA-binding function. It enhances the transcription of MHC-II genes through interaction with transcription regulatory proteins, forming a stable enhanceosome that can bind to the regulatory module of MHC-II promoters (26, 27). CIITA appears to be a vulnerable target for viral modulation of MHC-II gene expression (17, 28). Li et al. demonstrated that the EBV-encoded immediate early protein BZLF1, which is the master regulator of the EBV lytic cycle, can bind to the CIITA promoter and strongly inhibit the transcription and constitutive expression of CIITA molecules (28). MHC-II molecules are constitutively expressed on professional antigen-presenting cells, which express CTIIA, but the expression can also be induced on other cells by IFN-γ through the induced transcription of CIITA. In this context, HCMV infection of U373 MG cells reduced the level of IFN-γ-induced CIITA transcripts by more than 10-fold, with a concordant inhibition of induced MHC-II DR synthesis (29). Similarly, varicella-zoster virus infection of human foreskin fibroblasts can prevent IFN-γ-induced CIITA expression and expression of MHC-II molecules (30). KSHV vIRF3 can inhibit both constitutive and IFN-γ-induced CIITA transcription (17), but in the light of the findings of our present work, it is possible that the action of vIRF3 on CTIIA might be most important for inhibiting the IFN-γ-induced CTIIA.
As MHC-II molecules have a long half-life compared to MHC-I molecules (31), inhibition of CIITA and thus MHC-II gene transcription would not be expected to have a rapid effect on preexisting MHC-II protein levels. Indeed, both in a previous study by Schmidt and colleagues (17) and in the present work, a relatively small downregulation of MHC-II protein was observed 48 h after expression of vIRF3 vectors. Interestingly, over a longer time course in the MJS cell line, inhibition of transcription was observed to be a very effective mechanism to modulate the expression of MHC-II proteins; after 5 to 7 days of expression of vIRF3, expression of the MHC-II DR protein on the cell surface was reduced by more than 10-fold (Fig. 3B). As vIRF3 is a KSHV latent protein that is consistently expressed in the KSHV-positive PEL line (25), we postulate that the effect of vIRF3 on MHC-II transcription, whether it is via CTIIA or not, is a major factor responsible for maintaining the phenotype of little or no MHC-II expression in these tumors. In our experiments with B cells, expression of vIRF3 had no effect (Akata-A3) or little effect (BCBL-1) on total MHC-II DR expression. One possible explanation for this could be that MHC-II DR is even more stable in B cells than MJS and epithelial cells, so that downregulation of total MHC-II DR would require a longer time; it is difficult to test this experimentally, as we found vIRF3 to be toxic over prolonged periods in these cells. Another possibility, which is not mutually exclusive, is that transcriptional regulation of MHC-II is not the major mechanism by which vIRF3 interferes with antigen presentation in B cells.
Three lines of evidence indicate that vIRF3 effects a CIITA-independent mechanism to modulate MHC-II antigen presentation. First, vIRF3 mediated significant downregulation of MHC-II DR in CIITA-expressing MJS cells that ectopically overexpress CIITA from a retroviral vector (Fig. 4). Second, EBV-transformed LCLs stably expressing vIRF3 showed a reduced ability to present MHC-II/peptide to CD4 T cells, even though no inhibition of CIITA transcripts was detected by QRT-PCR (data not shown). Finally, ectopic overexpression of vIRF3 in the BCBL-1 PEL line did not alter the levels of CIITA transcripts but did reduce MHC-II transcription, expression, and recognition by CD4 T cells (Fig. 7). This CIITA-independent inhibition of the transcription of MHC-II elements in BCBL-1 cells by vIRF3 sheds some light on the nature of the mechanism, pointing to an alternative regulation of MHC-II element transcription. However, the data obtained from LCLs (Fig. 6) suggest that this is not the only CIITA-independent mechanism, as here vIRF3 inhibited the MHC-II antigen presentation function in the absence of any effect on MHC-II expression. The utilization of dual molecular mechanisms impacting antigen presentation is not unique to vIRF3. For example, EBV-encoded BZLF1 inhibits CIITA transcription (28) but also downregulates the surface expression of CD74 and T cell recognition in a CIITA-independent manner (14).
