

Abstract
All characterized whitefly-transmitted geminiviruses (begomoviruses) with origins in the New World (NW) have bipartite genomes composed of a DNA-A and DNA-B component. Recently, an NW begomovirus lacking a DNA-B component was associated with tomato leaf curl disease (ToLCD) in Peru, and it was named Tomato leaf deformation virus (ToLDeV). Here, we show that isolates of ToLDeV associated with ToLCD in Ecuador and Peru have a single, genetically diverse genomic DNA that is most closely related to DNA-A components of NW bipartite begomoviruses. Agroinoculation of multimeric clones of the genomic DNA of three ToLDeV genotypes (two variants and a strain) resulted in the development of tomato leaf curl symptoms indistinguishable from those of ToLCD in Ecuador and Peru. Biological properties of these ToLDeV genotypes were similar to those of Old World (OW) monopartite tomato-infecting begomoviruses, including lack of sap transmissibility, phloem limitation, a resistance phenotype in tomato germplasm with the Ty-1 gene, and functional properties of the V1 (capsid protein) and C4 genes. Differences in symptom phenotypes induced by the ToLDeV genotypes in tomato and Nicotiana benthamiana plants were associated with a highly divergent left intergenic region and C4 gene. Together, these results establish that ToLDeV is an emergent NW monopartite begomovirus that is causing ToLCD in Ecuador and Peru. This is the first report of an indigenous NW monopartite begomovirus, and evidence is presented that it emerged from the DNA-A component of a NW bipartite progenitor via convergent evolution and recombination.
INTRODUCTION
The family Geminiviridae is a large and diverse group of plant-pathogenic viruses that possess a small circular single-stranded DNA (ssDNA) genome encapsidated within characteristic twinned, quasi-isometric virions (1–4). Four genera of geminiviruses are currently recognized, based upon genome structure, host range, and type of insect vector (5). The genus Begomovirus contains viruses transmitted by the whitefly Bemisia tabaci, and many of these viruses cause economically important diseases of dicotyledonous crop plants in tropical and subtropical regions throughout the world (6–8). Begomoviruses have the largest number of species in the family (and probably of any type of virus), and many have been described within the past approximately 20 years (5). This makes begomoviruses a remarkably successful group of emerging viruses (1, 7), and this can be attributed to a number of factors. First, indigenous begomoviruses are widely distributed in noncultivated plants (e.g., weeds), and these serve as progenitors of crop-infecting viruses. Second, population explosions and the expanded geographical range of the invasive polyphagous B biotype of B. tabaci have facilitated the introduction of indigenous begomoviruses into crop plants. Third, the begomovirus genome has the capacity to evolve rapidly via mutation, pseudorecombination, recombination, and acquisition of new DNA components and satellites (1, 4, 7–9). Finally, the worldwide cultivation of susceptible crop plants (e.g., cassava, cotton, cucurbits, and tomato), often in monoculture and overlapping plantings, has resulted in the exposure of potential host plants to high populations of whiteflies and a diversity of begomoviruses (8, 10).
There are two types of begomoviruses: those with a monopartite genome (a single genomic DNA of approximately 2.9 kb) and those with a bipartite genome (two DNA components, each approximately 2.6 kb, referred to as DNA-A and DNA-B) (1, 3–5). Monopartite begomoviruses originated in the Old World (OW), and many are associated with satellite DNAs that are either required for development of typical disease symptoms (betasatellites) or have no apparent effect or modulate disease symptoms (alphasatellites) (1). In the absence of a betasatellite, many monopartite begomoviruses can infect and cause symptoms in the experimental host, Nicotiana benthamiana; however, only those that infect tomato cause typical disease symptoms in the natural host without a betasatellite (1). All indigenous New World (NW) begomoviruses have a bipartite genome, as do a relatively small number (approximately 15%) of OW viruses (11). Bipartite begomoviruses do not require satellite DNAs for induction of typical symptoms, but they can be found coinfecting plants (1, 4). Within the past approximately 20 years, the genomic complexity of begomoviruses in the NW has increased with (i) the introduction of the OW monopartite begomovirus Tomato yellow leaf curl virus (TYLCV) (12) and (ii) recent reports of alphasatellites associated with bipartite begomoviruses in Latin America (13, 14). However, monopartite begomoviruses indigenous to the NW have not been reported (1).
The genomic DNA of monopartite begomoviruses and the DNA-A component of bipartite begomoviruses are homologous and have a similar genome organization that includes the V1/AV1, C1/AC1, C2/AC2, C3/AC3, and C4/AC4 genes, which encode the capsid protein (CP), replication-associated protein (Rep), transactivator protein (TrAP), replication enhancer protein (REn), and C4/AC4 proteins, respectively (3, 15). However, there are also differences between these viral DNAs. The genomic DNA of monopartite begomoviruses is larger (approximately 2.9 kb) than the DNA-A components of NW bipartite begomoviruses (approximately 2.6 kb), and it has an additional virion-sense gene (V2), which encodes the precoat protein (note that DNA-A components of OW bipartite begomoviruses have a homologous AV2 gene). The CP of NW bipartite begomoviruses has a distinctive N-terminal PWRsMaGT motif (lowercase letters indicate variable amino acid residues) (16). There are also differences in the function of homologous gene products. The CP of monopartite begomoviruses is required for systemic infection, whereas CP is not required for systemic infection by many bipartite species (4, 17). In monopartite begomoviruses, the C4 is a pathogenicity determinant that can play a role in movement and suppression of host defense responses. In many NW bipartite begomoviruses, the homologous AC4 does not play a role in pathogenicity (4, 18). The evolution of the bipartite genome, by the acquisition of the DNA-B component, probably occurred following the appearance of monopartite species. The DNA-B component has two genes, BV1 and BC1, that encode proteins involved in nucleocytoplasmic transport (nuclear shuttle protein [NSP]) and cell-to-cell movement (movement protein [MP]), respectively (4). The differences between genome structure and gene function of monopartite and bipartite begomoviruses are also reflected in their biological properties. Monopartite begomoviruses are phloem limited, induce stunting and leaf curl symptoms, and are not sap transmissible, whereas many bipartite begomoviruses infect phloem and nonphloem tissues, induce leaf curling, crumpling, and mosaic/mottling symptoms and are sap transmissible.
The cultivated tomato (Solanum lycopersicum) has proven well suited for the emergence and evolution of bipartite and monopartite begomoviruses. Indeed, at least 67 species of tomato-infecting begomoviruses have been described, which is the largest number infecting any crop (5, 19). In the OW, monopartite begomoviruses predominate, although there are a few bipartite species, whereas in the NW only bipartite species have been described (1, 6, 8, 19–21). However, the finding that DNA-A components of some bipartite tomato-infecting begomoviruses are infectious has blurred the distinction between the monopartite and bipartite genomes. For example, when delivered by agroinoculation, the DNA-A component of the OW bipartite begomovirus Tomato yellow leaf curl Thailand virus (TYLCTHV) induces disease symptoms in tomato and N. benthamiana, although not as severe as in the presence of the DNA-B component (22). In nature, the TYLCTHV DNA-A component can be found causing leaf curl disease of tomato in association with the DNA-B component or a betasatellite (22, 23). Thus, TYLCTHV can be viewed as an intermediate between a monopartite and bipartite begomovirus. In contrast, DNA-A components of NW bipartite tomato-infecting begomoviruses are infectious only in the experimental host N. benthamiana (24, 25), and there are no reports of these components infecting tomato, either experimentally or in nature, in the absence of a DNA-B component.
In the mid-1990s, a leaf curl disease of tomatoes in Peru appeared following outbreaks of B. tabaci (6, 26, 27). In 2003, a similar disease was described in Ecuador. Disease symptoms included stunting and distorted growth, upward curling of leaves, and swelling and purpling of veins. A begomovirus was the suspected causal agent, but it was also noted that the symptoms were unusual with respect to those typically induced by NW begomoviruses, in particular, the absence of mottling, mosaicism, and yellowing. The DNA-A component of a putative new begomovirus species was associated with this disease (26, 27), and the name Tomato leaf deformation virus (ToLDeV) was proposed (26). However, Koch's postulates were not fulfilled for ToLCD, nor was a DNA-B component identified.
Here, we show that ToLDeV isolates associated with ToLCD in Ecuador and Peru have a single, genetically divergent genomic DNA that is most closely related to DNA-A components of NW bipartite begomoviruses. The development of leaf curl symptoms in tomato plants, following agroinoculation with the cloned genomic DNA of ToLDeV, fulfill Koch's postulates for this disease and confirmed the monopartite nature of the viral genome. In addition, the biological properties and functional properties of the V1 (CP) and C4 genes are similar to those of OW monopartite tomato-infecting begomoviruses. Together, these results establish that ToLDeV is a bone fide indigenous NW monopartite begomovirus, and evidence is presented that it evolved from the DNA-A component of a bipartite progenitor via convergent evolution and recombination.
MATERIALS AND METHODS
Virus sources, DNA extraction, and detection of begomovirus DNA.
In 1998, leaf samples were collected from tomato plants with stunting and upward leaf curling symptoms associated with a new virus-like disease from two fields (98-1 and 98-2) in the Ica province of Peru. These samples were sent to UC Davis and tested for begomovirus infection by squash blot hybridization analysis (28) and PCR with degenerate begomovirus DNA-A and DNA-B primer pairs (29).
In July 2010, a survey of commercial tomato fields in two provinces of Peru, Ica (8 fields) and Lima (2 fields), was conducted for leaf curl symptoms and whiteflies. In each field, a minimum of 20 plants at 3 to 4 locations were examined for leaf curl symptoms, and representative samples were collected. The presence and relative abundance (high, medium, or low) of adult whiteflies were estimated in each field by examining the underside of at least 10 leaves. Twenty-five samples with leaf curl symptoms were collected, and sap from each sample was prepared and applied onto Agdia absorption strips (Agdia, Elkhart, IN). The strips were dried and then transported to UC Davis. In Ecuador, surveys of commercial tomato fields were conducted in the provinces of Guayas and Santa Helena in August and September 2010. Sixty-eight samples with leaf curl symptoms were collected, dried in a plant press, and stored at room temperature.
Total genomic DNA was extracted from leaf samples collected in Peru in 1998 (frozen tissue) and 2010 (dried plant sap on absorption strips) as described by Dellaporta et al. (30), whereas the method of Doyle and Doyle (31) was used for the samples collected in Ecuador. PCR with the degenerate DNA-A primer pair PAL1v1978 and PAR1c496 was used to detect begomovirus infection (29). PCR-amplified DNA fragments were purified with the QIAquick gel extraction kit (Qiagen, Germantown, MD) and directly sequenced.
Three approaches were used to detect a begomovirus DNA-B component and satellite DNAs. First, PCR with various degenerate DNA-B primer pairs (PCRc1/PBL1v2040, PCRc1/PBL1v2039, PCRc154/PBL1v2040, and PCRc154/PBL1v2039) and the universal primer pair Beta01/Beta02 was performed (29, 32). Second, restriction enzyme digestion analysis of circular DNA molecules generated by rolling circle amplification (RCA) with Φ-29 DNA polymerase (TempliPhi; GE Healthcare, Piscataway, NJ) was used. The third approach was DNA gel blot hybridization analysis of total genomic DNA with the cloned DNA-B component of the bipartite begomovirus Bean dwarf mosaic virus (BDMV) under conditions of low stringency. Controls were total genomic DNA extracted from leaves of uninfected and BDMV-infected N. benthamiana plants.
