

Abstract
Hepatitis C virus (HCV) nonstructural protein 5A (NS5A) is involved in regulating viral replication through its direct interaction with the HCV RNA-dependent RNA polymerase. NS5A also alters infected cell metabolism through complex interactions with numerous host cell proteins. NS5A has furthermore been suggested to act as a transcriptional activator, although the impact on viral replication is unclear. To study this, HCV NS5A variants were amplified from hepatic tissue from an HCV-infected patient, and their abilities to activate gene transcription were analyzed in a single-hybrid yeast (Saccharomyces cerevisiae) model. Different variants isolated from the same patient displayed different transactivational activities. When these variants were inserted into the HCV subgenomic replicon system, they demonstrated various levels of RNA replication, which correlated with their transactivational activities. We showed that the C-terminal fragment of NS5A was localized to the nucleus and that a functional NS5A nuclear localization signal and cellular caspase activity were required for this process. Furthermore, nuclear localization of NS5A was necessary for viral replication. Finally, we demonstrate that nuclear NS5A binds to host cell promoters of several genes previously identified as important for efficient HCV RNA replication, inducing their transcription. Taken together, these results demonstrate a new mechanism by which HCV modulates its cellular environment, thereby enhancing viral replication.
INTRODUCTION
Hepatitis C virus (HCV) infection is characterized by a high frequency of chronicity and is responsible for chronic infection of approximately 130 million carriers—i.e., 2.2% of the worldwide population (1). Chronic HCV infection is responsible for chronic hepatitis, a major risk factor in the development of cirrhosis and hepatocellular carcinoma (HCC) or primary liver cancer, which occurs in 1 to 4% of cirrhotic patients every year (2, 3). Chronic HCV infection has become the principal cause of HCC in Japan, and modeling of the ongoing epidemics predicts a similar trend in Europe (4, 5).
HCV is an enveloped, positive-strand RNA virus with a cytoplasmic life cycle. HCV replication is catalyzed by an RNA-dependent RNA polymerase (RdRp). It takes place within a membranous web (replication complex) located close to perinuclear membranes, in close association with the nonstructural HCV proteins and a number of host cell factors that play an important role in this process (6, 7). The precise functions of HCV nonstructural protein 5A (NS5A) remains largely unclear. However, it has been shown to be a pleiotropic serine phosphoprotein, involved in the regulation of RdRp function as well as in a number of interactions with host cell mechanisms (8). Studies have also suggested a role for NS5A in the establishment of chronicity of HCV infection and in procarcinogenic events (9).
Among its numerous properties, the HCV NS5A protein has been shown to bear transcriptional activation properties in yeast (Saccharomyces cerevisiae) and mammalian cells (10–13). These properties are borne by a transcriptional activation domain, comprised of two acidic regions followed by a proline-rich region spanning amino acids 2135 to 2331 of the protein (reference strain HCV-J) (11). In an infected individual, HCV exists as a quasispecies, i.e., a complex mixture of genetically distinct but closely related variants (14, 15). We previously demonstrated that different NS5A variants isolated from serum samples of HCV-infected patients carry different levels of transcriptional activation in a yeast single-hybrid model (16, 62).
In infected cells, HCV NS5A is localized predominantly in the cytoplasm and the perinuclear region, despite the presence of a functional nuclear localization signal (NLS) in its C-terminal region (17, 18). Studies have shown that a small amphipathic α-helix at the N terminus of NS5A acts as an endoplasmic reticulum (ER) membrane retention signal (19). Accordingly, experimental NS5A mutants lacking the N-terminal region almost exclusively localize in the nucleus. It has also been demonstrated in vitro that cellular caspases can cleave the full-length NS5A to produce N-terminally deleted products that translocate to the nucleus and function as transcriptional activators (18, 20, 21). A recent study using the infectious HCV clone JFH1 has demonstrated that N-terminally truncated forms of NS5A are produced during viral infection and translocate to the nucleus (22). However, the physiological relevance of caspase-mediated cleavage and nuclear localization of NS5A fragments remains unknown.
The goal of this study was to assess the role of NS5A transcriptional activation in HCV replication and the influence of NS5A cleavage and translocation into the nucleus in this process.
MATERIALS AND METHODS
Human liver tissue sample.
The human liver biopsy specimen used in this study was obtained from our local liver tissue repository (Plateforme de Ressources Biologiques, Hôpital Henri Mondor). The patient had given written informed consent in accordance with French legislation. A material transfer agreement was signed between our laboratory and the repository. The biopsy specimen was from an 82-year-old male with untreated chronic HCV genotype 1b infection. The METAVIR inflammatory activity/fibrosis score was A3F3.
Cells.
NG cells were obtained from Abbott Laboratories (Abbott Park, IL). This cell line, highly permissive to HCV RNA replication, was derived from Huh-7 cells carrying a stably maintained replicon treated with alpha interferon, as previously described (23). These cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Antibodies.
Mouse anti-NS5A antibody OBT 1222 was purchased from AbD Serotec (Kidlington, United Kingdom); mouse anti-NS5A antibody Ab 11.1 was a gift from Darius Moradpour (University of Lausanne, Switzerland), rabbit anti-NS5A antibody was a gift from Eberhard Hildt (Robert Koch Institute, Berlin, Germany); sheep anti-NS5A antibody was a gift from Mark Harris (University of Leeds, United Kingdom). Anticalnexin and anti-ASH2L antibodies were purchased from Cell Signaling (Beverly, MA). Alexa Fluor-conjugated secondary antibodies were purchased from Molecular Probes (Life Technologies, Ltd., Paisley, United Kingdom). The antiactin antibody was purchased from Sigma-Aldrich (St. Louis, MO).
Plasmid construction. (i) HCV subgenomic replicon shuttle vector.
A subgenomic replicon shuttle vector (genotype 1b, strain Con1) was obtained from Abbott Laboratories (24, 25). This shuttle vector contains two unique restriction sites to facilitate the cloning of patient-derived NS5A sequences: a single NotI site present within NS4B, 97 nucleotides (nt) upstream of NS5A, and a PacI site present within NS5A, 18 nt upstream of NS5B. NS5A originating from pHCV-N (genotype 1b, accession no. AF139594) was cloned into this vector to create SGR-Nim. Cloning of NS5A sequences isolated from patients was performed as described previously (24).
(ii) Isolation and cloning of quasispecies variants.
Total hepatic RNA was isolated from a single liver biopsy specimen from a patient infected with HCV genotype 1b using the PARIS kit (Ambion, Austin, TX), according to the manufacturer's protocol. cDNA was synthesized from isolated RNA using a Superscript III reverse transcription kit (Life Technologies, Carlsbad, CA). Five NS5A quasispecies variants were amplified from the patient's cDNA using a nested PCR technique described previously (24). Flanking restriction sites NotI and PacI, together with the adaptive mutation S2204I, were introduced by PCR into all of the amplified NS5A sequences. Variants were cloned into the NotI-PacI site of the subgenomic replicon shuttle vector and verified by sequencing.
(iii) NS5A AR2 region swapping.
AR2 regions from strong and weak transactivation NS5A variants were swapped using a PCR-based swapping strategy adapted from Deminie et al. (26). Primers were named A to F according to Deminie et al.: A-NS5A-swap (TAGCGGCCGGGAGCAGCAAACGACGTCCTC), B-NS5A-swap (CAAAGAAGGCGCAGACAACTGGCTAGC), C-NS5A-swap (TTCCCCCCAGCGATGCCCATATGG), D-NS5A-swap (GGAAGCTTGTCCGGCTCGTGGCTAAGG), E-NS5A-swap (GCTATCCAGTTGTCTGCGCCTTCTTGT), and F-NS5A-swap (CCCATATGGGCATCGCTGGGGGGA).
(iv) Site-directed mutagenesis.
Site-directed mutagenesis was performed using QuikChange II XL (Agilent Technologies, Santa Clara, CA), according to the manufacturer's instructions. The NS5A NLS was invalidated by mutagenesis as previously described by Yeh et al. (27) using the following primers: CCCCTCCAATACCACCTCCAGGGGGAATGGGGACGGTTGTCCTAACAG (forward) and CTGTTAGGACAACCGTCCCCATTCCCCCTGGAGGTGGTATTGGAGGGG (reverse). Using this approach, the nuclear localization signal was mutated (boldface) from PPPRRKRTVVLTESTL to PPPGGMGTVVLTESTL. Similarly, site-directed mutagenesis was performed to introduce D154E, D154A, or D154S mutations to invalidate the caspase cleavage site in NS5A, as described by Kalamvoki et al. (28). The following primers, adapted to the HCV genotype 1b sequence, were used: D154E, ATTCTTCACAGAGGTGGAAGGGGTGCGGC (forward) and TAAGAAGTGTCTCCACCTTCCCCACGCCG (reverse); D154A, TTCACAGAGGTGGCTGGGGTGCGGCTG (forward) and CAGCCGCACCCCAGCCACCTCTGTGAA (reverse); and D154S, ATTCTTCACAGAGGTGAGTGGGGTGCGGCTGCAC (forward) and GTGCAGCCGCACCCCACTCACCTCTGTGAAGAAT (reverse).
Lentiviral vector production.