It is important to note from both this study and our previous work that surface MHC-II expression levels do not necessarily correlate with the MHC-II antigen presentation function. In one sense, this is unsurprising, as viral modulation of antigen presentation is known to target multiple points in the processing pathways, some of which might qualitatively alter the MHC/peptide complexes without necessarily affecting the quantity of MHC-II molecules at the cell surface. Functional T cell assays are exquisitely sensitive to such qualitative changes that are not detected in routine biochemical assays. In the present study, there was a substantial reduction in CD4 T cell recognition at 2 days after expression of vIRF3, when surface MHC-II levels were barely affected. Although this observation might be less relevant to the latent expression pattern of vIRF3 in KSHV-positive tumor cells, where the longer-term effects of CTIIA inhibition may be effective, we speculate that it could well be important in the early stage of KSHV infection of nonmalignant cells to prevent their recognition and elimination by KSHV-specific CD4 T cells.
T-cell responses to KSHV-encoded latent antigens and lytic antigens have been identified in both healthy immunocompetent donors and immunocompromised patients (32–37). Although generally weak, KSHV-specific CD8 T cells responses are more frequent, diverse, and strong in asymptomatic KSHV-infected patients than Kaposi's sarcoma patients (38). Very recently, Sabbah et al. found that the responses in healthy KSHV-infected donors to the KSHV latent antigens were most frequently directed to LANA-derived peptides (16). Interestingly, most of the LANA-specific T cell clones generated were CD4 lymphocytes. It might therefore be speculated that in order to establish a persistent infection (and also to generate malignancies), it is necessary for KSHV-infected cells to evade not only CD8 responses, for which various mechanisms have been identified (7, 8, 21, 39, 40), but also CD4 T cell responses.
Generally, effector CD4 T cells can be generated during both chronic and acute infection with herpesviruses and are important for controlling the infection through both cytotoxic and cytokine-dependent mechanisms. Many herpesviruses encode multiple immunoevasins which together target MHC-II processing and presentation pathways. There is now accumulating evidence to suggest that the same may be true for KSHV. A recent study showed that KSHV infection of human primary endothelial cells inhibits IFN-γ-induced expression of the MHC-II molecule at the transcriptional level via soluble mediators, including cytokines released from KSHV-infected endothelial cells (41). This is likely to be a mechanism separate from that identified in the present study, as we have observed the effects of vIRF3 on T cell recognition in the absence of effects on MHC-II expression.
Multiple functions have been ascribed to vIRF3. For example, it binds to IRF5 and inhibits interferon-responsive transcription (42); it can potently inhibit the transcription induced by overexpression of p53 and, consequently, inhibit apoptosis (25); it has been reported to be associated with the c-myc suppressor, which in turn stimulates c-myc-mediated transcription and consequently contributes to B cell growth (43); and it can specifically interact with the hypoxia-inducible factor 1 (HIF-1) alpha subunit, leading to HIF-1 stabilization and transcriptional activation (44). Ultimately, vIRF-3 was shown to be indispensable to the proliferation of PEL cells in culture and has been proposed to be a predominant factor in the pathogenesis of PEL cells (45). Indeed, in our study we tried to knock down vIRF3 expression in PEL cells with small interfering RNA or short hairpin RNA in an attempt to reverse the immune-modulatory phenotype of vIRF3, but we were unable to generate a stable line with lower vIRF3 expression (data not shown). To circumvent this problem, we took advantage of the fact that some PEL lines contained a small population of cells with high surface MHC-II expression and lower vIRF3 expression (Fig. 1). Interestingly, the variations in MHC-II and vIRF3 expression were dynamic, as when the high- and low-expressing sorted populations were returned to culture, they reverted to the mean level of expression within 7 days (data not shown). This phenomenon probably reflects the fact that a critical level of vIRF3 expression is required for continued growth of the cells. However, if the mechanism of vIRF3 regulation were to be elucidated, it might be targeted therapeutically in PELs; reduced vIRF3 expression might both impair their growth and/or survival and also render the tumors susceptible to LANA-specific CD4 effector T cells.