Obtaining full-length begomovirus DNA clones.
DNA extracts in which begomovirus DNA was detected by PCR were used for RCA as described previously (33). The RCA products were digested with various restriction enzymes to identify sites for obtaining full-length clones. Putative full-length clones were obtained by digesting DNA generated by RCA with restriction enzymes that linearized the viral DNA, purifying the approximately 2.6-kb fragment and cloning into pGEM-11Zf(+) (Promega, Madison, WI). Recombinant plasmids were identified by restriction enzyme digestion.
Sequencing and phylogenetic analyses.
DNA sequencing was conducted with the ABI 3730 DNA analyzer (Applied Biosystems). Sequence alignments were performed with the MUSCLE program in Mega 5 (34). The aligned nucleotide sequences were edited and exported as nexus files. For each data set, phylogenies were constructed with a Bayesian approach performed with MrBayes 3.2 (35). The best-fit model of nucleotide substitution for each data set was determined with the program ModelTest (36). The analyses were carried out by running 2 million generations, and trees were sampled every 100th generation, resulting in 20,000 trees. After discarding the first 10% of samples as burn-in, the remaining 18,000 trees were used for calculating posterior probabilities in the consensus tree. Trees were generated in Treeview (37) and rooted with the curtovirus Beet severe curly top virus (BSCTV).
Detection of potential recombination events was carried out with the recombination detection program (RDP) version 3.0 (38) and various data sets, including one with the complete nucleotide sequences of 180 begomoviruses (5). Similarity plot analysis was performed with SimPlot software 3.5.1 (39), using the Jukes-Cantor model, a window size of 200 nucleotides (nt), and a step size of 20 nt.
Analysis of genetic diversity and selection pressure.
Nucleotide diversity (π) was estimated with Tajima's test (40) by using DnaSP 5.10 (41). Codon-specific positive selection was determined by Bayesian posterior probability analysis under the Ny98 fitness regimen (42) (0<ω1<1, ω2 = 1, and ω3>1), as described and performed within MrBayes 3.2 (35). The distributions of synonymous and nonsynonymous mutations were analyzed with the SNAP program (www.hiv.lanl.gov) (43). The hypothesis of independent evolution in overlapping genes was tested as described by Zaaijer et al. (44). Variation was assessed by determining the entropy values for individual amino acids and nucleotides by using the BioEdit software (45). An alignment of AC4/C4 amino acid sequences was generated with the MUSCLE program in Jalview (46). Jalview was used to edit the alignment and determine conserved regions.
Production of multimeric clones for agroinoculation.
Multimeric clones were generated in pCAMBIA1300 (47) as previously described (48). For the PA98-1-1 and PA98-2-1 clones from samples collected in Peru in 1998, an approximately 1.6-kb EcoRI-HindIII fragment containing the intergenic region (IR) was cloned to generate a 0.6-mer. Full-length monomers were released with HindIII and cloned into HindIII-digested 0.6-mers to generate 1.6-mers. For the PA10-3 clone from the sample collected in Peru in 2010, the same strategy was used, except an approximately 2.4-kb EcoRI-HindIII fragment was cloned to generate a 0.9-mer. A 1.9-mer was generated following ligation with the full-length monomer. Recombinant plasmids with the multimeric clones were identified by restriction enzyme digestion and transformed into competent Agrobacterium tumefaciens cells (strain C58C1) by using the freeze-thaw method (49).
Determination of infectivity, host range, and response of tomato germplasm with the Ty-1 resistance gene.
Infectivity of the multimeric clones was initially assessed by agroinoculation of tomato (cultivar Glamour) and N. benthamiana plants with the needle puncture method (25). Host range determination was performed by agroinoculation of tomato (cultivar Glamour), Datura stramonium, N. benthamiana, N. glutinosa, N. tabacum cv. Samsum, Phaseolus vulgaris cv. Topcrop, Cucurbita pepo cv. Small sugar, Chenopodium quinoa, and C. amaranticolor plants. Infection was determined 18 to 21 days postinoculation (dpi), based upon symptom development and detection of viral DNA by PCR analysis with the PAR1c496 and ToLC-v1730 primer pair, which directs the amplification of an approximately 1.1-kb DNA fragment from these clones.
Tomato germplasm with the Ty-1 begomovirus resistance gene (LA3473 [C. M. Rick Tomato Genetics Resource Center, UC Davis] and cultivar Dominator [Seminis Vegetable Seeds, Woodland, CA]) and without the gene (LA3474 and cultivar Glamour) were agroinoculated as described above. Controls were uninfected plants and plants agroinoculated with the monopartite begomovirus TYLCV-Israel (TYLCV-IL) from the Dominican Republic (TYLCV-IL[DO]) (50) and the bipartite begomovirus Tomato mosaic Havana virus (ToMHV) from Guatemala (T. Kon and R. L. Gilbertson, unpublished data). Inoculated plants were maintained in a controlled environment growth chamber (25°C), and disease symptoms were assessed 21 to 25 dpi.
Assessment of viral DNA levels.
Total genomic DNA was extracted from leaf tissue, and DNA gel blot hybridization analysis was conducted as previously described (50). Newly emerged leaves were collected from infected plants 21 and 60 dpi, total genomic DNA was extracted, and DNA gel blot hybridization analysis was conducted with a probe composed of the recombinant plasmids pPA98-1-1, pPA98-2-1, and pPA10-3 labeled with [α-32P]dCTP by nick translation.
Sap transmission and immunolocalization studies.
Leaves of N. benthamiana plants at the 4- to 6-leaf stage were rub inoculated with sap as previously described (28). Sap was prepared with frozen leaf tissue from the 1998 samples and from symptomatic leaf tissue from N. benthamiana plants agroinoculated with the multimeric PA10-3 clone. Controls were N. benthamiana plants inoculated with sap prepared with symptomatic leaf tissue from common bean plants infected with the sap-transmissible BDMV (positive control) or 0.1 M phosphate buffer (pH 7.2) alone (negative control). Three independent experiments were performed with five plants inoculated per treatment per experiment.
Immunolocalization experiments were conducted with leaf and petiole tissues collected from symptomatic tomato plants (cultivar Glamour) 4 weeks after agroinoculation with the multimeric PA98-1-1 and PA98-2-1 clones. The antibody used for immunolocalization was raised against the Escherichia coli-expressed CP of the bipartite begomovirus Chino del tomate virus (51). Controls were equivalent tissues from uninfected plants probed with the CP antiserum and from infected plants probed with preimmune antiserum. Tissue preparation, sectioning, and immunolocalization were performed as previously described (52). Sections were visualized with a phase-contrast light microscope (DM5000 B; Leica). Images were recorded with the Leica Application Suite (LAS) 3.6 software.
Mutational analysis.
Mutations were introduced into the V1 (CP), C1 (Rep), C4, and C5 genes of the PA98-1-1, PA98-2-1, and PA10-3 clones. The recombinant plasmids pPA98-1-1.6, pPA98-2-1.6, and pPA10-3-1.9 were used as the templates for site-directed mutagenesis with phosphorylated primers (primers sequences are available upon request). The mutations in the V1 and C1 genes were single-nucleotide deletions in the start codons (V1, ATG→A-G at nt 176 or 177 [PA10-3], and C1, CAT→C-T at nt 2456 or 2458 [PA10-3]). The mutations in the C4 and C5 genes were single-nucloetide changes that abolished the start codons (C4, CAT→CGT at nt 2300 or 2301 [PA10-3]; C5, CAT→CAG at nt 824 [PA98-1-1], 825 [PA98-2-1], and 826 [PA10-3]), but were silent in the overlapping C1 and V1 genes, respectively (Fig. 1). The PCR was performed according to the protocol of the Phusion site-directed mutagenesis kit (Finnzymes, Espoo, Finland). Following mutagenesis, the PCR-amplified DNA components were self-ligated, digested with HindIII, and ligated into HindIII-digested pGEM-11Zf(+). Recombinant plasmids were recovered, and the presence of the introduced mutation (as well as the lack of any other introduced mutations) was verified by sequencing. Monomers confirmed to have the mutations were used to generate multimeric clones for agroinoculation as described elsewhere.
Fig 1.
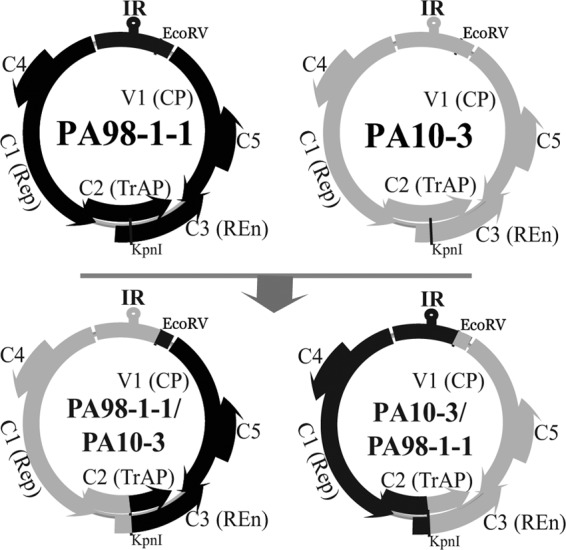
Organization of the genomic DNA of the PA98-1-1 (black) and PA10-3 (gray) isolates of ToLDeV cloned from plants with symptoms of tomato leaf curl disease collected in Peru in 1998 and 2010, respectively. The IR and viral genes, including V1, C1, C2, and C3, predicted to express the CP, Rep, transactivator (TrAP), and replication enhancer (REn) proteins, respectively, are shown. Chimeras generated between the genomic DNAs of these isolates by exchanging an approximately 1.5-kb EcoRV/KpnI fragment with the IR and C4 and C1 genes are shown beneath the line.
Replication was assessed in N. benthamiana leaf discs as previously described (50). Infectivity and symptomatology of mutants in agroinoculated tomato (cultivar Glamour) and N. benthamiana plants were assessed as described previously. The presence of the introduced mutation was assessed for c4 and c5 mutants in selected tomato and N. benthamiana plants by sequencing PCR-amplified DNA fragments with the mutated region of the gene. The stability of the c4 mutants was assessed by grafting uninfected tomato scions onto infected root stocks. Three weeks after grafting, the scions were assessed for symptoms, and the presence of the mutation was determined as described elsewhere.
Construction of chimeras.
Chimeras were generated between the PA98-1-1 and PA10-3 clones in the recombinant plasmids pPA98-1-1 and pPA10-3, respectively. The approximately 1.5-kb KpnI/EcoRV fragments with the left intergenic region (LIR) and C1 and C4 genes were exchanged (Fig. 1). This resulted in the generation of the chimeras pPA98-1-1/PA10-3 and pPA10-3/PA98-1-1 (the second name indicates the source of the approximately 1.5-kb fragment).
Nucleotide sequence accession numbers.
The sequences determined in our analyses for these ToLDeV isolates were deposited with GenBank and assigned accession numbers (in parentheses) as follows: EA10-LE3-5K (JX501510), PA98-1-1 (JX501502), PA98-2-1 (JX501503), PA98-2-TM8 (JX501507), PA98-2-TM13 (JX501508), PA98-2-TM5 (JX501509), PA10-3 (JX501504), PA10-S8 (JX501505), and PA10-S9 (JX501506).
RESULTS
ToLDeV is associated with ToLCD in Peru and Ecuador.