NS5A variants bearing the adaptive mutation S2204I were cloned into pLenti6.3/V5-TOPO (Life Technologies), and the nucleotide sequence of all constructs was verified by DNA sequencing. Recombinant lentiviral vector particles were synthesized by using the ViraPower HiPerform lentiviral expression system (Life Technologies), according to the manufacturer's instructions. Briefly, lentiviral vector particles were produced by transient transfection of 5 × 106 293FT cells with pLenti vectors containing different NS5A sequences and the ViraPower packaging mix using Lipofectamine 2000 (Life Technologies). The titers of the particles produced were determined on Huh7.5 cells according to the manufacturer's instructions.
Transient replication assay.
Replicon shuttle vector DNA was linearized and purified by phenol-chloroform extraction. One microgram of purified DNA was used as a template for RNA synthesis using the T7 Megascript kit (Life Technologies), according to the manufacturer's instructions. Synthesized RNA was purified using RNeasy minikit (Qiagen, Hilden, Germany).
Monolayers of NG cells seeded in 48-well plates (2 × 104 cells/well) were transfected with 0.25 μg RNA/well using TransIT-mRNA reagent (Mirus Bio LLC, Madison, WI) in Opti-MEM reduced serum medium (Life Technologies) containing 5% fetal calf serum. Transfected cells (6 wells per construct) were divided into two equal samples: 3 wells harvested at 4 h posttransfection and 3 wells harvested at 96 h posttransfection. Cells were washed and then lysed in cell culture lysis buffer (Promega Corporation, Madison, WI), and the resultant luciferase activity was measured with a Mithras LB940 luminometer (Berthold Technologies, Bad Wildbad, Germany) in conjunction with luciferin substrate (Promega Corporation). All luciferase activity measurements were done in triplicate. The replication efficiency for each replicon clone was calculated as described previously (24).
trans-complementation assays.
Monolayers of NG cells seeded in 48-well plates (104 cells/well) were transduced with lentiviral particles at a multiplicity of infection (MOI) of 1.0. Forty-eight hours later, cells were transfected with replicon RNA using TransIT-mRNA reagent as described above. Cells were harvested at 4 h and 96 h posttransfection, and luciferase activity was measured. Replication efficiency was calculated for each replicon clone as described previously (24) and normalized to the protein concentration at 96 h.
Yeast simple hybrid assay.
Nucleotide sequences encoding the NS5A transactivation domain (nt 6405 to 6981 of the subgenomic replicon) of selected variants were amplified by PCR as described previously by Pellerin et al. (16) and cloned into the BamHI-EcoRI sites of the yeast expression vector pGBT9 (Clontech, TaKaRa Bio, Inc., Shiga, Japan) to generate a fusion protein consisting of the NS5A transcriptional activation domain and the GAL4 DNA-binding domain. The resulting plasmids were transformed into Y187 yeast using a Yeast Maker yeast transformation kit (Clontech). Transformants were grown on tryptophan-deficient (Trp−) synthetic dropout plates for 3 days at 30°C and screened by PCR.
Transcriptional activation of NS5A-GAL4 fusions was measured by a quantitative luminescent β-galactosidase assay based on the Galacto Star kit (Tropix, Bedford, MA), according to the manufacturer's protocol using an automated Mithras LB940 luminometer. All assays were repeated with three independent transformants for each construct, each analyzed in duplicate.
Immunofluorescence.
For indirect immunofluorescence studies using confocal microscopy, NG cells (0.5 × 105 cells/well) cultured in LabTek 4-chamber slides (Thermo Fisher Scientific, Inc., Waltham, MA) were transduced with lentivirus particles at MOI of 1.5. Forty-eight hours postransduction, cells were fixed with ice-cold methanol at room temperature for 5 min, washed with phosphate-buffered saline (PBS), and incubated for 2 h at room temperature with rabbit anti-NS5A polyclonal antibody (29). Bound antibody was detected with antirabbit fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Sigma-Aldrich), and cellular DNA marked with 1 μM To-Pro-3 DNA stain (Life Technologies). Samples were analyzed with a confocal laser-scanning microscope (Leica DMRE-7/TCSSP2; Wetzlar, Germany) in conjunction with a 63× oil immersion objective and 633-nm HeNe and 488-nm argon lasers. Confocal image z-stacks were obtained using optical slice intervals of 0.2 μm from the bottom to top of the cell, with three scans made per slice. The mean of the signal was recorded, and Imaris software (Bitplane AG, Zurich, Switzerland) was used to reconstruct three-dimensional (3D) image projections. 3D objects were counted in order to measure the proportion of NS5A in the nucleus of 20 randomly chosen cells per condition in duplicate experiments.
For indirect immunofluorescence studies and proximity-dependent DNA ligation studies using epifluorescence microscopy, transduced NG cells or cells harboring the stable subgenomic replicon I389-neo/NS3-3′/5.1 (30) were fixed with 2% paraformaldehyde in PBS for 10 min and permeabilized using ice-cold methanol at −20°C for 10 min, washed with PBS, and incubated for 1 h at room temperature with 10% normal goat serum–0.3% Triton X-114 in PBS. The primary antibody was diluted in a mixture of PBS, 1% bovine serum albumin (BSA), and 0.3% Triton X-114 and incubated for 1 h at room temperature. For regular indirect immunofluorescence studies, bound antibodies were detected with an Alexa Fluor-conjugated secondary antibody (Life Technologies), and cellular DNA was stained with 1 μM To-Pro-3 (Life Technologies) or DAPI (4′,6-diamidino-2-phenylindole) dilactate (Life Technologies). Slides were mounted using an antifading solution (Prolong Gold; Life Technologies), and images were captured using a Zeiss Axioskop 40 microscope in conjunction with a Zeiss MRm Axiocam and Axiovision V40 4.7 software. For proximity-dependent DNA ligation studies, the Duolink in situ assay (Olink Bioscience, Uppsala, Sweden) was used according to the manufacturer's instructions. Briefly, after primary antibody incubation, the PLA probe anti-mouse Plus and PLA probe anti-rabbit Minus were diluted in PBS containing 1% BSA and 0.3% Triton X-114 and added to the cells for 1 h at 37°C. Cells were incubated with a ligase for 30 min at 37°C and then incubated with a polymerase and green fluorescent-labeled nucleotides for 100 min at 37°C. Cellular DNA was stained with 1 μM To-Pro-3, and slides were mounted using the antifading agent provided in the kit. Images were captured using a Zeiss Axioskop 40 microscope in conjunction with a Zeiss MRm Axiocam and Axiovision V40 4.7 software.
Immunoblotting.
Cells were lysed with Promega cell culture lysis reagent, separated by SDS-PAGE, and immobilized onto Hybond ECL nitrocellulose membrane (GE Healthcare, Chalfont St-Giles, United Kingdom). Bound polypeptides were detected using sheep anti-NS5A antibody (kindly provided by Mark Harris, University of Leeds, United Kingdom), rabbit anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (Abcam, Cambridge, United Kingdom), or mouse antiactin antibody (Sigma-Aldrich, St. Louis, MO). Immunodetection was achieved by enhanced chemiluminescence (GE Healthcare) in conjunction with horseradish peroxidase-conjugated anti-sheep or anti-mouse antibodies (Sigma-Aldrich). Chemiluminescent signals were detected and quantified using an Image Quant Las 4000 miniscanner and Image Quant software (GE Healthcare).
Caspase inhibition assays.
NG cells were treated with the pan-caspase inhibitor z-VAD-fmk (R&D Systems, Inc., Minneapolis, MN) at 10 μM or 20 μM final concentrations 2 h prior to transfection with replicon RNA. Four hours posttransfection, culture medium was changed and fresh z-VAD-fmk was added at the appropriate concentrations.
Real-time quantitative gene expression assay.
Total RNA was extracted from NG cells transduced with the appropriate lentiviral expression vector particles (72 h postransduction) using a PARIS RNA isolation kit (Ambion). RNA quality and quantity were determined using a 2100 Bioanalyser and RNA Nano Chips (Agilent Technologies, Santa Clara, CA). RNA integrity number was calculated using the Agilent software, and samples displaying an RNA integrity number below 6 were discarded.
cDNA was synthesized using a high-capacity cDNA synthesis kit (Applied Biosystems, Life Technologies). Quantitative PCR (qPCR) was performed on pooled cDNA from two independent experiments using Applied Biosystems 7300 Thermal Cycler and TaqMan reagents (Applied Biosystems) (see Table 1 for primer information).
Table 1.