In summary, this study has characterized aspects of the MHC-II immune modulation function of vIRF3 that extend our understanding of the roles of this protein in infection of nonmalignant target cells in asymptomatic persistence and in the KSHV-associated malignancy PEL.
ACKNOWLEDGMENTS
This work was supported by grants from the Medical Research Council, London, United Kingdom (G0901755).
We thank Alan Rickinson for critical and helpful discussions during this study.
Footnotes
Published ahead of print 28 February 2013
REFERENCES
- 1. Chang Y, Cesarman E, Pessin M, Lee F, Culpepper J, Knowles D, Moore P. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 2. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 4. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay M, Clauvel J, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 5. Flore O, Rafii S, Ely S, O'Leary JJ, Hyjek EM, Cesarman E. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588–592 [DOI] [PubMed] [Google Scholar]
- 6. Offermann MK. 2007. Kaposi sarcoma herpesvirus-encoded interferon regulator factors. Curr. Top. Microbiol. Immunol. 312:185–209 [DOI] [PubMed] [Google Scholar]
- 7. Coscoy L, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U. S. A. 97:8051–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ishido S, Wang C, Lee B-S, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zuo J, Rowe M. 2012. Herpesviruses placating the unwilling host: manipulation of the MHC class II antigen presentation pathway. Viruses 4:1335–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tomazin R, Boname J, Hegde NR, Lewinsohn DM, Altschuler Y, Jones TR, Cresswell P, Nelson JA, Riddell SR, Johnson DC. 1999. Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat. Med. 5:1039–1043 [DOI] [PubMed] [Google Scholar]
- 11. Hegde NR, Tomazin RA, Wisner TW, Dunn C, Boname JM, Lewinsohn DM, Johnson DC. 2002. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J. Virol. 76:10929–10941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Odeberg J, Plachter B, Branden L, Soderberg-Naucler C. 2003. Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR α-chain. Blood 101:4870–4877 [DOI] [PubMed] [Google Scholar]
- 13. Temme S, Eis-Hubinger AM, McLellan AD, Koch N. 2010. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J. Immunol. 184:236–243 [DOI] [PubMed] [Google Scholar]
- 14. Zuo J, Thomas WA, Haigh TA, Fitzsimmons L, Long HM, Hislop AD, Taylor GS, Rowe M. 2011. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 7:e1002455 doi:10.1371/journal.ppat.1002455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ressing ME, van Leeuwen D, Verreck FAW, Gomez R, Heemskerk B, Toebes M, Mullen MM, Jardetzky TS, Longnecker R, Schilham MW, Ottenhoff THM, Neefjes J, Schumacher TN, Hutt-Fletcher LM, Wiertz EJHJ. 2003. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. U. S. A. 100:11583–11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabbah S, Jagne YJ, Zuo J, de Silva T, Ahasan MM, Brander C, Rowland-Jones S, Flanagan KL, Hislop AD. 2012. T-cell immunity to Kaposi sarcoma-associated herpesvirus: recognition of primary effusion lymphoma with LANA-specific CD4+ T cells. Blood 119:2083–2092 [DOI] [PubMed] [Google Scholar]
- 17. Schmidt K, Wies E, Neipel F. 2011. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex class II expression. J. Virol. 85:4530–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leung CS, Haigh TA, Mackay LK, Rickinson AB, Taylor GS. 2010. Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc. Natl. Acad. Sci. U. S. A. 107:2165–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson JP, Demmer-Dieckmann M, Meo T, Hadam MR, Riethmüller G. 1981. Surface antigens of human melanoma cells defined by monoclonal antibodies. I. Biochemical characterization of two antigens found on cell lines and fresh tumors of diverse tissue origin. Eur. J. Immunol. 11:825–831 [DOI] [PubMed] [Google Scholar]