The survey of commercial tomato fields conducted in Peru in July 2010 revealed ToLCD incidences of 20% and 5% in the Ica and Lima provinces, respectively. Whitefly populations in these fields were low, with <10% of plants having adult insects. Surveys conducted in Ecuador in August and September 2010 revealed disease incidences of 5 to 10% and 20% in the Guayas and Santa Helena provinces, respectively, and low whitefly populations. The relatively low incidence of ToLCD and whiteflies was attributed to these surveys being conducted outside the main tomato-growing season (October to April).
The degenerate DNA-A primer pair PAL1v1978/PAR1c496 directed the amplification of the expected-size 1.1-kb fragment from the two samples collected from the Ica province in 1998, consistent with previous results from 1998 (data not shown). The 1.1-kb fragment was also amplified from 13 of 20 and 1 of 5 samples collected from the Ica and Lima provinces in 2010, respectively, and from 44 of 68 samples collected from Ecuador in 2010. Comparisons performed with sequences of representative 1.1-kb DNA fragments from these samples revealed highest identities with sequences of DNA-A components of NW bipartite begomoviruses (data not shown). Thus, many of the tomato plants with leaf curl symptoms from Ecuador and Peru were infected with a begomovirus, but the finding that some samples were not infected showed that these symptoms can be caused by other factors (53).
Several lines of evidence indicated that a DNA-B component and satellite DNAs were not present in the ToLCD samples from Ecuador and Peru. First, in PCR assays performed with degenerate DNA-B primer pairs and a universal betasatellite primer pair, no DNA fragments were amplified, whereas the expected-size fragments were amplified from the controls. Second, restriction enzyme digestion of DNA(s) generated by RCA from samples collected in Peru in 1998 and 2010 and Ecuador in 2010 revealed one or more fragments totaling approximately 2.6 kb, indicating a single begomovirus genomic DNA. For the 44 begomovirus-positive samples from Ecuador, digestion of RCA DNAs with the 4-base-cutting enzyme MspI generated the same pattern of fragments totaling approximately 2.6 kb, whereas fragments generated from an equivalent analysis of a tomato sample infected with a bipartite begomovirus from Brazil added up to approximately 5.2 kb (data not shown). Finally, no DNA-B component was detected in DNA gel blot hybridization analyses of total genomic DNA from the ToLCD samples collected in Peru in 1998, whereas the expected hybridization signal was detected in total genomic DNA from N. benthamiana leaves infected with the bipartite begomovirus BDMV (data not shown). Together, these results indicated that ToLCD in Ecuador and Peru is associated with a single begomovirus genomic DNA, and our results are in agreement with the results of Marquez-Martin et al. (26).
A single HindIII site, identified in the begomovirus genomic DNA in the ToLCD samples from Peru, was used to obtain five putative full-length clones from the two samples collected in Ica in 1998 (one from the 98-1 sample [PA98-1-1] and four from the 98-2 sample [PA98-2-1, -TM5, -TM8, and -TM13]) and three clones each from a different sample collected in the same location in 2010 (PA10-3, -S8, and -S9). A similar strategy was used to obtain 28 putative full-length clones from the Ecuador samples. The complete nucleotide sequences of these clones were determined, and comparisons revealed identities ranging from 92% to 100% with the DNA-A component of ToLDeV (ToLDeV [PE:03], accession number GQ334472). These identities are above the threshold for defining distinct begomovirus species (89%) and within the range for strains (<94%) and variants (>94%) of a species (5). Thus, these are a collection of genetically diverse ToLDeV clones.
Relationships among ToLDeV isolates associated with ToLCD in Ecuador and Peru and other begomoviruses.
Comparisons performed with the complete nucleotide sequence of the PA98-1-1 clone from Peru revealed identities of 98 to 99% with sequences of ToLDeV (PE:03) and all 28 clones from Ecuador, 95% with sequences of the four PA98-2 clones, and 92% with those of the three PA10 clones (Table 1). Based upon the 94% threshold value for begomovirus strains, the PA98-1-1 clone, all 28 clones from Ecuador, and ToLDeV (PE:03) are isolates of a ToLDeV variant (named the PA98-1 variant); the four PA98-2 clones (with 99% identities) are isolates of another variant (named the PA98-2 variant); the three clones from the 2010 samples (with 99% identities) are isolates of a strain (named the PA10 strain). Collectively, the variants and strain are referred to as the ToLDeV genotypes. Because of the high degree of nucleotide sequence identity among the 28 isolates from Ecuador, the sequence of a representative isolate (EA10-LE3-5K) was used in subsequent analyses.
Table 1.
Nucleotide identities for total, IR, LIR, and RIR sequences and amino acid identities and similarities for genes of ToLDeV isolates from Ecuador and Peru and those of the most closely related begomovirusesa
| ToLDeV isolate or begomovirus species | Yr collected | Geographic locationb | % identity (% similarity for amino acids) for region |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total nt | IR/CR nt | LIR nt | RIR nt | V1 |
C1 |
C2 |
C3 |
C4 |
C5 |
|||||||||
| nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | nt | aa | |||||||
| PA98-1-1 | 1998 | PE | ||||||||||||||||
| ToLDeV [PE:03] | 2003 | PE | 98 | 97 | 98 | 97 | 98 | 99 (99) | 99 | 99 (99) | 98 | 96 (96) | 97 | 96 (97) | 99 | 99 (99) | 98 | 94 (97) |
| EA10-LE3-5K | 2010 | EC | 99 | 99 | 99 | 99 | 99 | 100 (100) | 99 | 99 (99) | 98 | 95 (96) | 99 | 98 (98) | 100 | 99 (99) | 99 | 98 (99) |
| PA98-2-1 | 1998 | PE | 95 | 92 | 85 | 98 | 98 | 98 (99) | 93 | 93 (96) | 96 | 94 (95) | 97 | 96 (98) | 91 | 86 (87) | 98 | 97 (98) |
| PA98-2-TM5 | 1998 | PE | 95 | 92 | 85 | 98 | 98 | 98 (99) | 93 | 93 (96) | 97 | 95 (95) | 97 | 97 (98) | 91 | 86 (87) | 98 | 97 (98) |
| PA98-2-TM8 | 1998 | PE | 95 | 92 | 85 | 97 | 98 | 98 (99) | 93 | 93 (95) | 97 | 95 (95) | 97 | 97 (98) | 91 | 86 (87) | 98 | 97 (98) |
| PA98-2-TM13 | 1998 | PE | 95 | 92 | 85 | 98 | 98 | 98 (99) | 93 | 93 (96) | 96 | 95 (95) | 97 | 96 (97) | 91 | 86 (87) | 98 | 97 (98) |
| PA10-3 | 2010 | PE | 92 | 84 | 65 | 98 | 98 | 99 (100) | 88 | 87 (92) | 98 | 95 (96) | 97 | 96 (97) | 86 | 78 (84) | 98 | 95 (98) |
| PA10-S8 | 2010 | PE | 92 | 84 | 66 | 97 | 98 | 99 (99) | 88 | 86 (91) | 98 | 95 (96) | 98 | 98 (98) | 86 | 78 (84) | 98 | 95 (98) |
| PA10-S9 | 2010 | PE | 92 | 84 | 66 | 98 | 98 | 99 (100) | 88 | 86 (91) | 98 | 95 (96) | 97 | 97 (98) | 86 | 78 (84) | 98 | 95 (98) |
| SbBMV [NOA] | ND | AR | 79 | 73 | ND | ND | 84 | 96 (91) | 82 | 76 (82) | 78 | 67 (74) | 84 | 81 (86) | 84 | 73 (81) | ND | ND |
| ToChLPV [MM1] | ND | MX | 78 | 66 | ND | ND | 78 | 83 (92) | 82 | 82 (88) | 83 | 73 (81) | 86 | 80 (88) | 87 | 78 (86) | ND | ND |
| ToRMV [Ube] | ND | BR | 78 | 70 | ND | ND | 84 | 90 (94) | 79 | 79 (88) | 75 | 67 (74) | 81 | 73 (88) | 84 | 70 (77) | ND | ND |
| ToMMV [Pda58] | 2005 | BR | 74 | 66 | ND | ND | 80 | 88 (94) | 79 | 80 (87) | 81 | 73 (80) | 84 | 79 (86) | 87 | 72 (89) | ND | ND |
| TGMV [YV] | ND | BR | 77 | 64 | ND | ND | 85 | 92 (95) | 79 | 79 (87) | 74 | 61 (72) | 78 | 75 (85) | 82 | 65 (74) | ND | ND |
| TYMLCV [Mer] | ND | BR | 77 | 68 | ND | ND | 84 | 92 (96) | 78 | 77 (86) | 80 | 73 (83) | 84 | 81 (86) | 82 | 62 (72) | ND | ND |
| MerMV [PR80] | ND | PR | 77 | 76 | ND | ND | 84 | 93 (97) | 77 | 74 (83) | 80 | 63 (71) | 83 | 78 (86) | 87 | 69 (80) | ND | ND |
| TbYCV [Pepper] | 2007 | CU | 77 | 63 | ND | ND | 83 | 92 (97) | 78 | 75 (84) | 76 | 65 (73) | 80 | 75 (81) | 84 | 72 (79) | ND | ND |
| CabLCJV [Cuc-3] | ND | JM | 77 | 72 | ND | ND | 84 | 91 (96) | 78 | 78 (88) | 76 | 67 (74) | 78 | 75 (84) | 82 | 67 (76) | ND | ND |
The next most closely related begomovirus DNAs were DNA-A components of New World bipartite begomoviruses. Underlined identities show divergent sequences that differentiate ToLDeV genotypes. Abbreviations are those defined by Fauquet at al. (5). ND, not determined.
AR, Argentina; BR, Brazil; CU, Cuba; EC, Ecuador; JM, Jamaica; MX, Mexico; PE, Peru; PR, Puerto Rico.
The genomic DNAs of the ToLDeV isolates ranged from 2,589 to 2,591 nt, which is typical for components of NW bipartite begomoviruses. The genome organization of these DNAs is similar to that of DNA-A components of NW bipartite begomoviruses, i.e., one virus-sense gene (V1) and no V2 gene and four complementary-sense genes (C1, C2, C3, and C4) (Fig. 1). The CP amino acid sequence has the N-terminal motif PWRsMaGT, which is characteristic of NW begomoviruses. An additional complementary-sense gene (C5) was identified that encodes a predicted protein of 105 aa.
The IR sequences (between the start codons of the C1 and V1 genes) of the ToLDeV isolates are approximately 300 nt and have the characteristic geminivirus stem-loop structure with the conserved nonanucleotide sequence TAATATT↓AC, the Rep high-affinity binding site, and the C1 TATA box (2). Interestingly, the ToLDeV genotypes have different Rep binding sites and inverted repeat sequences (Fig. 2A). The PA98-1 variant has an imperfect reiterated site (GGTGTACTGGAGT [iteron is underlined]) and an ACACC inverted repeat, the PA98-2 variant has a GGGGTAAAGGGGT reiterated site and ACCCC inverted repeat, and the PA10 strain has a GGTAGAACGGTAA reiterated site and TTACC inverted repeat (Fig. 2A). The ToLDeV genotypes also have different Rep iteron-related domains (IRD). The PA98-1 variant has the GGTGT iteron and FKIN IRD motif, the PA98-2 variant has the GGGGT iteron and KRFKIS motif, and the PA10 strain has the GGTAG iteron and KRFQIS motif (Fig. 2B). These results were consistent with the ToLDeV variant/strain designations and revealed an unusually high degree of genetic diversity in the IR sequences of these genotypes.
Fig 2.