RT-qPCR TaqMan probes for gene expression analysis
| Primers | Gene accession no. | Assay identification no. | Gene product or function |
|---|---|---|---|
| GAPDH | NM_002046.3 | Hs99999905_m1 | Glyceraldehyde-3-phosphate dehydrogenase |
| CDC42 | NM_001791.3 | Hs03044122_g1 | Cell division cycle 42 (GTP binding protein, 25 kDa) |
| COPZ1 | NM_016057.1 | Hs00255433_m1 | Coatomer protein complex, subunit zeta 1 |
| CSK | NM_001127190.1 | Hs01062579_g1 | c-src tyrosine kinase |
| CYP1A1 | NM_000499.3 | Hs01054797_g1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 |
| DDX3X | NM_001356.3 | Hs00606179_m1 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 3, X-linked |
| DICER1 | NM_030621.3 | Hs00998582_g1 | Dicer 1, RNase type III |
| EIF2S3 | NM_001415.3 | Hs00831673_gH | Eukaryotic translation initiation factor 2, subunit 3 gamma, 52 kDa |
| FBXL2 | NM_012157.3 | Hs00247211_m1 | F-box and leucine-rich repeat protein 2 |
| HAMP | NM_021175.2 | Hs01057160_g1 | Hepcidin antimicrobial peptide |
| JAK1 | NM_002227.2 | Hs01026996_m1 | Janus kinase 1 |
| LTΒ | NM_009588.1 | Hs00242739_m1 | Lymphotoxin beta (TNF superfamily, member 3) |
| MAP2K7 | NM_145185.2 | Hs00178198_m1 | Mitogen-activated protein kinase kinase 7 |
| NFKB2 | NM_001077493.1 | Hs00174517_m1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 2 (p49/p100) |
| NUAK2 | NM_030952.1 | Hs00388292_m1 | NUAK family, SNF1-like kinase, 2 |
| PI4KA | NM_058004.2 | Hs00176931_m1 | Phosphatidylinositol 4-kinase |
| PI4KB | NM_002651.1 | Hs00356327_m1 | Phosphatidylinositol 4-kinase, catalytic, beta |
| PPIA | NM_021130.3 | Hs99999904_m1 | Peptidylprolyl isomerase A (cyclophilin A) |
| RELA | NM_001145138.1 | Hs00153294_m1 | v-rel reticuloendotheliosis viral oncogene homolog A |
| SLC12A4 | NM_001145961.1 | Hs00957122_m1 | Solute carrier family 12 (potassium/chloride transporters), member 4 |
| SLC12A5 | NM_001134771.1 | Hs00221168_m1 | Solute carrier family 12 (potassium-chloride transporter), member 5 |
| TBXA2R | NM_201636.2 | Hs00169054_m1 | Thromboxane A2 receptor |
| TRAF2 | NM_021138.3 | Hs00184186_m1 | TNF receptor-associated factor 2 |
| TXNIP | NM_006472.3 | Hs01006900_g1 | Thioredoxin interacting protein |
| VAPA | NM_194434.2 | Hs00427749_m1 | VAMP (vesicle-associated membrane protein)-associated protein A, 33 kDa |
| VAPB | NM_004738.3 | Hs00191003_m1 | VAMP (vesicle-associated membrane protein)-associated protein B and C |
| VRK1 | NM_003384.2 | Hs00177470_m1 | Vaccinia related kinase 1 |
| IL-8 | NM_000584.2 | Hs99999034_m1 | Interleukin 8 |
| PLK1 | NM_005030.3 | Hs00983233_g1 | Polo-like kinase 1 |
ChIP.
For chromatin immunoprecipitation (ChIP), NG cells were transduced with lentiviral vector particles at an MOI of 1.5. Forty-eight hours postransduction, cells were fixed with 1% formaldehyde and lysed in a mixture of 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), and 0.1% NP-40 containing protease inhibitors (Roche Applied Science, Indianapolis, IN). The nuclei were recovered and lysed in a mixture containing 50 mM Tris-HCl (pH 8), 10 mM EDTA, and 1% SDS, with protease inhibitors, and DNA was sheared by sonication. Sheared chromatin was precleared by incubation with protein G Mag Sepharose beads (GE Healthcare) and incubated with rabbit anti-NS5A antibody. The precipitated DNA was purified by phenol-chloroform extraction and amplified by PCR using the following primers: interleukin-8 (IL-8), 5′-GTTGTAGTATGCCCCTAAGAG-3′ (forward) and 5′-CTCAGGGCAAACCTGAGTCATC-3′ (reverse); lymphotoxin B (LTΒ), 5′-TACGGGCCTCTCTGGTACAC-3′ (forward) and 5′-ATATTCCCTCACCCCACCAT-3′ (reverse); NUAK2, 5′-CCTGAAGTTGCTGCTGTGAA-3′ (forward) and 5′-CCTGAAGGCCTAGAGAACACA-3′ (reverse); MAP2K7, 5′-AACGAGGTTCCAGGAATGC-3′ (forward) and 5′-GAAGGATGACGCCACCTAGA-3′ (reverse); TRAF2, 5′-GGGAAGGGACCCAATTAGC-3′ (forward) and 5′-CAGCCCTCAGGAAGCTGTAG-3′ (reverse); and FBXL2, 5′-TGGGGTGAGTGTCGTTTTATC-3′ (forward) and 5′-CGTATCTGGTCCATCCTCTCA-3′ (reverse). DNA immunoprecipitated from cells not expressing NS5A was used as a negative control.
Statistics.
Statistical comparisons of the data were performed using a Mann-Whitney test where appropriate. P values of <0.05 were considered statistically significant. All error bars indicate standard errors of the mean (SEM).
RESULTS
Naturally occurring NS5A variants are associated with different transcriptional activation capacities in vitro.
To examine the transcriptional activation properties of different NS5A quasispecies variants within the same patient, five NS5A full-length variants were amplified from the liver of a patient infected with HCV genotype 1b. Their sequences are shown in Fig. 1. Amino acid differences were observed over the full NS5A sequence, but were more frequent between amino acids 210 and 450, a region spanning the NS5A transactivation domain. In order to verify our previous findings that naturally occurring NS5A variants are associated with different transcriptional activation capacities in vitro (16), the NS5A transactivation domain (underlined in Fig. 1) was reamplified by PCR. The amplified domains, which were confirmed to be identical in sequence to the full-length variants, were subsequently cloned into the pGBT9 yeast expression vector to generate fusion proteins consisting of the NS5A transcriptional activation domain fused to the GAL4 DNA-binding domain (Fig. 2A). The capacity of each fusion protein to activate lacZ transcription (and thus β-galactosidase expression) was assayed in yeast. As shown in Fig. 2B, the five tested variants (v1 to v5) exhibited markedly different transcriptional activities in vitro, with the weakest activator (NS5A-v5) exhibiting transcriptional activities approximately 1 log below the activity of the strongest activator (NS5A-v1).
Fig 1.

Amino acid sequence alignment of isolated NS5A variants. Amino acid sequences of NS5A quasispecies variants (v1 to v5) isolated from the liver of a patient infected with HCV genotype 1b aligned with the SGR-Nim NS5A sequence used as a reference sequence. Numbers represent amino acid positions within NS5A. The underlined sequence represents the NS5A transactivation domain; the shaded sequence represents the NLS. Lowercase letters in the consensus sequence denote variability among variants; capital letters represent conserved amino acids among variants.
Fig 2.
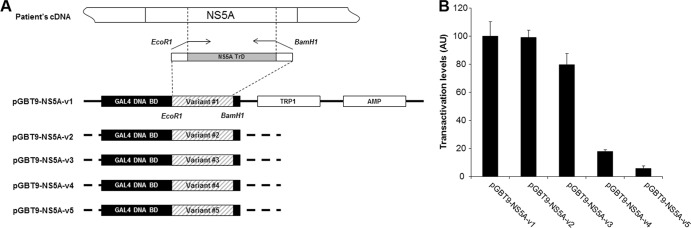
Transactivation properties of NS5A variants. (A) Schematic representation of yeast one-hybrid constructs encoding NS5A-v1 to -v5. The NS5A transcriptional activation domains (NS5A TrD) were amplified from the patient's cDNA and cloned into plasmid pGBT9, in order to generate fusion proteins containing the GAL4 DNA-binding domain (BD) (pGBT9-NS5A-v1 to -v5). (B) Yeast one-hybrid vectors pGBT9-NS5A-v1 to -v5 were transfected into yeast strain Y187. The ability of the hybrid proteins to activate transcription of the LacZ gene was analyzed by quantification of β-galactosidase activity. The results are expressed as mean ± standard error of the mean (SEM) transactivation levels obtained from at least three independent experiments. AU, arbitrary units.
Naturally occurring NS5A variants are associated with different HCV RNA replication efficiencies in vitro.
To test the hypothesis that NS5A mutations naturally occurring in vivo could be associated with different replication capacities of the corresponding variants, the five distinct NS5A variants described above (v1 to v5) were cloned into a subgenomic replicon shuttle vector (24) containing a luciferase reporter gene, and the replication capacity of the corresponding replicons was measured (Fig. 3A). Western blotting revealed that at 96 h posttransfection, cells transfected with the different constructs exhibited different levels of NS5A expression, suggesting that different NS5A variants could be associated with different replication capacities of the corresponding replicons (Fig. 3B). To rule out a loss of affinity of the anti-NS5A antibody related to sequence differences, we analyzed NS5A expression of v3, v4, and v5 4 h posttransfection by Western blotting (Fig. 3C). We also demonstrated identical stability of NS5A protein from v1 and v5 (Fig. 3D). Thus, we confirmed that the absence of NS5A signal observed at 96 h in Fig. 3B was solely due to the lower replication level of the corresponding replicons. To assess the replication efficiencies of replicons harboring the different NS5A variant sequences, we quantified the luciferase activity from replicon-transfected cell extracts (Fig. 3E). Different replication levels were observed, including higher (v1), nearly identical (v2 and v3), and lower (v4 and v5) replication efficiencies compared to the wild-type (wt) subgenomic replicon (Fig. 3E). In the latter variants, the replication level was close to that of a replicon containing a deficient RNA polymerase. We further demonstrated that, in contrast to v1, v5 was unable to overcome the degradation of input RNA at 48 h, although it expressed luciferase and NS5A at 4 h (Fig. 3F). Together, these results demonstrate that naturally occurring NS5A variants are associated with different RNA replication efficiencies in vitro.
Fig 3.