- 20. Bornkamm GW, Berens C, Kuklik-Roos C, Bechet J-M, Laux G, Bachl J, Korndoerfer M, Schlee M, Holzel M, Malamoussi A, Chapman RD, Nimmerjahn F, Mautner J, Hillen W, Bujard H, Feuillard J. 2005. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 33:e137 doi:10.1093/nar/gni137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zuo J, Thomas W, van Leeuwen D, Middeldorp JM, Wiertz EJHJ, Ressing ME, Rowe M. 2008. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 82:2385–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee SP, Thomas WA, Murray RJ, Khanim F, Kaur S, Young LS, Rowe M, Kurilla M, Rickinson AB. 1993. HLA A2.1-restricted cytotoxic T cells recognizing a range of Epstein-Barr virus isolates through a defined epitope in latent membrane protein LMP2. J. Virol. 67:7428–7435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brander C, Suscovich T, Lee Y, Nguyen PT, O'Connor P, Seebach J, Jones NG, van Gorder M, Walker BD, Scadden DT. 2000. Impaired CTL recognition of cells latently infected with Kaposi's sarcoma-associated herpes virus. J. Immunol. 165:2077–2083 [DOI] [PubMed] [Google Scholar]
- 24. Cunningham C, Barnard S, Blackbourn DJ, Davison AJ. 2003. Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J. Gen. Virol. 84:1471–1483 [DOI] [PubMed] [Google Scholar]
- 25. Rivas C, Thlick A-E, Parravicini C, Moore PS, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75:429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reith W, LeibundGut-Landmann S, Waldburger J-M. 2005. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 5:793–806 [DOI] [PubMed] [Google Scholar]
- 27. Ting JP-Y, Trowsdale J. 2002. Genetic control of MHC class II expression. Cell 109:S21–S33 [DOI] [PubMed] [Google Scholar]
- 28. Li D, Qian L, Chen C, Shi M, Yu M, Hu M, Song L, Shen B, Guo N. 2009. Down-regulation of MHC class II expression through inhibition of CIITA transcription by lytic transactivator Zta during Epstein-Barr virus reactivation. J. Immunol. 182:1799–1809 [DOI] [PubMed] [Google Scholar]
- 29. Le Roy E, Muhlethaler-Mottet A, Davrinche C, Mach B, Davignon J-L. 1999. Escape of human cytomegalovirus from HLA-DR-restricted CD4+ T-cell response is mediated by repression of gamma interferon-induced class II transactivator expression. J. Virol. 73:6582–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abendroth A, Slobedman B, Lee E, Mellins E, Wallace M, Arvin AM. 2000. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J. Virol. 74:1900–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mackay LK, Long HM, Brooks JM, Taylor GS, Leung CS, Chen A, Wang F, Rickinson AB. 2009. T cell detection of a B-cell tropic virus infection: newly-synthesised versus mature viral proteins as antigen sources for CD4 and CD8 epitope display. PLoS Pathog. 5:e1000699 doi:10.1371/journal.ppat.1000699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lepone L, Rappocciolo G, Knowlton E, Jais M, Piazza P, Jenkins FJ, Rinaldo CR. 2010. Monofunctional and polyfunctional CD8+ T cell responses to human herpesvirus 8 lytic and latency proteins. Clin. Vaccine Immunol. 17:1507–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Amodio E, Goedert JJ, Barozzi P, Riva G, Firenze A, Bonura F, Viviano E, Romano N, Luppi M. 2011. Differences in Kaposi sarcoma-associated herpesvirus-specific and herpesvirus-non-specific immune responses in classic Kaposi sarcoma cases and matched controls in Sicily. Cancer Sci. 102:1769–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guihot A, Dupin N, Marcelin A-G, Gorin I, Bedin A-S, Bossi P, Galicier L, Oksenhendler E, Autran B, Carcelain G. 2006. Low T cell responses to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J. Infect. Dis. 194:1078–1088 [DOI] [PubMed] [Google Scholar]