Genetic diversity among ToLDeV isolates collected from Peru in 1998 (PA98-1-1, PA98-2-1, PA98-2-TM5, PA98-2-TM8, and PA98-2-TM13), 2003 (ToLDeV [PE:03]; GenBank accession number GQ335572), and 2010 (PA10-3, PA10-S8, and PA10-S9), and Ecuador in 2010 (EA10-LE3-5K). (A) An alignment of intergenic region nucleotide sequences with the Rep binding site. The sequence and orientation of the Rep binding site iteron and inverted repeat are indicated with boxes and arrows, respectively; the TATA box of the C1 (Rep) gene is indicated by a horizontal bracket. Vertical brackets show isolates with identical iteron and inverted repeat sequences and that correspond to the PA98-1 and PA98-2 variants and the PA10 strain of ToLDeV. (B) An alignment of N-terminal Rep protein amino acid sequences with the IRD. The IRD is shown in bold and underlined, and the Rep motifs I, II, and III and GRS are shown in boxes. Vertical brackets show isolates with identical IRD sequences and that correspond to the PA98-1 and PA98-2 variants and the PA10 strain of ToLDeV. (C) A similarity plot analysis of total nucleotide sequences of the genomic DNA of isolates of ToLDeV collected from Peru in 1998 and 2010 and Ecuador in 2010. The PA98-1 variant data are for isolates PA98-1-1 and EA10-LE3-5K, the PA98-2 variant data are for isolates PA98-2-1, -TM5, -TM8, and -TM13, and the PA10 strain data are for isolates PA10-3, -S8, and -S9.
The Simplot analysis grouped the ToLDeV isolates into the PA98-1 and PA98-2 variants and strain PA10 and revealed that the majority of the sequence divergence is in the IR and the C1 and C4 genes (Fig. 2C). This was in agreement with the divergent IR and IRD sequences (Fig. 2A and B) and was further supported by results of comparisons performed with IR and individual gene sequences (Table 1). Here, the right IR sequences were almost identical (>97% identity), whereas the left IR sequences were highly divergent (Table 1), especially for isolates of the PA10 strain (65 to 66% identity). The V1, C5, C2, and C3 genes were highly conserved among the three genotypes (≥95% nucleotide and amino acid identities), whereas the C1 and C4 genes were more divergent (Table 1). In comparisons with the PA98-1 variant, the PA98-2 variant had C1/Rep and C4 nucleotide/amino acid identities of 93/93% and 91/86%, respectively, whereas those of the PA10 strain variant were 88/86 to 87% and 86/78%, respectively.
Comparisons performed with the complete nucleotide sequences of the ToLDeV isolates and those of other begomoviruses revealed highest identities with DNA-A components of NW bipartite begomoviruses from Latin America (Table 1). The highest identities were with Soybean blistering mosaic virus (SbBMV) from Argentina (79 to 86%), Tomato chino La Paz virus (ToChLPV) from Mexico (78 to 81%), and Merremia mosaic virus from Puerto Rico (77 to 81%). Similar results were obtained in comparisons made with IR and individual gene sequences (Table 1 and data not shown). An exception was with the C4 genes. In this case, a BLAST search performed with the sequence of the C4 gene of the PA10 strain revealed highest identities (91 to 92%) with C4 genes of some OW monopartite tomato-infecting begomoviruses (e.g., Tomato leaf curl Mali virus, Tomato leaf curl Cameroon virus, and Tomato curly stunt virus) (data not shown) and slightly lower identities (89 to 90%) with AC4 sequences of NW bipartite begomoviruses (e.g., Rhynchosia golden mosaic virus and Sida golden mosaic virus). The C4 sequence of the PA98-1 variant had highest identities (88 to 89%) with AC4 and C4 sequences, whereas that of the PA98-2 variant had highest identities (89%) with AC4 sequences and slightly lower identities (88%) with C4 sequences (data not shown).
Results of the phylogenetic analyses performed with the complete and C4 nucleotide sequences of the ToLDeV isolates and those of the most closely related begomovirus components/genomes are presented in Fig. 3. In the analysis with the complete nucleotide sequences, NW and OW begomoviruses were placed in two strongly supported clades (Fig. 3A). The ToLDeV isolates were placed in a strongly supported clade (the ToLDeV clade), which was contained within the NW begomovirus clade. The ToLDeV clade had three groups: one with isolates of the PA98-1 variant, one with isolates of the PA98-2 variant, and the other with isolates of the PA10 strain. The sequence of the DNA-A component of ToChLPV was most closely related to those of the ToLDeV isolates and was placed in a strongly supported group (100% credibility value) with the ToLDeV clade (Fig. 3A). The sequences of DNA-A components of other tomato- and weed-infecting bipartite begomoviruses, mostly from South America (Argentina, Bolivia, Brazil, and Venezuela), were the next most closely related and were placed in a large strongly supported group (100% credibility value) with the ToLDeV clade and ToChLPV (Fig. 3A).
Fig 3.

Phylogenetic consensus trees showing the relationships among ToLDeV isolates collected from Peru in 1998 (PA98-1-1, PA98-2-1, PA98-2-TM5, PA98-2-TM8, and PA98-2-TM13), 2003 (ToLDeV [PE:03]; GenBank accession number GQ335572), and 2010 (PA10-3, PA10-S8, and PA10-S9) and Ecuador in 2010 (EA10-LE3-5K) and the most closely related genomes of New World (DNA-A component) and Old World begomoviruses based on alignments of total nucleotide (A) and C4 gene (B) sequences. Phylogenetic analyses were performed with MrBayes 3.2 (35). Branch strengths were evaluated by Bayesian posterior probabilities. Nodes with clade credibility values of >60% are shown. The curtovirus BSCTV was used as an outgroup. The bar indicates length of horizontal branches corresponding to the rate of substitution/nucleotide. The following sequences were obtained from GenBank and used for comparisons and phylogenetic analysis: Abutilon mosaic virus-[Hawaii] (AbMV-[US:HW]) and AbMV-India [AbMV-IN]; African cassava mosaic Burkina Faso virus (ACMVBFV-[BF:OUa:08]); Cabbage leaf curl Jamaica virus-[Jamaica] (CabLCJV [JM:Cuc-3]); Merremia mosaic virus (MerMV)-[Puerto Rico] (MerMV-[PR]), MerMV-[PR:PR80-H3] and MerMV-Venezuela (MerMV-[VE:Trujillo]); Pepper leaf curl virus-[Thailand] (PepLCV-[TH]); Potato yellow mosaic virus-Trinidad (PYMV-[TT]) and PYMV-[VE]; Sida golden mosaic Liguanea virus-[Florida] (SiGMV-[US:FL:09]) and Sida golden mosaic virus-[Florida] (SiGMV-[US:FL]); Sida micrantha mosaic virus-[Bolivia] (SiMMV-[BO:Rho:07]); Sida mosaic Alagoas virus-[Brazil] (-[BR:BgV02A.C61]); Sida mosaic Bolivia virus-[Bolivia] (-[BO:1:07]); Soybean blistering mosaic virus-[Argentina] (SbBMV-[AR:NOA]), Tobacco yellow crinkle virus-[Cuba] (TbYCV-[CU:Pepper:07]), Tomato chino La Paz virus-[Mexico] (ToChLPV [MX:MM1]) and (ToChLPV-[MX:Pepper:BC:08]); Tomato curly stunt virus-[South Africa] (ToCSV-[ZA]); Tomato golden mosaic virus-[Brazil] (TGMV-[BR:YV]); Tomato leaf curl Arusha virus-[Tanzania] (ToLCArV-[TZ:Kili:AFTT23]); Tomato leaf curl Diana virus-[Madagascar] (-[MG:Namakely]); Tomato leaf curl Ghana virus-[Cameroon] (ToLCGV-[CM: TOS2B1F1]); ToLCGV [CM: TOS2B1F4]); Tomato leaf curl Guangdong virus-[China] (TYLCGuV [CN:Guangdong]); Tomato leaf curl Madagascar virus (ToLCMGV [MG:Morondova:01]), Tomato leaf curl Mali virus-[Mali] (ToLCMLV-[ML]); Tomato leaf distortion virus-[Brazil] (ToLDV-[BR:Pda4:05]); Tomato mild mosaic virus-[Brazil] (ToMMV-[BR:Pda58:05]); Tomato mosaic Havana virus-[Cuba] (ToMHV-[CU:Quivican]); Tomato mottle virus-[Florida] (ToMoV-[US:FL]); Tomato rugose mosaic virus-[Brazil] (ToRMV-[BR:Ube]); Tomato severe mottle Argentina virus-[Argentina] (-[AR:Pichanal:08]); Tomato yellow leaf curl China virus-[China] (TYLCCNV-[CN:Y43]); Tomato yellow leaf curl Mali virus-[Mali] (TYLCMLV-[ML]); Tomato yellow leaf curl Thailand virus-[Thailand] (TYLCTHV-[TH]); Tomato yellow leaf curl Vietnam virus-[Vietnam] (TYLCVNV-[VN:Hanoi]); Tomato yellow leaf curl virus-[Dominican Republic] (TYLCV-[DO]) and TYLCV-[PR]; Tomato yellow margin leaf curl virus-[Venezuela] (TYMLCV-[VE:Merida]). The positions of the ToLDeV isolates from Ecuador and Peru are indicated by brackets.
The phylogenetic relationship revealed by the analysis of the AC4/C4 sequences was incongruent, as a clear division between NW and OW begomoviruses was not revealed. Moreover, the ToLDeV isolates were not placed into a single clade. The ToLDeV variants were placed in a strongly supported clade with two groups, one with PA98-1 isolates and the other with the PA98-2 isolates (Fig. 3B). The isolates of the PA10 strain were placed in a different clade, which included C4 sequences of OW monopartite tomato-infecting begomoviruses from Africa and the AC4 sequence of a cassava-infecting bipartite begomovirus from Burkina Faso (Fig. 3B). Although this clade had a lower credibility value (70%), this relationship was in agreement with the BLAST search results. The clades with the ToLDeV genotypes were included in a large strongly supported clade (99% credibility value) that included AC4 sequences of NW bipartite begomoviruses from South America (Fig. 3B). Another large and strongly supported clade (96% credibility value) included AC4/C4 sequences of other NW bipartite and OW monopartite begomoviruses (Fig. 3B).
Together, these results indicated that the genome of ToLDeV is composed of a single genetically divergent genomic DNA that is most closely related to the DNA-A components of NW bipartite begomoviruses. However, these results did not reveal the genetic mechanism(s) underlying the diversity in the ToLDeV genome or resolve the etiology of ToLCD.
Recombination and mutation have driven the emergence and evolution of the ToLDeV genome.
The results of the Simplot analysis and sequence comparisons provided evidence of recombination in the ToLDeV genome, i.e., a divergent LIR/C1/C4 region embedded within the highly conserved remainder of the genome. Inspection of the IR/C1/C4 sequence alignment confirmed the high degree of sequence divergence in this region and revealed potential recombination junctions in the IR and C1 gene, respectively (see Fig. S1 in the supplemental material). Furthermore, the distribution of nucleotide differences in this divergent region correlated with the ToLDeV genotypes (see Fig. S1). Considered together with the different Rep binding sites and IRDs (Fig. 2A and B), these results provided evidence of at least two recombination events in the evolution of these genotypes. Unexpectedly, the RDP3 analyses with different data sets (including one with 180 full-length sequences of bipartite and monopartite begomoviruses) did not reveal convincing evidence of recombination in the ToLDeV genome (i.e., P values of >10−4). However, this may have been due to the absence of parental sequences in the database (note that ToLDeV is the first begomovirus described from Peru).