Impact of HCV NS5A transactivation properties on replication efficiency of subgenomic HCV replicons harboring different NS5A quasispecies variants. (A) Schematic representation of subgenomic replicon constructs encoding NS5A-v1 to -v5. NS5A variant sequences were cloned into a subgenomic replicon shuttle vector. Solid triangles indicate the introduction of cell culture adaptive mutation S2204I. An open triangle indicates the introduction of the GDD-to-GND mutation in the NS5B RNA-dependent RNA polymerase. SGR-NS5A-vX are subgenomic replicons carrying NS5A variant sequence vX; SGR-Nim is the wt replicon (24); SGR-GND is the nonreplicative control replicon. NTR, nontranslated region; EMCV, encephalomyocarditis virus; IRES, internal ribosome entry site; HDV, hepatitis delta virus. (B) NS5A protein expression in NG cells transfected with subgenomic replicons encoding different NS5A variants as analyzed by Western blotting at 96 h posttransfection. Actin expression was used as a loading control. Bands corresponding to 56-kDa NS5A or 42-kDa actin are indicated. (C) NS5A protein expression in NG cells transfected with subgenomic replicons encoding NS5A-v3, -v4, and -v5 was analyzed by Western blotting at 4 and 96 h posttransfection. Actin expression was used as a loading control. Bands corresponding to 56-kDa NS5A or 42-kDa actin are indicated. (D) NG cells transduced with NS5A-v1 and -v5 lentiviral vectors were treated with cycloheximide (CHX) and analyzed for NS5A expression by Western blotting at 0 and 96 h posttreatment. (E) Replication efficiency of HCV subgenomic replicons carrying different NS5A variants was analyzed in the cell lysates as detailed in panel B. Luciferase activities were assayed in transfected cells, and the replication efficiency was calculated as a percentage of that obtained with replicon SGR-Nim (represented by the dashed line). The data are indicative of mean ± SEM replication efficiencies obtained from 7 independent experiments carried out in triplicate. (F) Replication efficiency of HCV subgenomic replicons carrying the v1 and v5 NS5A variants was analyzed as a time course in the cell lysates. Luciferase activities were assayed in transfected cells and are presented as a percentage of that obtained at 4 h. Data are indicative of mean ± SEM replication efficiencies obtained from 4 independent experiments. (G) Relationship between the transcriptional activation capacities of NS5A variants and the replication efficiencies of the corresponding subgenomic replicons. Data points represent the mean transactivational capacities and replication efficiencies for each variant.
HCV RNA replication efficiency in vitro correlates with NS5A variant transcriptional activation capacity.
As shown in Fig. 3G, we found a statistically significant correlation (R2 = 0.985, P < 0.01) between the transactivation capacities of NS5A variants in vitro and the in vitro replication efficiency of the corresponding replicons. In order to assess whether the relationship between NS5A transactivation and RNA replication is causal, and not just the result of the requirement for identical clusters of amino acidic residues or posttranslational modifications, we expressed NS5A-v1 in trans in cells harboring the replication-deficient variant SGR-NS5A-v5. Such expression significantly increased SGR-NS5A-v5 replication efficiency 3-fold (P = 0.016), whereas SGR-NS5A-v5 replication efficiency remained unchanged when the putative caspase target site mutated at position 154 (NS5A-v1/D154E) was coexpressed in trans (Fig. 4). Since it has been shown previously that NS5A expressed alone does not restore RNA replication (31, 61), this result suggests that the observed rescue is due to NS5A transactivation.
Fig 4.
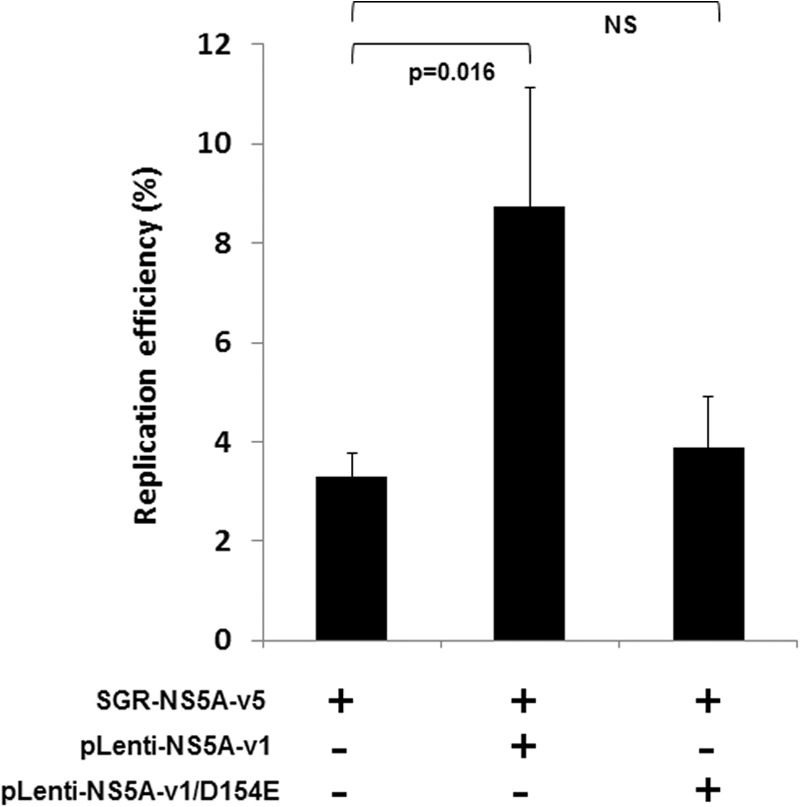
trans-complementation of a replication-deficient replicon with a strong transactivator NS5A sequence. trans-complementation assays were performed on cells harboring the SGR-NS5A-v5 replication-deficient replicon. trans-complementation was tested with pLenti-NS5A-v1 and pLenti-NS5A-v1/D154E. Values are expressed as the percentage of replication efficiency, with 100% obtained with SGR-NS5A-v1 used as the reference.
NS5A transactivation properties and the resulting effect on RNA replication are borne by NS5A acidic region 2.
Sequence analysis of the NS5A transactivation domains from v1 to v5 showed no relationship between specific amino acid residues and the transactivation capacities of the corresponding variants (Fig. 1). We assessed the role of the global charge of the NS5A protein, which has been shown to influence its transcriptional activation capacities (16). Strong transactivation capacities were associated with a lower charge than weak transactivation capacities (average of −13.2 for v1 to v3 versus −12.3 for v4 and v5). The difference was greater when analyzing the sequence of the transactivating domains only (−7.2 versus −5.5, respectively). A similar charge difference was observed in the AR2 sequence (−9.2 versus −8.5, respectively, whereas the charge in NS5A acidic region 1 (−6.5) and the number of proline residues within the proline-rich region did not differ among the different variants. This observation suggested that AR2 is the main driver of NS5A transactivation and its effect on HCV replication.
To experimentally confirm this hypothesis, we swapped the AR2 region sequences of strongly and weakly transactivating NS5A variants (v1 and v5, respectively). Transcriptional activation by these chimeras was measured using the yeast single-hybrid system, and the replication capacity of the corresponding subgenomic replicons was assessed (Fig. 5). As shown in Fig. 5, both transcriptional activation and HCV RNA replication were strongly reduced when the AR2 region of the strong transactivator variant was replaced by that of the weak transactivator variant. Conversely, transactivation was significantly enhanced when the AR2 region of the weak transactivator was replaced by that of the strong transactivator, but the replication capacity of the corresponding variant was only modestly affected. Together, these observations suggest that the NS5A AR2 region plays an important, although not exclusive, role in the transactivation properties of the NS5A protein and in HCV replication.
Fig 5.
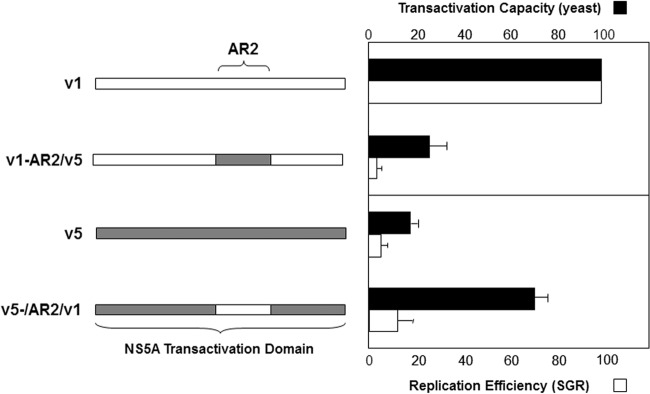
Role of the AR2 region in NS5A transcriptional activity and HCV replication efficiency. The AR2 region of the NS5A transactivation domain of strongly (NS5A-v1) and weakly (NS5A-v5) transactivating NS5A variants were swapped (left panel). The effects of these exchanges on NS5A transactivation capacities and RNA replication were studied in the yeast one-hybrid system (black bars) and in the subgenomic replicon model (white bars), respectively. Error bars indicate SEM. The results are indicative of three independent experiments.
A subset of HCV NS5A protein translocates to the nucleus.