- 35. Bihl F, Narayan M, Chisholm JV, Henry LM, Suscovich TJ, Brown EE, Welzel TM, Kaufmann DE, Zaman TM, Dollard S, Martin JN, Wang F, Scadden DT, Kaye KM, Brander C. 2007. Lytic and latent antigens of the human gammaherpesviruses Kaposi's sarcoma-associated herpesvirus and Epstein-Barr virus induce T-cell responses with similar functional properties and memory phenotypes. J. Virol. 81:4904–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Woodberry T, Suscovich TJ, Henry LM, Martin JN, Dollard S, O'Connor PG, Davis JK, Osmond D, Lee T-H, Kedes DH, Khatri A, Lee J, Walker BD, Scadden DT, Brander C. 2005. Impact of Kaposi sarcoma-associated herpesvirus (KSHV) burden and HIV coinfection on the detection of T cell responses to KSHV ORF73 and ORF65 proteins. J. Infect. Dis. 192:622–629 [DOI] [PubMed] [Google Scholar]
- 37. Guihot A, Oksenhendler E, Galicier L, Marcelin A-G, Papagno L, Bedin A-S, Agbalika F, Dupin N, Cadranel J, Autran B, Carcelain G. 2008. Multicentric Castleman disease is associated with polyfunctional effector memory HHV-8-specific CD8+ T cells. Blood 111:1387–1395 [DOI] [PubMed] [Google Scholar]
- 38. Lambert M, Gannage M, Karras A, Abel M, Legendre C, Kerob D, Agbalika F, Girard P-M, Lebbe C, Caillat-Zucman S. 2006. Differences in the frequency and function of HHV8-specific CD8 T cells between asymptomatic HHV8 infection and Kaposi sarcoma. Blood 108:3871–3880 [DOI] [PubMed] [Google Scholar]
- 39. Kwun HJ, da Silva SR, Shah IM, Blake N, Moore PS, Chang Y. 2007. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mimics Epstein-Barr virus EBNA1 immune evasion through central repeat domain effects on protein processing. J. Virol. 81:8225–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glaunsinger B, Ganem D. 2004. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 13:713–723 [DOI] [PubMed] [Google Scholar]
- 41. Butler LM, Jeffery HC, Wheat RL, Long HM, Rae PC, Nash GB, Blackbourn DJ. 2012. Kaposi's sarcoma-associated herpesvirus inhibits expression and function of endothelial cell major histocompatibility complex class II via suppressor of cytokine signaling 3. J. Virol. 86:7158–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wies E, Hahn AS, Schmidt K, Viebahn C, Rohland N, Lux A, Schellhorn T, Holzer A, Jung JU, Neipel F. 2009. The Kaposi's sarcoma-associated herpesvirus-encoded vIRF-3 inhibits cellular IRF-5. J. Biol. Chem. 284:8525–8538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lubyova B, Kellum MJ, Frisancho JA, Pitha PM. 2007. Stimulation of c-Myc transcriptional activity by vIRF-3 of Kaposi sarcoma-associated herpesvirus. J. Biol. Chem. 282:31944–31953 [DOI] [PubMed] [Google Scholar]
- 44. Shin YC, Joo C-H, Gack MU, Lee H-R, Jung JU. 2008. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 to induce vascular endothelial growth factor expression. Cancer Res. 68:1751–1759 [DOI] [PubMed] [Google Scholar]
- 45. Wies E, Mori Y, Hahn A, Kremmer E, Sturzl M, Fleckenstein B, Neipel F. 2008. The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 111:320–327 [DOI] [PubMed] [Google Scholar]