Genetic diversity values revealed the greatest variation in the ToLDeV C4 gene, followed by the IR, the C1 gene, and the V1 gene (Table 2). A Bayesian analysis revealed strong constrained selection in the C1 and V1 genes, i.e., negative selection for 78 and 93% of sites, respectively, whereas the C4 gene showed less constrained selection, with 50% of sites under negative selection (Table 2). The C4 gene showed the highest level of positive selection (20.6% of sites) compared with the C1 (10.4%) and V1 (4.2%) genes. Moreover, the finding that 20.6% of sites in the C4 gene were under positive selection compared with only 9.5% of sites in the overlapping region of the C1 gene (Table 2) revealed differential evolutionary pressure on these genes.
Table 2.
Nucleotide diversities and evolutionary analysis results for selected regions of the ToLDeV genome
| Sequencea | Nucleotide diversity (π)b | Bayesian estimation of positively selected sites in coding regionsc |
|---|---|---|
| IR | 0.07472 | |
| C4 | 0.09084 | ωN = 1.000 (1.000), piN = 0.324 (0.252) |
| ω+ = 2.028 (1.750), pi+ = 0.206 (0.167) | ||
| ω− = 0.285 (0.237), pi− = 0.470 (0.498) | ||
| C1 (Rep) | 0.07572 | ωN = 1.000 (1.000), piN = 0.123 (0.118) |
| ω+ = 1.546 (1.343), pi+ = 0.104 (0.093) | ||
| ω− = 0.038 (0.033), pi− = 0.774 (0.777) | ||
| Overlapping C1 (Rep)/C4 region | 0.08961 | ωN = 1.000 (1.000), piN = 0.090 (0.060) |
| ω+ = 2.858 (1.934), pi+ = 0.095 (0.082) | ||
| ω− = 0.118 (0.111), pi− = 0.812 (0.829) | ||
| V1 (CP) | 0.01577 | ωN = 1.000 (1.000), piN = 0.027 (0.021) |
| ω+ = 5.817 (5.301), pi+ = 0.042 (0.040) | ||
| ω− = 0.022 (0.019), pi− = 0.931 (0.932) |
The IR database was derived from 29 sequences, 5 from GenBank and 24 from this study, including those of the 9 full-length clones from Peru and Ecuador and 15 DNA fragments amplified from samples collected from Peru in 1998 and 2010 by PCR with the 1730v/496c primer pair. The databases for the C4, C1 (Rep), and V1 (CP) genes, as well as the overlapping region of the C4/C1 genes, were derived from 11 sequences, two from GenBank plus those of the nine full-length clones from this study.
Tajima's test was implemented by using DnaSP 5.10.
Omega (ω) values (ratio of nonsynonymous [dN] and synonymous [dS] substitutions) for the negatively selected (dN/dS < 1), neutral (dN/dS = 1), and positively (dN/dS > 1) selected sites ω−, ωN, and ω+, respectively. The proportions of the site categories (piN, pi+, and pi−) associated with each omega value are presented. Median values are shown in parentheses.
The overlapping C1/C4 region consists of 84 codons, with the first nucleotide position in C1 codons corresponding to the third position in C4 codons (C1-1/C4-3), the second position in C1 codons corresponding to the first position in C4 codons (C1-2/C4-1), and the third position in C1 codons corresponding to the second position in C4 codons (C1-2/C4-3) (Fig. 4A). Evolutionary constraints in this region were assessed by determining the entropy values for nucleotide and amino acid positions (Fig. 4B). Variation at the C1-2/C4-1 nucleotide sites, which had the highest probability of altering the amino acid sequences of both proteins, was the lowest (cumulative entropy value of 2.4) compared with values of 12.3 and 16.1 for the C1-1/C4-3 and C1-3/C4-2 positions, respectively. Furthermore, amino acid changes in the Rep protein were largely determined by nucleotide substitutions at the C1-1/C4-3 sites, whereas those in the C4 protein were determined by substitutions at the C1-3/C4-2 sites (Fig. 4B). The SNAP analysis, which plots the cumulative occurrence of synonymous and nonsynonymous substitutions, showed that the C1 gene accumulated more synonymous mutations, whereas the C4 gene accumulated more nonsynonymous mutations (Fig. 4C). Together, these results revealed positive and negative selection in the overlapping sequences of the C4 and C1 genes, respectively, thereby allowing for more rapid evolution of the C4 protein while minimizing amino acid changes in the more conserved Rep protein.
Fig 4.
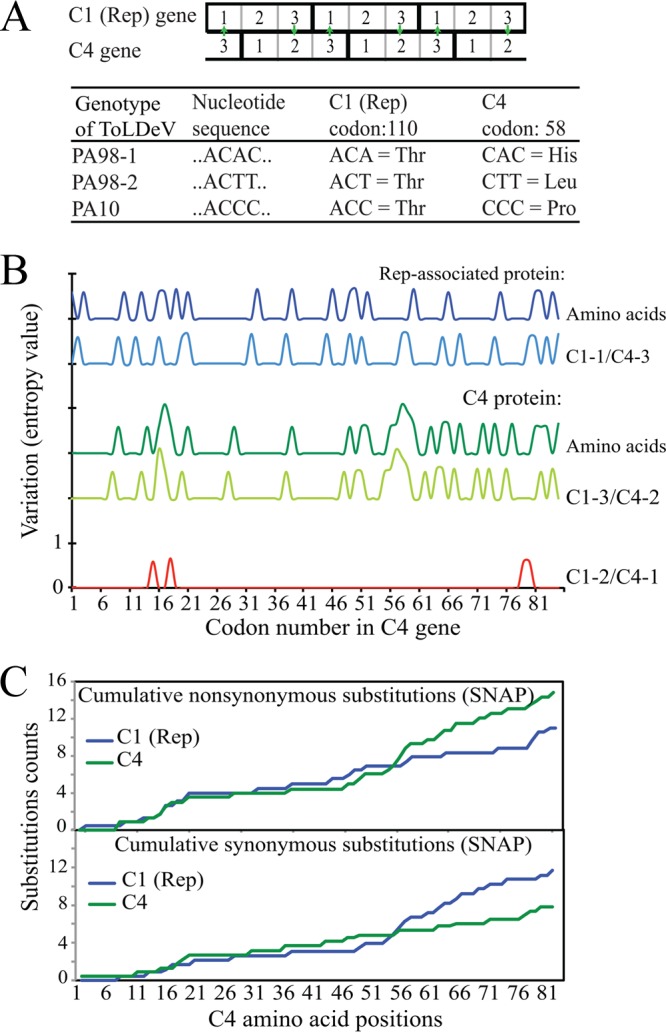
Mutations in the reading frames of the overlapping C1 and C4 genes in the ToLDeV genome result in different rates of amino acid changes in the conserved Rep and more divergent C4 proteins, respectively. (A) In this overlapping region, nucleotide substitutions in the third position of the C1 codon are likely to be synonymous mutations, whereas the same substitutions in the second position of the C4 codon are more likely to be nonsynonymous mutations (arrows). For example, a nucleotide substitution in codon 110 of the C1 gene results in a synonymous mutation, whereas the same mutation results in a nonsynonymous mutation in codon 58 of the C4 gene. (B) Levels of nucleotide and amino acid variation relative to the nucleotide positions of the codons in the reading frames of the overlapping C1 and C4 genes. Variation in nucleotides of the three codons are color coded and are shown in relation to the amino acid variations in the Rep and C4 proteins. (C) Results of a SNAP analysis (43), showing cumulative synonymous/nonsynonymous codon changes from the 5′ (N termini [left]) to the 3′ (C termini [right]) of the C1 (Rep) and C4 (C4) genes in the overlapping region. Nonsynonymous mutations in C4 codons, resulting in positive selection, are found at the 3′ end of the gene, whereas these mutations are synonymous in the overlapping codons of the C1 gene and show purifying selection or neutrality.
Inspection of the AC4/C4 amino acid alignment revealed a high level of variability among these sequences, especially in the C terminus (see Fig. S2 in the supplemental material). This is in agreement with a SNAP analysis that showed a higher proportion of nonsynonomous sites in the 3′ region of the ToLDeV C4 gene (positions 50 to 84) (data not shown). There was no evidence that the C4 genes of the ToLDeV genotypes (or AC4 genes of NW bipartite begomoviruses) were acquired by recombination with an OW begomovirus (i.e., extensive regions of shared amino acids), and this was in agreement with RDP results, which did not reveal such a recombination event (note that these potential OW begomovirus parental sequences were in some of the data sets used in these analyses). For example, although the C4 of the PA10 strain had slightly higher identities with C4 sequences of some OW monopartite begomoviruses, the alignment revealed a similar level of divergence (approximately 24% of residues) for OW C4 and NW AC4 sequences. The amino acid differences were also distributed throughout the sequence, and there were residues in the PA10 C4 sequence that were either unique (4 residues), found mostly in NW sequences (6 residues), or found mostly in OW sequences (3 residues) (see Fig. S2). Together, these results suggest that the diversity in the AC4/C4 genes arose through independent mutational events and that the relatively high level of amino acid sequence identity among the ToLDeV C4 proteins and those of OW monopartite begomoviruses are an example of convergent or parallel evolution, as defined in terms of the independent evolution of a given phenotype or trait (54).
The single genomic DNA of ToLDeV is infectious and induces leaf curl symptoms in tomato and N. benthamiana.
Tomato plants individually agroinoculated with the multimeric clones of the PA98-1-1 and PA98-2-1 variants and the PA10-3 strain developed stunting and upward leaf curling and swollen purple veins, whereas plants inoculated with the empty vector control did not develop symptoms (Fig. 5A). Importantly, these symptoms were indistinguishable from those associated with ToLCD in the field in Ecuador and Peru. N. benthamiana plants agroinoculated with these clones developed stunting, leaf curling, and vein swelling (Fig. 5B). In both plant species and for all three clones, disease symptoms developed 9 to 12 dpi and rates of infectivity were high (92 to 100%). The presence of viral DNA in leaves of representative symptomatic plants was confirmed by PCR with the PAR1c496/ToLCv1730 primer pair. These results fulfilled Koch's postulates for ToLCD and provided conclusive evidence that ToLDeV is a monopartite begomovirus.
Fig 5.

Disease symptoms induced by the cloned genomic DNAs of three genotypes of ToLDeV. The PA98-1-1 and PA98-2-1 variants came from samples of tomato leaf curl disease collected in 1998, whereas the PA10-3 strain came from a sample collected in 2010. From left to right are shown the empty vector (EV) control and symptoms induced by the PA98-1-1 and PA98-2-1 variants and PA10-3 strain in tomato (cultivar Glamour) (A) and Nicotiana benthamiana plants (B) at 50 and 25 days after agroinoculation, respectively. (C) Viral DNA levels in newly emerged leaves of infected tomato plants (cultivar Glamour) at 21 and 60 days postagroinoculation with the ToLDeV genotypes, as determined by DNA gel blot hybridization with a probe composed of a mixture of the cloned DNA components of these genotypes. The numbers 1 and 2 represent individual plants. M, the size marker (in kb). (D) Biomass of tomato plants determined 50 days after agroinoculation with the EV control, the PA98-1-1 and PA98-2-1 variants, and the PA10-3 strain. Biomass is presented as a percentage relative to the EV control. Bars with the same letters are not significantly different (P > 0.05) according to Tukey's test.