Immunofluorescence analysis of cells harboring the stable subgenomic replicon I389-neo/NS3-3′/5.1 (30) by means of confocal microscopy and 3D reconstruction showed that on average 11.6% ± 2.2% of NS5A protein localized to the nucleus (Fig. 6A). To confirm these results in the absence of viral replication, we cloned NS5A-v1 or NS5A-v5 into a lentivirus vector (Fig. 6B) and observed that 14.0% ± 2.8% and 16.3% ± 3.1% of NS5A proteins localized to the nucleus in cells transduced with these lentiviruses, respectively (Fig. 6C). In addition, we used a proximity-dependent DNA ligation assay (32) to demonstrate that the presence of NS5A was less than 40 nm from the nuclear protein ASH2L (Fig. 7A), a component of the nuclear Set1 histone methyltransferase complex (33), in NG cells harboring the stable subgenomic replicon (Fig. 7B). Using this assay (Fig. 7C to F), we observed that NS5A colocalized with this nuclear protein, thus confirming that a proportion of NS5A resides in the nucleus.
Fig 6.

Subcellular localization of NS5A. (A) Cells harboring the subgenomic replicon I389-neo/NS3-3′/5.1 were analyzed by confocal microscopy using anti-NS5A antibody in conjunction with anti-rabbit FITC-conjugated secondary antibody (green). Nuclei were stained with To-Pro-3 (red). Three-dimensional representations were reconstructed from image z-stacks. Scale bar, 10 μm. *, non-replicon-harboring cells. The yellow arrow shows where the 3D reconstructed image was cropped to display the cell sections. The yellow pictogram indicates the direction of sight. (B) Schematic representation of lentivirus vectors carrying NS5A-v1 and -v5 and β-galactosidase. Filled triangles indicate the presence of cell culture adaptive mutation S2204I. (C) Confocal analysis of cells transduced with Lenti-NS5A-v1 or -v5 was carried out as described for panel A. Scale bar, 10 μm. *, nontransduced cells. The yellow arrow shows where the 3D reconstructed image was cropped to display the cell sections. The yellow pictogram indicates the direction of sight.
Fig 7.

Epifluorescence analysis of proximity ligation assays. Cells harboring the subgenomic replicon I389-neo/NS3-3′/5.1 were analyzed by epifluorescence microscopy, as described in Materials and Methods, using an anti-NS5A antibody (antibody [Ab] 1877, IgG2A isotype, from AbD Serotec) in conjunction with anti-mouse Alexa Fluor 594-conjugated secondary antibody (red). ER (calnexin) or nuclear (ASH2L, IgG2A isotype) protein markers were detected using an anti-rabbit Alexa Fluor 488-conjugated secondary antibody (green). Nuclei were stained with DAPI (blue). (A) ASH2L expression was localized in all cell nuclei. A proportion of NS5A was shown to colocalize with ASH2L in the nucleus. (B) As shown in the merged image, NS5A mainly colocalized with ER, except for some spots present in the nucleus. Cells harboring the subgenomic replicon I389-neo/NS3-3′/5.1 were analyzed by epifluorescence microscopy after proximity ligation assays (Duolink InSitu). (C) Proximity ligation was performed using a mouse anti-NS5A antibody (OBT 1222) and a rabbit anti-NS5A antibody (29) as positive controls to confirm the expression of NS5A in this cell line. In SGR-harboring cells, the fluorescent dots were mainly localized in the cytoplasm, with a small proportion in the nucleus. (D) Proximity ligation was performed using a mouse anti-NS5A antibody (OBT 1222) and a rabbit anticalnexin antibody. This analysis confirmed the presence of NS5A in the cytoplasm, in close proximity to ER proteins (calnexin). (E) Proximity ligation was performed using a mouse anti-NS5A antibody (OBT 1222) and a rabbit anti-ASH2L antibody. This analysis showed the presence of a proportion of NS5A proteins in close proximity to nuclear proteins in the nucleus. (F) As a negative control, proximity ligation was performed using PLA probes only. Nuclei were visualized using To-Pro-3 staining.
HCV RNA replication efficiency in vitro is dependent upon nuclear translocation of NS5A, as a result of cellular caspase activity and NLS-driven nuclear import.
We next assessed the previously documented respective roles of caspase activity and the NLS on NS5A translocation to the nucleus and HCV RNA replication. Mutation of the putative caspase recognition site at position 154 (D154E) (Fig. 8A) completely abolished nuclear translocation of NS5A (Fig. 8B and Fig. 9F). In order to investigate the importance of caspase cleavage of NS5A in HCV replication, cells harboring subgenomic replicons were treated with the pan-caspase inhibitor z-VAD-fmk and replication capacity was analyzed. As shown in Fig. 8C, inhibition of cellular caspase activity substantially reduced, but did not abrogate, replicon replication. To verify that this result was not due to a nonspecific effect of caspase activity inhibition, we analyzed the replicative capacity of an SGR mutated in the putative N-terminal caspase cleavage site (D154E, D154A, or D154S) of NS5A (Fig. 8C). These mutations resulted in the complete loss of subgenomic replicon replication (Fig. 8C).
Fig 8.

Role of caspase cleavage in NS5A nuclear localization and HCV RNA replication. (A) Schematic representation of lentivirus vectors carrying mutated NS5A-v1. The closed triangle indicates mutation of the caspase recognition site at amino acid position 154. LTR, long terminal repeat. (B) Confocal analysis of cells transduced with the mutated NS5A-v1 lentiviruses was carried out as described in the legend to Fig. 4. Scale bar, 10 μm. *, nontransduced cells. The yellow arrow shows where the 3D reconstructed image was cropped to display the cell sections. The yellow pictogram indicates the direction of sight. (C) Effect of caspase cleavage of NS5A on HCV replicon replication. (Upper panel) Schematic representation of three subgenomic replicons encoding NS5A-v1 with a mutated caspase cleavage site. An open triangle denotes mutation of the caspase recognition site at amino acid position 154; a closed triangle indicates the presence of the cell culture adaptive mutation S2204I. (Lower panel) Replication efficiency of the indicated subgenomic replicons was analyzed in the presence or absence of the caspase inhibitor z-VAD-fmk at the indicated concentrations. The data represent the mean ± SEM replication efficiencies obtained from 3 independent experiments carried out in triplicate.
Fig 9.

Epifluorescence analysis of proximity ligation assays. Cells were transduced with the NS5A-v1 lentivirus (A and B) or the mutated NS5A-v1 lentiviruses Lenti-NS5A-v1/NLSmut (C and D) and pLenti-NS5A-v1/D154E (E and F). In panels A, C, and E, proximity ligation was performed using a mouse anti-NS5A antibody (OBT 1222) and a rabbit anti-NS5A antibody (29) as positive controls to confirm the expression of NS5A in this transduced cells. In panels B, D, and F, proximity ligation was performed using a mouse anti-NS5A antibody (OBT 1222) and a rabbit anti-ASH2L antibody. This analysis confirmed the presence of a small proportion of NS5A in cell nuclei and indicated that NS5A translocation to the nucleus is reduced or abolished by impairing NS5A NLS or caspase cleavage.
To assess the role of the NS5A NLS in NS5A nuclear translocation, we invalidated the NLS by site-directed mutagenesis, as previously described (27) (Fig. 10A). After verifying that the constructs expressed similar amounts of NS5A (data not shown), immunofluorescence analyses showed that abolition of the NLS reduced NS5A translocation to the nucleus by 2.5-fold (down to 5.61% ± 0.9%) (Fig. 10B). This result was confirmed by the proximity ligation assay in cells expressing the NLS-mutated NS5A (Fig. 9D). These results indicate that the NLS of NS5A is functional and plays an important role in NS5A translocation to the nucleus; however, passive nuclear translocation of NS5A could also occur in this system.
Fig 10.

Contribution of NS5A nuclear localization signal to HCV RNA replication. (A) Schematic representation of lentivirus vectors carrying mutated NS5A-v1. The open triangle denotes invalidation of the NS5A NLS. (B) Confocal analysis of cells transduced with the mutated NS5A-v1 lentiviruses was carried out as described in the legend to Fig. 4. Scale bar, 10 μm. *, nontransduced cells. The yellow arrow shows where the 3D reconstructed image was cropped to display the cell sections. The yellow pictogram indicates the direction of sight. (C) Schematic representation of subgenomic replicons encoding NS5A-v1 or -v2 containing mutated NLS. An open triangle denotes invalidation of the NS5A NLS, and a closed triangle indicates the presence of the cell culture adaptive mutation S2204I. (D) Western blotting of NS5A expression in cells transfected with NLS-mutated and nonmutated v1 and v2 replicons at 4 and 96 h posttransfection. Actin expression was used as a loading control. Bands corresponding to 56-kDa NS5A or 42-kDa actin are indicated. (E) Replication efficiency of HCV subgenomic replicons carrying NS5A variants encoding mutated NLS was analyzed in the cell lysates 96 h posttransduction. The replication efficiencies were calculated as described in the legend to Fig. 3C. The data are indicative of the mean ± SEM replication efficiencies obtained from 7 independent experiments carried out in triplicate. The data were normalized to the corresponding nonmutated controls (v1 or v2). Statistical significance was analyzed by means of a Mann-Whitney test.
To investigate whether NLS-mediated NS5A nuclear localization is important in HCV RNA replication, we analyzed the impact of NS5A NLS invalidation on replication of the subgenomic replicons. Replicons encoding two different NS5A variants associated with high replication levels (v1 and v2) in which the NLS of NS5A was mutated (Fig. 10C) exhibited reduced NS5A expression after 96 h posttransfection (Fig. 10D, lower panel), despite a similar expression at 4 h (Fig. 10D, upper panel), and a corresponding reduction in replicon replication in vitro (Fig. 10E). These results demonstrate the importance of a functional NS5A NLS for efficient replication of the HCV subgenomic replicon. However, invalidation of the NLS did not completely abolish replication, in line with our findings that a proportion of NS5A translocates to the nucleus in the absence of a functional NLS (Fig. 10B).
Taken together, these results demonstrate that caspase-mediated cleavage of NS5A, its release from the endoplasmic reticulum, and its subsequent localization to the nucleus driven by its NLS are essential for the replication of subgenomic HCV replicons. They are in line with the correlation between HCV replication efficiency and the NS5A transcriptional activation capacities.
Different NS5A transcriptional activation properties are associated with differential expression of host cell genes essential for HCV RNA replication.
To better understand the mechanisms involved in regulation of HCV replication by NS5A transcriptional activation, the expression of 28 cellular genes previously shown to be essential for HCV replication (listed in Table 1) was analyzed in cells harboring replicons carrying an NS5A variant with high transcriptional and replication capacity (v1) and with low transcriptional and replication capacities (v5). When host gene expression of cells harboring SGR-NS5A-v1 or SGR-NS5A-v5 was analyzed by real-time qPCR (RT-qPCR) screening, no significant difference in gene expression profiles was observed (data not shown). Because the large number of untransfected cells in these transient assays could have masked differences, we made use of our lentivirus vectors expressing NS5A-v1 or NS5A-v5 (Fig. 6B), allowing the transduction of a high proportion of cells and thus increasing signal-to-noise ratios. Analysis of host cell gene expression in lentivirus-transduced cells revealed significant differences in the expression of 5 of the 28 genes analyzed. As shown in Fig. 11, the genes coding for interleukin 8 (IL-8), lymphotoxin beta (LTΒ), and SNF-like kinase 2 (NUAK2) were upregulated by Lenti-NS5A-v1, whereas they were either weakly upregulated (IL-8 and NUAK2) or downregulated (LTΒ) by Lenti-NS5A-v5. Expression of β-galactosidase using an identical lentiviral backbone had no impact on the expression of these genes, indicating that our observations were specifically due to the expression of NS5A. In addition, the MAP2K7 and TRAF2 genes were downregulated by Lenti-NS5A-v5, whereas their levels were not significantly changed by Lenti-NS5A-v1. Together, these results suggest that NS5A variants with different transcriptional activation properties differentially regulate transcription of the host cell genes previously shown as being required for viral replication and that this may impact HCV RNA replication.
Fig 11.
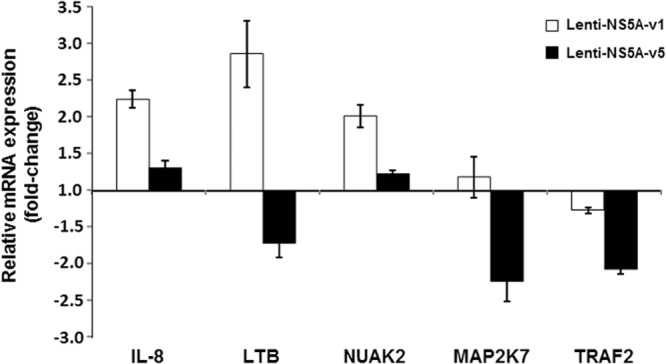
Gene expression analyses of cells expressing NS5A variants. mRNA expression in NG cell cultures transduced with Lenti-NS5A-v1 and Lenti-NS5A-v5. The expression levels of the indicated genes were determined by quantitative real-time PCR from the pooled RNA of three independent experiments. The results were normalized to expression of GAPDH, and the mean ± SEM mRNA quantities are expressed in relation to the values from control cells transduced with Lenti-β-gal.
NS5A is recruited to host cell promoters.
The deregulation of host cell gene expression observed above might be due either to a direct interaction of NS5A with the promoter sequence of these genes or to an indirect regulation involving NS5A protein interactions. To investigate whether nuclear NS5A was capable of binding to the promoters of the genes identified above, we performed chromatin immunoprecipitation assays (ChIP) on nuclear extracts from cells transduced with lentiviruses encoding NS5A-v1 or NS5A-v5. Such experiments revealed that both NS5A-v1 and NS5A-v5 could bind specifically to the promoters of IL-8, LTΒ, and NUAK2 but were absent from the promoters of MAP2K7, TRAF2, and FBXL2 (Fig. 12). The latter gene was included as a negative control, as its expression was unaltered by NS5A in previous experiments (data not shown). These data suggest that the regulation of MAP2K7 and TRAF2 by NS5A occurs via indirect mechanisms, which do not require the presence of NS5A on their respective promoters. Taken together, these results strongly suggest that NS5A directly interacts with the promoters of IL-8, LTΒ, and NUAK2 and that regulation of their expression through its intrinsic transactivation properties is important in modulating HCV replication.
Fig 12.

Chromatin immunoprecipitation analyses of cells expressing NS5A-v1 and -v5. Chromatin was immunoprecipitated using an anti-NS5A antibody from NG cells transduced with lentiviruses expressing NS5A-v1 or NS5A-v5. Immunoprecipitated chromatin and input DNA were amplified using primers spanning the promoters of the indicated genes. The PCR products were analyzed by agarose gel electrophoresis. Cells transduced with Lenti-β-gal were used as a control. The promoter of FBXL2 was amplified as a negative control. Numbers on the left indicate sizes in base pairs of the DNA ladder.
DISCUSSION
We studied the effect of NS5A transcriptional activation on HCV RNA replication using a subgenomic HCV replicon model in an Huh-7-derived cell line (24). In order to avoid the accumulation of compensatory mutations and to allow the use of nonreplicating variants, we decided to use a transient system instead of stably established cell clones. Significantly different replication capacities were observed for replicons bearing NS5A variants with quantitatively different transactivation properties, despite similar levels of NS5A expression. Importantly, we observed a significant correlation between NS5A transcriptional activation and the replication capacity of the corresponding subgenomic HCV replicon, indicating that NS5A transactivation could play a role in modulating viral replication. The role of NS5A transactivation in regulating HCV replication was further supported by the partial rescue of a replication-deficient replicon harboring an NS5A sequence with weak transactivating capacity by the coexpression in trans of an NS5A variant with strong transactivation capacities.
It has been demonstrated that adaptive mutations in the NS5A gene significantly increase the replication capacity of replicons through as yet unknown mechanisms (31, 34). Although none of our NS5A variants carried any of these previously identified substitutions, our results suggest that the mutations carried by these variants played a similar role, mediated by the associated transcriptional activation properties. Since this domain of NS5A is not well structured (35), mutations in this area are unlikely to alter the structure of the protein. As we suggested in our previous work (16) and confirmed here, it is likely that a change in the physical properties of this domain, such as its global charge, is involved in the modulation of its transactivation properties.
The native NS5A protein is retained in the endoplasmic reticulum via a sequence located in the N-terminal domain 1. To be able to directly exert transcriptional activation functions, the NS5A transcriptional activation domain must translocate to the nucleus. Immunofluorescence confocal microscopy, 3D reconstruction analysis, and proximity-dependent DNA ligation assays in cells harboring a stable subgenomic HCV replicon indeed revealed the presence of a subset of NS5A in the nucleus. This was further confirmed by similar analyses of cells transduced with lentiviral vectors expressing NS5A-v1 and NS5A-v5 (i.e., a strong and a weak transactivator, respectively), showing that both variants could translocate to the nucleus regardless of their transactivation potential and that the difference in replication between the two variants was not due to defective nuclear localization.
NS5A carries a small amphipathic α-helix in its N terminus that functions as an ER membrane retention signal (19). To be able to translocate into the nucleus, NS5A must be released from this anchor and transported to the nucleus, possibly with the help of an NLS. Previous studies have demonstrated that NS5A can be cleaved by cellular caspases to produce N-terminally truncated fragments (20, 21). Other reports have suggested that N-terminally truncated forms of NS5A can translocate to the nucleus with the help of a functional NLS located in its C-terminal region (11, 36). We therefore examined the influence of caspase cleavage and NLS-mediated translocation on NS5A relocalization and HCV replication in our model.
We first studied the effects of a pan-caspase inhibitor or a mutation in the putative N-terminal caspase cleavage site on NS5A translocation to the nucleus. Pan-caspase inhibition partially inhibited NS5A relocalization into the nucleus, while mutation of the cleavage site resulted in a complete loss of nuclear NS5A localization, without any alteration of NS5A expression levels. These results strongly suggest that caspase-mediated release of NS5A from its N-terminal ER membrane anchor is indispensable for its translocation into the nucleus. Importantly, mutation of the conserved caspase cleavage site of NS5A, which prevented NS5A nuclear localization, also resulted in a complete loss of replication of the corresponding subgenomic replicons. These results were different from the recent observation of reduced JFH1 replication in Huh-7.5 cells overexpressing the N-terminally truncated NS5A (22). This difference could be due to the different amounts of nuclear NS5A in the two systems. Indeed, as previously published, we observed that all NS5A relocalized within the nucleus when expressed as a caspase-cleaved truncated form and this resulted in high levels of cell death (data not shown). In contrast, only 10 to 15% of nuclear NS5A is observed when the full-length NS5A is expressed. Furthermore, Sauter et al. did not characterize the transactivation potential of the NS5A protein in the JFH1 model.