Distinct symptom phenotypes are induced in tomato and N. benthamiana plants by the genomic DNAs of the ToLDeV genotypes.
Tomato plants agroinoculated with the PA98-1-1 variant developed severe leaf curl symptoms but relatively little stunting. By approximately 30 dpi, the symptoms in newly emerging leaves were attenuated (Fig. 5A), and this recovery phenotype continued for up to 60 dpi. Severe leaf curl symptoms also developed in tomato plants agroinoculated with the PA98-2-1 variant, but these plants showed stunting and little or no recovery (Fig. 5A). Tomato plants agroinoculated with the PA10-3 strain developed more severe stunting and leaf curl symptoms than plants inoculated with either variant, and infected plants did not recover (Fig. 5A). Similar results were observed in agroinoculated N. benthamiana plants; here, the symptoms induced by the PA10-3 strain were substantially more severe than those induced by either variant (Fig. 5B).
The recovery phenotype in plants infected with begomoviruses is a type of defense response and is associated with reduced levels of viral DNA (55, 56). Results of DNA gel blot hybridization analyses indicated that viral DNA levels were similar and relatively high for all three genotypes at 21 dpi, when infected plants showed severe leaf curl symptoms (Fig. 5C). However, by 60 dpi, when plants infected with the PA98-1-1 variant had undergone recovery, viral DNA levels were reduced in these plants compared with those in plants infected with the PA98-2-1 variant and PA10-3 strain, which had not recovered (Fig. 5C). These results were observed in three independent experiments and suggest that the recovery phenotype is associated with a reduced level of viral DNA.
Finally, the differential pathogenicity of these ToLDeV genotypes was quantified by determining the biomass of infected tomato plants. Regardless of the ToLDeV genotype, the biomass of infected plants was significantly less than that of the empty vector control plants. The biomass of plants infected with the highly virulent PA10-3 strain was significantly less than that of plants infected with the PA98-1-1 or PA98-2-1 variants (Fig. 5D).
ToLDeV has a narrow host range and a resistant symptom phenotype in tomato germplasm with the Ty-1 gene.
The host range of the ToLDeV genotypes (the PA98-1-1 and PA98-2-1 variants and the PA10-3 strain) was identical (Table 3). Three types of symptom phenotypes were observed: symptomatic infection, symptomless infection, and no infection (no symptoms and no viral DNA detected in newly emerged leaves). Disease symptoms developed only in tomato and N. benthamiana plants, whereas symptomless infections were detected in the other solanaceous species (Table 3). Chenopodium quinoa, Chenopodium amaranticolor, Cucurbita pepo cv. Small sugar pumpkin, and Phaseolus vulgaris cv. Topcrop plants were not infected. These results indicated that the host range of ToLDeV is limited to solanaceous plants.
Table 3.
Infectivity, partial host range, and symptomatology of three genotypes of ToLDeV from Perua
| Host plant | ToLDeV genotypeb | No. infected/no. inoculated (% infected)c | Symptom(s)d |
|---|---|---|---|
| Solanum lycopersicum cv. Glamour | PA98-1-1 | 49/50 (98) | dg, s, ulc, vp, vs |
| PA98-2-1 | 47/50 (94) | dg, s, ulc, vp, vs | |
| PA10-3 | 46/50 (92) | dg, ss, ulc, vp, vs | |
| Nicotiana benthamiana | PA98-1-1 | 28/28 (100) | s, dlc, vs |
| PA98-2-1 | 28/28 (100) | s, dlc, vs | |
| PA10-3 | 28/28 (100) | s, svs, ulc | |
| N. tabacum cv. Samsum | PA98-1-1 | 15/18 (83) | si |
| PA98-2-1 | 10/18 (56) | si | |
| PA10-3 | 11/19 (58) | si | |
| N. glutinosa | PA98-1-1 | 16/16 (100) | si |
| PA98-2-1 | 14/14 (100) | si | |
| PA10-3 | 13/14 (93) | si | |
| Datura stramonium | PA98-1-1 | 18/19 (95) | si |
| PA98-2-1 | 15/18 (83) | si | |
| PA10-3 | 10/19 (53) | si | |
| Chenopodium quinoa | PA98-1-1 | 0/13 (0) | ni |
| PA98-2-1 | 0/12 (0) | ni | |
| PA10-3 | 0/16 (0) | ni | |
| C. amaranticolor | PA98-1-1 | 0/13 (0) | ni |
| PA98-2-1 | 0/13 (0) | ni | |
| PA10-3 | 0/19 (0) | ni | |
| Cucurbita pepo cv. Small Sugar | PA98-1-1 | 0/16 (0) | ni |
| PA98-2-1 | 0/16 (0) | ni | |
| PA10-3 | 0/19 (0) | ni | |
| Phaseolus vulgaris cv. Topcrop | PA98-1-1 | 0/12 (0) | ni |
| PA98-2-1 | 0/12 (0) | ni | |
| PA10-3 | 0/17 (0) | ni |
Viruses were delivered by agroinoculation. Infectivity was determined 18 to 21 days postagroinoculation and based on symptom development (in tomato and N. benthamiana) and detection of viral DNA by PCR with the PAR1c496/ToLC-v1730 primer pair.
Genotypes included the PA98-1 and PA98-2 variants, represented by the PA98-1-1 and PA98-2-1 isolates, respectively, and the PA10 strain, represented by the PA10-3 isolate.
Results represent data from at least three independent experiments.
Symptom abbreviations: dg, distorted growth; dlc, downward leaf curling; ni, no infection (no symptoms and no viral DNA detected in newly emerged leaves); si, symptomless infection; s, stunting; ss, severe stunting; ulc, upward leaf curling; vp, vein purpling; vs, vein swelling; svs, severe vein swelling.
The Ty-1 gene confers resistance to monopartite begomoviruses, such as TYLCV, but not to many bipartite begomoviruses (57). In control experiments, cultivar Dominator plants (with Ty-1) agroinoculated with TYLCV-IL[DO] did not develop symptoms, whereas all cultivar Glamour plants (without Ty-1) developed yellow leaf curl symptoms. In contrast, all cultivar Dominator and cultivar Glamour plants agroinoculated with the bipartite begomovirus ToMHV developed leaf crumpling and yellow mottling symptoms. In experiments with the ToLDeV genotypes, LA3473 and cultivar Dominator plants (with Ty-1) agroinoculated with each of the three ToLDeV genotypes did not develop symptoms (10 to 21 plants inoculated/genotype). In contrast, all LA3474 and cultivar Glamour plants (without Ty-1) agroinoculated with the ToLDeV genotypes developed symptoms of stunting and leaf curl (10 to 16 plants inoculated/genotype). These results established that the Ty-1 gene confers resistance to the three ToLDeV genotypes (i.e., none of the genotypes overcame this resistance) and supported the notion that this gene confers resistance to monopartite but not bipartite begomoviruses.
Biological properties of ToLDeV are similar to those of monopartite begomoviruses.
Tomato and N. benthamiana seedlings rub inoculated with sap prepared from leaf tissue from tomato plants with leaf curl symptoms collected in 1998 or from leaves of symptomatic N. benthamiana plants infected with the ToLDeV PA10-3 strain did not develop symptoms, nor was viral DNA detected in newly emerged leaves (15 total plants inoculated in three independent experiments). In contrast, 14 of 15 N. benthamiana plants inoculated with sap from leaves infected with the sap-transmissible bipartite begomovirus BDMV developed stunting and leaf curling, crumpling, and mosaic symptoms. Immunolocalization experiments performed with petioles and leaves from ToLDeV-infected tomato plants revealed virus only in phloem-associated cells, e.g., companion and phloem parenchyma cells (Fig. 6). No signal was detected in petiole and leaf tissues of infected plants probed with preimmune antiserum or in uninfected plants probed with CP antiserum (Fig. 6 and data not shown). These results indicated that ToLDeV is phloem limited, which is fully consistent with the lack of sap transmissibility.
Fig 6.
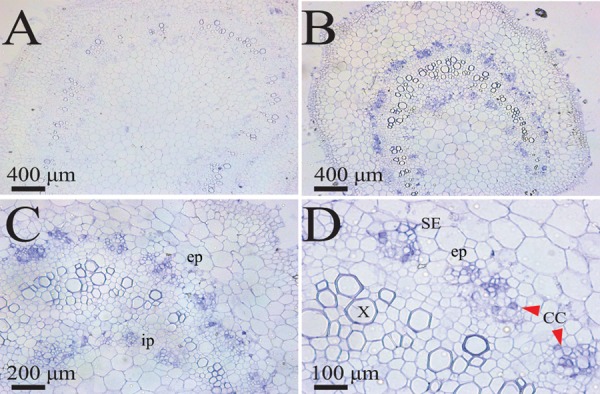
Immunolocalization of ToLDeV in petiole tissues from tomato plants with leaf curl disease symptoms 4 weeks after agroinoculation with the cloned genomic DNAs of the PA98-1-1 and PA98-2-1 variants. (A) Section of a petiole from a tomato plant agroinoculated with the empty vector control and probed with begomovirus CP antiserum. (B and C) Sections of a leaf petiole from a ToLDeV-infected plant, probed with CP antiserum and showing signal over cells of the internal and external phloem. (D) Section of a leaf petiole from a ToLDeV-infected plant showing signal over companion cells and sieve elements. Abbreviations: cc, companion cells; ep, external phloem; ip, internal phloem; se, sieve element; x, xylem.
Mutational analyses revealed functional homology of the ToLDeV V1 and C4 genes with monopartite begomovirus counterparts.
In leaf disc replication assays, DNA gel blot hybridization analyses revealed replication of the wild-type genomic DNA of all three ToLDeV genotypes, including open circular double-stranded DNA (ocDNA) and single-stranded DNA (ssDNA) forms. Similar levels of replication, as well as DNA forms, were detected in leaves agroinfiltrated with the v1, c4, and c5 mutants of all three genotypes (Fig. 7A). Replication was not detected for the c1 mutants, consistent with the requirement of Rep for viral DNA replication (2).
Fig 7.

Replication competence and infectivity of ToLDeV mutants. (A) Results of replication assays in Nicotiana benthamiana leaf discs for ToLDeV wild-type (wt) and v1, c1, c4, and c5 mutants of the genomic DNAs of three genotypes of ToLDeV: the PA98-1-1 and PA98-2-1 variants and the PA10-3 strain. M, size marker (in kb). (B to G) Disease symptoms induced by c4 mutants of the three ToLDeV genotypes in tomato (cultivar Glamour) and N. benthamiana plants. From left to right, the empty vector control and symptoms induced by the c4 mutants and wild type in tomato plants for the PA98-1-1 variant (B), PA98-2-1 variant (C), and PA10-3 strain (D) and in N. benthamiana plants for the PA98-1-1 variant (E), PA98-2-1 variant (F), and PA10-3 strain (G).
The c1 (Rep) and v1 (CP) mutants did not systemically infect tomato or N. benthamiana plants, based upon lack of symptom development and failure to detect viral DNA in newly emerged leaves by PCR (Table 4). Tomato plants agroinoculated with the c4 mutants developed no symptoms or an attenuated symptom phenotype (Fig. 7B to D), and PCR analyses revealed that only plants with the attenuated symptoms were infected (Table 4). Sequencing of the PCR-amplified fragment with the C4 gene from selected plants with the attenuated symptom phenotype confirmed the presence of the mutation. N. benthamiana plants agroinoculated with the c4 mutants developed no obvious symptoms (Fig. 7E to G), but PCR analyses revealed symptomless infections in most plants (Table 4). Mutation of the C5 gene had no effect on viral infectivity and symptom development, with the exception of the PA10-3 c5 mutant, which induced less severe symptoms in N. benthamiana plants (Table 4). Sequencing of the PCR-amplified fragment with the C5 gene from selected symptomatic tomato and N. benthamiana plants agroinoculated with the c5 mutant confirmed the presence of the mutation. Together, these results indicate that (i) Rep and CP are required for systemic infection, (ii) C4 is a pathogenicity factor, and (iii) C5 does not play a major role in pathogenicity.
Table 4.
Infectivity and symptomatology of the mutants and wild type of three genotypes of ToLDeV from Perua
| Mutation | ToLDeV genotypeb |
N. benthamiama |
S. lycopersicon cv. Glamour |
Graftingd | ||||
|---|---|---|---|---|---|---|---|---|
| No. of symptomatic plants/total inoculated | Symptom(s)c | No. of infected plants/total inoculated | No. of symptomatic plants/total inoculated | Symptom(s)c | No. of infected plants/total inoculated | |||
| v1 | PA98-1-1 | 0/14 | ni | 0/14 | 0/15 | ni | 0/15 | NT |
| PA98-2-1 | 0/14 | ni | 0/14 | 0/17 | ni | 0/17 | NT | |
| PA10-3 | 0/16 | ni | 0/16 | 0/13 | ni | 0/13 | NT | |
| c1 | PA98-1-1 | 0/14 | ni | 0/14 | 0/15 | ni | 0/15 | NT |
| PA98-2-1 | 0/14 | ni | 0/14 | 0/14 | ni | 0/14 | NT | |
| PA10-3 | 0/16 | ni | 0/16 | 0/14 | ni | 0/14 | NT | |
| c4 | PA98-1-1 | 0/23 | si | 23/23 | 20/45 | ni, or mlc and ms | 30/45 | 0/14 |
| PA98-2-1 | 0/22 | si | 20/22 | 10/49 | ni, or mlc and ms | 12/49 | 8/8 | |
| PA10-3 | 0/17 | si | 17/17 | 15/45 | ni, or mlc and ms | 15/45 | 0/8 | |
| c5 | PA98-1-1 | 14/14 | dlc, s, vs | 14/14 | 9/9 | dg, s, ulc, vp, vs | 9/9 | NT |
| PA98-2-1 | 15/15 | dlc, s, vs | 15/15 | 9/9 | dg, s, ulc, vp, vs | 9/9 | NT | |
| PA10-3 | 19/19 | mulc, s, vs | 19/19 | 21/21 | dg, ss, ulc, vp, vs | 21/21 | NT | |
| Wild type | PA98-1-1 | 14/14 | dlc, s, vs | 14/14 | 14/15 | dg, s, ulc, vp, vs | 14/15 | NT |
| PA98-2-1 | 14/14 | dlc, s, vs | 14/14 | 13/15 | dg, s, ulc, vp, vs | 13/15 | NT | |
| PA10-3 | 15/15 | s, svs, ulc | 15/15 | 14/15 | dg, ss, ulc, vp, vs | 14/15 | NT | |
Viruses were delivered by agroinoculation. Infectivity was determined 14 to 21 days postagroinoculation and based on symptom development and detection of viral DNA by PCR with the PAR1c496/ToLC-v1730 primer pair. Results represent data from at least three independent experiments.
Genotypes include the PA98-1 and PA98-2 variants, represented by the PA98-1-1 and PA98-2-1 isolates, respectively, and the PA10 strain, represented by the PA10-3 isolate.
Symptom abbreviations: dg, distorted growth; dlc, downward leaf curling; mlc, mild leaf curling; ms, mild stunting; mulc, mild upward leaf curling; ni, no infection (no symptoms and no viral DNA detected in newly emerged leaves); si, symptomless infection; s, stunting; ss, severe stunting; ulc, upward leaf curling, vp, vein purpling; vs, vein swelling; svs, severe vein swelling.
Number of tomato plants with grafted scions in which symptoms reverted to wild type. NT, not tested.
The stability of the c4 mutation was assessed by transmission through tomato plants following grafting. Uninfected scions (cultivar Glamour) grafted onto root stocks infected with the PA98-1-1 (14 plants) or PA10-3 c4 (8 plants) mutants became infected and developed the attenuated symptoms associated with the c4 mutation. Sequencing of the PCR-amplified C4 fragment from these scions confirmed the presence of the mutation (Table 4). In contrast, all scions grafted onto root stocks infected with the PA98-2-1 c4 mutant (8 plants) developed wild-type symptoms (stunting and severe leaf curling), and sequencing of the PCR-amplified C4 fragment from these scions revealed that the c4 mutation had reverted to the wild-type sequence (Table 4). These results are consistent with C4 functioning as a pathogenicity factor, and they reveal positive selection pressure on the C4 gene during infection.
Infectivity and symptomatology of chimeric genomes generated between two ToLDeV genotypes.
The role of the divergent region of the ToLDeV genome was further investigated by generating chimeras between the PA98-1-1 variant, which induces less severe symptoms and shows the recovery phenotype, and the PA10-3 strain, which induces severe symptoms and shows no recovery (Fig. 1 and 5A). The PA98-1-1/PA10-3 chimera composed of the PA98-1-1 variant with the 1.5-kb fragment with the IR/C4/C1 sequences from the PA10-3 strain induced symptoms in tomato and N. benthamiana plants that were more severe than those induced by the PA98-1-1 variant, but less severe than those induced by the PA10-3 strain, and recovery was less pronounced (Fig. 8). The PA10-3/PA98-1-1 chimera induced symptoms that were less severe than those induced by the PA10-3 strain but more severe than those induced by the PA98-1-1 variant. Plants infected with this chimera showed a partial recovery phenotype (Fig. 8). Together, these results support the hypothesis that this divergent region of the viral genome, including the C4 gene, plays a role in the pathogenicity of ToLDeV.
Fig 8.

Disease symptoms induced by the cloned DNA genome of the ToLDeV PA98-1-1 variant, PA10-3 strain, and chimeras generated between these genotypes 21 and 32 days after agroinoculation of Nicotiana benthamiana and tomato plants, respectively. Symptoms induced in N. benthamiana plants by the empty vector control (A), the PA98-1-1 variant (B), the PA98-1-1/PA10-3 chimera (C), the PA10-3 strain (D), and the PA10-3/PA98-1-1 chimera (E) are illustrated. Symptoms induced in tomato plants by the empty vector control (F), the PA98-1-1 variant (G), the PA98-1-1/PA10-3 chimera (H), the PA10-3 strain (I), and the PA10-3/PA98-1-1 chimera (J) are also shown. For a schematic representation of the wild-type and chimeric genomes, see Fig. 1.
DISCUSSION
ToLCD in Ecuador and Peru is caused by an indigenous NW monopartite begomovirus.
Based upon the known genome structure of NW begomoviruses, it was presumed that ToLDeV would have a bipartite genome (26). The finding that ToLDeV isolates associated with ToLCD in Ecuador and Peru have a single genomic DNA, with no associated DNA-B component or satellite DNAs, confirmed and extended the results of Marquez-Martin et al. (26) and indicated an unconventional etiology for the disease. Here, we established that ToLDeV is an indigenous NW monopartite begomovirus and is the causal agent of ToLCD in Ecuador and Peru. The induction of leaf curl symptoms in tomato, indistinguishable from those observed in the field, by the cloned genomic DNA of ToLDeV fulfilled Koch's postulates for ToLCD and confirmed the monopartite nature of the viral genome. The finding that ToLDeV has biological properties similar to those of OW monopartite tomato-infecting begomoviruses, e.g., lack of sap transmission, phloem limitation, a resistance symptom phenotype in tomato germplasm with the Ty-1 gene, and functional properties of the V1 and C4 genes, provided further evidence for a monopartite genome. Several lines of evidence indicated that ToLDeV originated in the NW, rather than being an introduced exotic OW monopartite begomovirus, like TYLCV. First, the size (approximately 2.6 kb) and organization of the ToLDeV genome, including the lack of a V2 gene, are similar to DNA-A components of NW bipartite begomoviruses. Second, ToLDeV has the characteristic NW begomovirus motif, PWRsMaGT, in the N terminus of the CP. Finally, sequence comparisons and phylogenetic analyses showed that ToLDeV was most closely related to DNA-A components of NW bipartite begomoviruses. Thus, these results established that ToLDeV is an indigenous NW monopartite begomovirus, which probably evolved from the DNA-A component of a local bipartite begomovirus progenitor.
Although the DNA-A components of some NW bipartite begomoviruses are infectious in the experimental host, N. benthamiana, these infections have not been found in nature and are not examples of NW monopartite begomoviruses. On the other hand, ToLDeV is a bona fide NW monopartite begomovirus, because it causes a disease under natural conditions and possesses biological properties that are typical of monopartite begomoviruses. However, the identification of ToLDeV raises the question of what factor(s) led to the evolution of a NW monopartite begomovirus at this time. It is generally believed that the first begomoviruses were monopartite with the bipartite genome appearing later, either through component duplication and diversification or acquisition and modification of satellite DNAs (1, 4, 58). Furthermore, because bipartite begomoviruses occur in NW and OW locations (1, 4, 11), it is likely the bipartite genome evolved before continental drift. The absence of indigenous monopartite begomoviruses in the NW can be explained by the predominance of a bipartite progenitor(s) on the landmasses that gave rise to the NW or a subsequent introduction event. Local evolution events then drove the emergence of new begomovirus species that reflected the genome structure of the virus(es) in a given geographical area, i.e., bipartite species in the NW (1, 5, 7, 20, 21, 59–61) and monopartite (1, 48) and, to a lesser extent, bipartite species in the OW (1, 11, 62). In addition, a few evolutionary events in the OW have involved changes in the viral genome structure, e.g., the aforementioned TYLCTHV, which can exist as a bipartite or monopartite virus, and the monopartite Sri Lankan cassava mosaic virus, which acquired a DNA-B component and became bipartite (9). However, the emergence of a monopartite begomovirus in the NW represents a new evolutionary direction, which probably occurred due to a specific combination of factors. First, extensive cultivation of tomato provided a highly susceptible host in which a monopartite begomovirus could evolve in the absence of a betasatellite. Second, a suitable local progenitor bipartite begomovirus existed in native or cultivated plants in Peru. Finally, the invasive and polyphagous B biotype of B. tabaci mediated the introduction of this local progenitor begomovirus into crop plants, such as tomato (6).
The evolution of ToLDeV likely started with the acquisition of indigenous bipartite begomoviruses by the polyphagous whitefly vector and delivery of virions into the phloem of tomato plants. During this process, DNA-A-containing virions are delivered, via the phloem, to replication-competent cells in the protophloem (51). Here, DNA-A infections are initiated, either in the absence of the DNA-B component or possibly following the loss of this component. Viral spread occurred via cell division or cell-to-cell movement mediated by DNA-A proteins (e.g., CP or C4). Nascent DNA-A containing virions were released into sieve elements, where they were acquired and spread by whiteflies. Further evolution of the monopartite genome occurred via mutation and recombination (see below). This evolutionary scenario gains support from the aforementioned limited infectivity of some NW DNA-A components (24, 25), and fits with the classical role of bottlenecks in the emergence and evolution of plant and animal virus populations during the process of within-host spread and host-to-host transmission (63).