Second, we assessed the role of the NS5A NLS and observed that its invalidation was associated with a dramatic reduction in the quantity of NS5A in the nucleus. However, a small proportion of NS5A was still detectable in the nucleus in the absence of a functional NLS, probably as a result of passive diffusion. Indeed, proteins up to 60 kDa have been shown to be able to passively diffuse through the nuclear pores, even in the absence of active nuclear import (37–39). Altogether, our results suggest that, although it is not absolutely required, the NLS of NS5A plays an important role in its nuclear translocation. In parallel, we found a significant reduction in the replication capacity of replicons in which the NS5A NLS was invalidated, highlighting the importance of a functional NS5A NLS for HCV RNA replication. The replication capacity of these modified replicons was not completely abolished, a finding in line with our observations that a subset of NS5A could still translocate to the nucleus by passive diffusion in the absence of a functional NLS. Therefore, caspase cleavage and nuclear translocation by means of the NLS are required for NS5A to exert its transcriptional activation properties and regulate HCV replication.
NS5A has been shown to modulate cellular responses to HCV infection through multiple, complex interactions with proteins involved in host cell signaling pathways related to interferon response, cell cycle, and apoptosis (40–42). Based on our findings, we hypothesized that direct regulation of host gene transcription by NS5A is a mechanism employed by HCV to regulate its replication. We analyzed the expression of a subset of cellular genes whose expression has been previously described to be required for efficient HCV RNA replication (43–51). Our results indicate that NS5A proteins with different transcriptional activation properties can differentially regulate the expression of several of these genes, including those coding for IL-8, LTB, and NUAK2, through a direct interaction with the promoters of these cellular genes.
Our observation of a modulation of IL-8 expression via NS5A transactivation is in keeping with previously published results (52). In addition, JFH1 infection was shown to be associated with enhanced expression of this proinflammatory cytokine (53, 54), and high levels of IL-8 have been reported in chronically infected patients (55). HCV RNA synthesis in replicon-harboring cells was shown to be inhibited by IL-8 small interfering RNA (siRNA) knockdown, whereas IL-8 protein levels correlated positively with HCV RNA levels in subgenomic and genomic replicon lines (54). However, IL-8 could have opposing antiviral and proviral effects, depending on the level of HCV replication, the cellular context, and whether the infection is acute or chronic (54). HCV infection triggers double-stranded RNA (dsRNA) signaling pathways that induce IL-8 expression via transcriptional activation, and mutations of the interferon-stimulated response element (ISRE)- and NF-κB-binding sites of the IL-8 promoter reduced and abrogated IL-8 transcription (56). Overall, the cross-regulation of IL-8 gene transcription and HCV replication could involve multiple pathways, including a direct transcriptional activation effect of NS5A, leading to the inhibition of IFN antiviral actions and the resulting enhancement of HCV replication.
Knockdown of the membrane protein LTB and of the NUAK2 kinase in Huh-7-derived cells containing a subgenomic replicon identical to the one used in our study correlated with a 60 to 80% decrease in HCV replication (46). Our data suggest that NS5A directly activates the transcription of both genes. LTB is a member of the tumor necrosis factor (TNF) superfamily which controls cell survival. Like IL-8, its signaling pathway leads to the activation of NF-κB. In contrast, little is known about the link between NUAK2 and HCV replication, although it is interesting to note that NUAK2 could be an NF-κB-regulated antiapoptotic gene (57). Interestingly, HCV infection has been described to increase the hepatic expression of LTΒ (58), the sustained expression of which has been shown to represent an important pathway in HCV-induced primary liver tumors (59).
Although we found that NS5A-v5, our weakest transactivator, decreased the abundance of TRAF2 mRNA, we observed no direct interaction between NS5A and the TRAF2 promoter. It has been reported that TRAF2 and NS5A directly interact and that this interaction results in an inhibition of NF-κB activation (60). Taken together, it is likely that NS5A modulates TNF receptor signaling through a direct interaction with its signaling complex rather than through repression of TRAF2 gene expression.
In conclusion, we observed that the ability of the HCV nonstructural protein NS5A to transactivate was significantly related to the replication efficiency of HCV replicons, while both properties depended upon efficient caspase-mediated release of NS5A and its subsequent NLS-mediated translocation to the nucleus. We also provided compelling evidence that NS5A binds directly to cell promoters involved in HCV replication regulation and modulates their activity. Altogether, these data support a new mechanism by which a nonstructural HCV protein can alter its cellular environment to modulate viral replication. Given the likelihood of a cellular compartmentalization of HCV quasispecies variants (16), this could be an essential mechanism by which HCV quasispecies regulate their survival at the body level.
ACKNOWLEDGMENTS
We thank Rakesh L. Tripathi and Akhteruzzaman Molla from Abbott Laboratories for kindly providing us with the p1071 plasmid and for comments on the manuscript. We thank Mark Harris from the University of Leeds (United Kingdom) and Eberhardt Hildt from the Robert-Koch-Institute, Berlin, Germany, for the gift of the sheep and rabbit anti-NS5A antibodies, respectively.
This study was supported by the National Agency for Research on AIDS and Viral Hepatitis (ANRS). M.A.M. was the recipient of a fellowship from the Higher Education Commission of Pakistan. M.R.I. is the recipient of a fellowship from the French Ministry of Research; M.R.H. was the recipient of a postdoctoral fellowship from the ANRS. H.L. was supported by a grant from ANRS/Fondation de France, by Institut National de la Santé et de la Recherche Médicale (Young Investigator Program), and by a grant from the European Community 6th Framework Program (Marie Curie International Reintegration Grant).
Footnotes
Published ahead of print 6 March 2013
REFERENCES
- 1. Lavanchy D. 2011. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17:107–115 [DOI] [PubMed] [Google Scholar]
- 2. Di Bisceglie AM. 1998. Hepatitis C. Lancet 351:351–355 [DOI] [PubMed] [Google Scholar]
- 3. Gordon SC, Bayati N, Silverman AL. 1998. Clinical outcome of hepatitis C as a function of mode of transmission. Hepatology 28:562–567 [DOI] [PubMed] [Google Scholar]
- 4. Aizawa Y, Shibamoto Y, Takagi I, Zeniya M, Toda G. 2000. Analysis of factors affecting the appearance of hepatocellular carcinoma in patients with chronic hepatitis C. Cancer 89:53–59 [PubMed] [Google Scholar]
- 5. Kiyosawa K, Umemura T, Ichijo T, Matsumoto A, Yoshizawa K, Gad A, Tanaka E. 2004. Hepatocellular carcinoma: recent trends in Japan. Gastroenterology 127:S17–S26 [DOI] [PubMed] [Google Scholar]
- 6. Mottola G, Cardinali G, Ceccacci A, Trozzi C, Bartholomew L, Torrisi MR, Pedrazzini E, Bonatti S, Migliaccio G. 2002. Hepatitis C virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology 293:31–43 [DOI] [PubMed] [Google Scholar]
- 7. Waris G, Sarker S, Siddiqui A. 2004. Two-step affinity purification of the hepatitis C virus ribonucleoprotein complex. RNA 10:321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Le Guillou-Guillemette H, Vallet S, Gaudy-Graffin C, Payan C, Pivert A, Goudeau A, Lunel-Fabiani F. 2007. Genetic diversity of the hepatitis C virus: impact and issues in the antiviral therapy. World J. Gastroenterol. 13:2416–2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Macdonald A, Harris M. 2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol. 85:2485–2502 [DOI] [PubMed] [Google Scholar]
- 10. Chung KM, Song OK, Jang SK. 1997. Hepatitis C virus nonstructural protein 5A contains potential transcriptional activator domains. Mol. Cells 7:661–667 [PubMed] [Google Scholar]
- 11. Kato N, Lan KH, Ono-Nita SK, Shiratori Y, Omata M. 1997. Hepatitis C virus nonstructural region 5A protein is a potent transcriptional activator. J. Virol. 71:8856–8859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi L, Zhang SL, Li K, Hong Y, Wang Q, Li Y, Guo J, Fan W-H, Zhang L, Cheng J. 2008. NS5ATP9, a gene up-regulated by HCV NS5A protein. Cancer Lett. 259:192–197 [DOI] [PubMed] [Google Scholar]
- 13. Tanimoto A, Ide Y, Arima N, Sasaguri Y, Padmanabhan R. 1997. The amino terminal deletion mutants of hepatitis C virus nonstructural protein NS5A function as transcriptional activators in yeast. Biochem. Biophys. Res. Commun. 236:360–364 [DOI] [PubMed] [Google Scholar]
- 14. Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, Gomez J. 1992. Hepatitis-C virus (HCV) circulates as a population of different but closely related genomes—quasi-species nature of HCV genome distribution. J. Virol. 66:3225–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weiner AJ, Brauer MJ, Rosenblatt J, Richman KH, Tung J, Crawford K, Bonino F, Saracco G, Choo QL, Houghton M, Han JH. 1991. Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology 180:842–848 [DOI] [PubMed] [Google Scholar]
- 16. Pellerin M, Lopez-Aguirre Y, Penin F, Dhumeaux D, Pawlotsky J-M. 2004. Hepatitis C virus quasispecies variability modulates nonstructural protein 5A transcriptional activation, pointing to cellular compartmentalization of virus-host interactions. J. Virol. 78:4617–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ide Y, Zhang L, Chen M, Inchauspe G, Bahl C, Sasaguri Y, Padmanabhan R. 1996. Characterization of the nuclear localization signal and subcellular distribution of hepatitis C virus nonstructural protein NS5A. Gene 182:203–211 [DOI] [PubMed] [Google Scholar]
- 18. Kalamvoki M, Mavromara P. 2004. Calcium-dependent calpain proteases are implicated in processing of the hepatitis C virus NS5A protein. J. Virol. 78:11865–11878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brass V, Bieck E, Montserret R, Wolk B, Hellings JA, Blum HE, Penin F, Moradpour D. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277:8130–8139 [DOI] [PubMed] [Google Scholar]
- 20. Goh P-Y, Tan Y-J, Lim SP, Lim SG, Tan YH, Hong WJ. 2001. The hepatitis C virus core protein interacts with NS5A and activates its caspase-mediated proteolytic cleavage. Virology 290:224–236 [DOI] [PubMed] [Google Scholar]
- 21. Satoh S, Hirota M, Noguchi T, Hijikata M, Handa H, Shimotohno K. 2000. Cleavage of hepatitis C virus nonstructural protein 5A by a caspase-like protease(s) in mammalian cells. Virology 270:476–487 [DOI] [PubMed] [Google Scholar]
- 22. Sauter D, Himmelsbach K, Kriegs M, Carvajal Yepes M, Hildt E. 2009. Localization determines function: N-terminally truncated NS5A fragments accumulate in the nucleus and impair HCV replication. J. Hepatol. 50:861–871 [DOI] [PubMed] [Google Scholar]
- 23. Lu L, Pilot-Matias TJ, Stewart KD, Randolph JT, Pithawalla R, He W, Huang PP, Klein LL, Mo H, Molla A. 2004. Mutations conferring resistance to a potent hepatitis C virus serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 48:2260–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tripathi RL, Krishnan P, He Y, Middleton T, Pilot-Matias T, Chen C-M, Lau DTY, Lemon SM, Mo H, Kati W, Molla A. 2007. Replication efficiency of chimeric replicon containing NS5A-5B genes derived from HCV-infected patient sera. Antiviral Res. 73:40–49 [DOI] [PubMed] [Google Scholar]
- 25. Yi M, Bodola F, Lemon SM. 2002. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology 304:197–210 [DOI] [PubMed] [Google Scholar]
- 26. Deminie CA, Emerman M. 1993. Incorporation of human immunodeficiency virus type 1 Gag proteins into murine leukemia virus virions. J. Virol. 67:6499–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yeh C-T, Chang M-H, Shyu W-C, Chang M-L, Yang P-Y, Tsao M-L, Lai H-Y. 2005. Characterization of a HCV NS5A protein derived from a patient with hepatoma. Biochem. Biophys. Res. Commun. 327:516–522 [DOI] [PubMed] [Google Scholar]
- 28. Kalamvoki M, Georgopoulou U, Mavromara P. 2006. The NS5A protein of the hepatitis C virus genotype 1a is cleaved by caspases to produce C-terminal-truncated forms of the protein that reside mainly in the cytosol. J. Biol. Chem. 281:13449–13462 [DOI] [PubMed] [Google Scholar]
- 29. Bürckstümmer T, Kriegs M, Lupberger J, Pauli EK, Schmittel S, Hildt E. 2006. Raf-1 kinase associates with hepatitis C virus NS5A and regulates viral replication. FEBS Lett. 580:575–580 [DOI] [PubMed] [Google Scholar]
- 30. Ahmed-Belkacem A, Ahnou N, Barbotte L, Wychowski C, Pallier C, Brillet R, Pohl RT, Pawlotsky JM. 2010. Silibinin and related compounds are direct inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Gastroenterology 138:1112–1122 [DOI] [PubMed] [Google Scholar]
- 31. Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79:3187–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. 2002. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 20:473–477 [DOI] [PubMed] [Google Scholar]
- 33. Lee JH, Tate CM, You JS, Skalnik DG. 2007. Identification and characterization of the human Set1B histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 282:13419–13428 [DOI] [PubMed] [Google Scholar]
- 34. Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974 [DOI] [PubMed] [Google Scholar]
- 35. Hanoulle X, Badillo A, Verdegem D, Penin F, Lippens G. 2010. The domain 2 of the HCV NS5A protein is intrinsically unstructured. Protein Pept. Lett. 17:1012–1018 [DOI] [PubMed] [Google Scholar]
- 36. Song J, Nagano-Fujii M, Wang F, Florese R, Fujita T, Ishido S, Hotta H. 2000. Nuclear localization and intramolecular cleavage of N-terminally deleted NS5A protein of hepatitis C virus. Virus Res. 69:109–117 [DOI] [PubMed] [Google Scholar]
- 37. Nigg EA. 1997. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature 386:779–787 [DOI] [PubMed] [Google Scholar]
- 38. Silver PA. 1991. How proteins enter the nucleus. Cell 64:489–497 [DOI] [PubMed] [Google Scholar]
- 39. Wang R, Brattain MG. 2007. The maximal size of protein to diffuse through the nuclear pore is larger than 60kDa. FEBS Lett. 581:3164–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arima N, Kao C-Y, Licht T, Padmanabhan R, Sasaguri Y, Padmanabhan R. 2001. Modulation of cell growth by the hepatitis C virus nonstructural protein NS5A. J. Biol. Chem. 276:12675–12684 [DOI] [PubMed] [Google Scholar]
- 41. Gale M, Jr, Kwieciszewski B, Dossett M, Nakao H, Katze MG. 1999. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol. 73:6506–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gale MJ, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. 1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230:217–227 [DOI] [PubMed] [Google Scholar]
- 43. Borawski J, Troke P, Puyang XL, Gibaja V, Zhao SC, Mickanin C, Leighton-Davies J, Wilson CJ, Myer V, CornellaTaracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither LA. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83:10058–10074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78:3480–3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79:13473–13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ng TI, Mo H, Pilot-Matias T, He Y, Koev G, Krishnan P, Mondal R, Pithawalla R, He W, Dekhtyar T, Packer J, Schurdak M, Molla A. 2007. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology 45:1413–1421 [DOI] [PubMed] [Google Scholar]
- 47. Randall G, Panis M, Cooper JD, Tellinghuisen TL, Sukhodolets KE, Pfeffer S, Landthaler M, Landgraf P, Kan S, Lindenbach BD, Chien M, Weir DB, Russo JJ, Ju J, Brownstein MJ, Sheridan R, Sander C, Zavolan M, Tuschl T, Rice CM. 2007. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. U. S. A. 104:12884–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Supekova L, Supek F, Lee J, Chen S, Gray N, Pezacki JP, Schlapbach A, Schultz PG. 2008. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 283:29–36 [DOI] [PubMed] [Google Scholar]
- 49. Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, Cordingley MG, Kukolj G. 2009. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387:5–10 [DOI] [PubMed] [Google Scholar]
- 51. Wang CF, Gale M, Keller BC, Huang H, Brown MS, Goldstein JL, Ye J. 2005. Identification of FBL2 as a geranylgeranylated required for hepatitis C cellular protein virus RNA replication. Mol. Cell 18:425–434 [DOI] [PubMed] [Google Scholar]
- 52. Polyak SJ, Khabar KSA, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. 2001. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J. Virol. 75:6095–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blackham S, Baillie A, Al-Hababi F, Remlinger K, You S, Hamatake R, McGarvey MJ. 2010. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J. Virol. 84:5404–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koo BC, McPoland P, Wagoner JP, Kane OJ, Lohmann V, Polyak SJ. 2006. Relationships between hepatitis C virus replication and CXCL-8 production in vitro. J. Virol. 80:7885–7893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Polyak SJ, Khabar KSA, Rezeiq M, Gretch DR. 2001. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J. Virol. 75:6209–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wagoner J, Austin M, Green J, Imaizumi T, Casola A, Brasier A, Khabar KS, Wakita T, Gale M, Jr, Polyak SJ. 2007. Regulation of CXCL-8 (interleukin-8) induction by double-stranded RNA signaling pathways during hepatitis C virus infection. J. Virol. 81:309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Legembre P, Schickel R, Barnhart BC, Peter ME. 2004. Identification of SNF1/AMP kinase-related kinase as an NF-kappaB-regulated anti-apoptotic kinase involved in CD95-induced motility and invasiveness. J. Biol. Chem. 279:46742–46747 [DOI] [PubMed] [Google Scholar]
- 58. Lowes KN, Croager EJ, Abraham LJ, Olynyk JK, Yeoh GC. 2003. Upregulation of lymphotoxin beta expression in liver progenitor (oval) cells in chronic hepatitis C. Gut 52:1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haybaeck J, Zeller N, Wolf MJ, Weber A, Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien PA, Thimme R, Blum H, Nedospasov SA, Zatloukal K, Ramzan M, Ciesek S, Pietschmann T, Marche PN, Karin M, Kopf M, Browning JL, Aguzzi A, Heikenwalder M. 2009. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell 16:295–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Park KJ, Choi SH, Lee SY, Hwang SB, Lai MM. 2002. Nonstructural 5A protein of hepatitis C virus modulates tumor necrosis factor alpha-stimulated nuclear factor kappa B activation. J. Biol. Chem. 277:13122–13128 [DOI] [PubMed] [Google Scholar]
- 61. Appel N, Herian U, Bartenschlager R. 2005. Efficient rescue of hepatitis C virus RNA replication by trans-complementation with nonstructural protein 5A. J. Virol. 79:896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Webster N, Jin JR, Green S, Hollis M, Chambon P. 1988. The yeast UASG is a transcriptional enhancer in human HeLa cells in the presence of the GAL4 trans-activator. Cell 52:169–178 [DOI] [PubMed] [Google Scholar]