The reason that more DNA-A components of NW bipartite begomoviruses have not evolved into monopartite tomato-infecting begomoviruses may be due to the lack of movement-related functions encoded by the DNA-B component and/or the AV2 gene. The functional homology of the ToLDeV CP and C4 with monopartite begomovirus counterparts indicates a key role for these genes in the evolution of this NW monopartite begomovirus. The essential role of the ToLDeV CP in systemic infection is probably due to nucleocytoplasmic transport and long distance movement functions. In bipartite begomoviruses, nucleocytoplasmic transport is provided by the NSP, whereas in monopartite viruses this function is provided by the CP (4, 17, 52). The likelihood that the ToLDeV CP provides this function is supported by the capacity of the CP of some NW begomoviruses to complement, enhance, and mask NSP function (4, 64, 65). The CP also plays a key role in the long distance movement of monopartite begomoviruses, and this has been associated with virion formation (52, 66). Thus, the CP probably plays a similar role in the long distance movement of ToLDeV. As a phloem-limited monopartite begomovirus, ToLDeV may not require the cell-to-cell movement function provided by the MP or, alternatively, this may be provided by C4 (52, 66). Finally, the findings that the C5 gene was not involved in pathogenesis and the V2 gene was absent indicated that neither gene played a role in the evolution of ToLDeV.
A number of lines of evidence indicated that the ToLDeV C4 is a pathogenicity factor. First, c4 mutants induced attenuated or no symptoms and showed reduced infectivity in tomato. Second, distinct symptom phenotypes were associated with the C4 genes of the ToLDeV genotypes. Finally, the reappearance of wild-type symptoms after graft transmission of the PA98-2 c4 mutant was associated with reversion of the mutation. Based on these results and studies of C4 function in OW begomoviruses (66–71), the ToLDeV C4 could be involved in movement and suppression of host defense responses. The recovery phenotype in begomovirus-infected plants is a host defense response that involves methylation of the viral genome and a reduction in viral titer and symptom severity (56, 69, 72). The differences among ToLDeV genotypes in virulence, viral DNA levels, and recovery phenotypes in infected plants suggest that the C4 functions as a suppressor of host defense responses. The high levels of amino acid identity among NW and OW AC4/C4 proteins (24) (Fig. 3B; see also Fig. S2 in the supplemental material) may indicate a common function(s) among these proteins (e.g., in defense suppression), and several lines of evidence indicated that some of these similarities have appeared via convergent evolution. However, it will be important to experimentally demonstrate a common function for these proteins.
Recombination and mutation have driven the emergence and evolution of the ToLDeV genotypes.
The genetic diversity and differential virulence of the ToLDeV genotypes revealed evidence of viral evolution in the direction of increased virulence (e.g., the highly virulent PA10 strain associated with ToLCD in Peru in 2010). Low levels of diversity and purifying selection in the V1 and C1 genes reflect the conserved functions of the CP and Rep proteins, whereas the higher level of diversity and positive selection in the C4 gene indicate a role in the evolution of ToLDeV. The majority of the genetic diversity in the ToLDeV genome was found in the LIR, C4, and C1 genes, which is a well-known recombinational hot spot in the genomes of bipartite and monopartite begomoviruses (73–75). Evidence for recombination in this region of the ToLDeV genome came from the Simplot analysis (Fig. 2C), the different Rep binding sites and IRD sequences, and the identification of potential recombination junctions (see Fig. S1 in the supplemental material). Recombination in this region may provide a selective advantage for ToLDeV genotypes, e.g., the acquisition of a C4 with strong defense suppression properties. The failure of the RDP analysis to detect this recombinant region of the ToLDeV genome shows the importance of using multiple approaches to detect recombination in begomovirus genomes.
The relatively recent emergence of ToLDeV (within the past 20 years), together with the relatively high degree of negative selection on begomovirus coding regions (76, 77) and the estimated mutation rates for these viruses (10−3 to 10−4 substitutions/site/year) (78, 79), suggests that the high level of genetic diversity in the ToLDeV genotypes was due to mutation and recombination. Thus, many of the nucleotide differences in the divergent region were from mutations that occurred prior to the recombination events. However, random mutations would not explain the different diversity values and evolutionary constraints identified in the overlapping sequences of the C1 and C4 genes, which encode the conserved Rep and less-conserved C4 proteins, respectively. The genetic analysis revealed that variation in the C4 protein was achieved through mutations that did not alter the Rep protein, whereas mutations that altered both proteins were rare (Fig. 4A and B). This finding was supported by SNAP and Bayesian analyses, which showed that positions in the 3′ end of the C4 gene, which were under adaptive evolution (positive selection), were under purifying selection in the overlapping portion of the C1 gene. This explains how convergent evolution in AC4 genes of NW bipartite begomoviruses can generate AC4/C4 proteins with functional similarity to C4 counterparts in OW monopartite begomoviruses while maintaining the more conserved Rep protein sequence. This strategy, which allows for maximizing the viral genome size and maintaining differential rates of mutation in overlapping genes, is used by a range of animal and plant-infecting viruses (44).
The emergence of an indigenous NW monopartite begomovirus is a new evolutionary direction and another example of the remarkable genetic flexibility of these viruses and their capacity to exploit niches in agricultural ecosystems. Definitive evidence that ToLDeV is a bone fide NW monopartite begomovirus came from the characterization of the viral genome and biological properties, and the completion of Koch's postulates for ToLCD. It will be interesting to identify the progenitor(s) of the ToLDeV genotypes and to further investigate the function(s) of the viral gene products. The finding that the Ty-1 gene confers resistance to ToLDeV and the identification of a time of year when virus and whitefly incidence are low can be used to develop an integrated pest management strategy for ToLCD (10).
Supplementary Material
ACKNOWLEDGMENTS
We thank the Servicio Nacional de Sanidad Agraria, Peru, for assistance in carrying out the July 2010 disease survey.
This research was funded in part by a grant from the United States Agency for International Development (USAID) as part of the Integrated Pest Management Collaborative Research Support Program (IPM-CRSP) through cooperative agreement EPPA-00-04-00016-00. T.A.M. was supported by fellowships from the Fulbright Program and the Organization of American States.
Footnotes
Published ahead of print 6 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00234-13.
REFERENCES
- 1. Briddon RW, Patil BL, Bagewadi B, Nawaz-ul Rehman MS, Fauquet CM. 2010. Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol. Biol. 10:97 doi:10.1186/1471-2148-10-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanley-Bowdoin L, Settlage SB, Orozco BM, Nagar S, Robertson D. 1999. Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation. CRC Crit. Rev. Plant Sci. 18:71–106 [PubMed] [Google Scholar]
- 3. Harrison BD, Robinson DJ. 1999. Natural genomic and antigenic variation in whitefly-transmitted geminiviruses (begomoviruses). Annu. Rev. Phytopathol. 37:369–398 [DOI] [PubMed] [Google Scholar]
- 4. Rojas MR, Hagen C, Lucas WJ, Gilbertson RL. 2005. Exploiting chinks in the plant's armor: evolution and emergence of geminiviruses. Annu. Rev. Phytopathol. 43:361–394 [DOI] [PubMed] [Google Scholar]
- 5. Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini FM, Zhou X. 2008. Geminivirus strain demarcation and nomenclature. Arch. Virol. 153:783–821 [DOI] [PubMed] [Google Scholar]
- 6. Morales FJ. 2010. Distribution and dissemination of begomoviruses in Latin America and the Caribbean: Bemisia, p 283–318 In Stansly PA, Naranjo SE. (ed), Bionomics and management of a global pest. Springer, London, United Kingdom [Google Scholar]
- 7. Rojas MR, Gilbertson RL. 2008. Emerging plant viruses: a diversity of mechanisms and opportunities, p 27–51 In Roossinck MJ. (ed), Plant virus evolution. Springer-Verlag, Berlin, Germany [Google Scholar]
- 8. Seal SE, vanden Bosch F, Jeger MJ. 2006. Factors influencing begomovirus evolution and their increasing global significance: implications for sustainable control. CRC Crit. Rev. Plant Sci. 25:23–46 [Google Scholar]
- 9. Saunders K, Salim N, Mali VR, Malathi VG, Briddon R, Markham PG, Stanley J. 2002. Characterisation of Sri Lankan cassava mosaic virus and Indian cassava mosaic virus: evidence for acquisition of a DNA B component by a monopartite begomovirus. Virology 293:63–74 [DOI] [PubMed] [Google Scholar]
- 10. Gilbertson RL, Rojas MR, Natwick ET. 2011. Development of integrated pest management (IPM) strategies for whitefly (Bemisia tabaci)-transmissible geminiviruses, p 323–356 In Thompson WMO. (ed), The whitefly, Bemisia tabaci (Homoptera: Aleyrodidae) interaction with geminivirus-infected host plants. Springer, Dordrecht, Netherlands [Google Scholar]
- 11. Ha C, Coombs S, Revill P, Harding R, Vu M, Dale J. 2008. Molecular characterization of begomoviruses and DNA satellites from Vietnam: additional evidence that the New World geminiviruses were present in the Old World prior to continental separation. J. Gen. Virol. 89:312–326 [DOI] [PubMed] [Google Scholar]
- 12. Lefeuvre P, Martin DP, Harkins G, Lemey P, Gray AJA, Meredith S, Lakay F, Monjane A, Lett JM, Varsani A, Heydarnejad J. 2010. The spread of Tomato yellow leaf curl virus from the Middle East to the world. PLoS Pathog. 6:e1001164 doi:10.1371/journal.ppat.1001164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fiallo-Olivé E, Martínez-Zubiaur Y, Moriones E, Navas-Castillo J. 2012. A novel class of DNA satellites associated with New World begomoviruses. Virology 426:1–6 [DOI] [PubMed] [Google Scholar]
- 14. Paprotka T, Metzler V, Jeske H. 2010. The first DNA 1-like α satellites in association with New World begomoviruses in natural infections. Virology 404:148–157 [DOI] [PubMed] [Google Scholar]
- 15. Gutierrez C. 1999. Geminivirus DNA replication. Cell. Mol. Life Sci. 56:313–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrison BD, Swanson MM, Fargette D. 2002. Begomovirus coat protein: serology, variation and functions. Physiol. Mol. Plant Pathol. 60:257–271 [Google Scholar]
- 17. Sudarshana MR, Wang HL, Lucas WJ, Gilbertson RL. 1998. Dynamics of bean dwarf mosaic geminivirus cell-to-cell and long-distance movement in Phaseolus vulgaris revealed, using the green fluorescent protein. Mol. Plant Microbe Interact. 11:277–291 [Google Scholar]
- 18. Pooma W, Petty ITD. 1996. Tomato golden mosaic virus open reading frame AL4 is genetically distinct from its C4 analogue in monopartite geminiviruses. J. Gen. Virol. 77:1947–1951 [DOI] [PubMed] [Google Scholar]
- 19. Polston JE, Anderson PK. 1997. The emergence of whitefly-transmitted geminiviruses in tomato in the Western hemisphere. Plant Dis. 81:1358–1369 [DOI] [PubMed] [Google Scholar]
- 20. Zhou YC, Noussourou M, Kon T, Rojas MR, Jiang H, Chen LF, Gamby K, Foster R, Gilbertson RL. 2008. Evidence of local evolution of tomato-infecting begomovirus species in West Africa: characterization of tomato leaf curl Mali virus and tomato yellow leaf crumple virus from Mali. Arch. Virol. 153:693–706 [DOI] [PubMed] [Google Scholar]
- 21. Fernandes JJ, Carvalho MG, Andrade EC, Brommonschenkel SH, Fontes EPB, Zerbini FM. 2006. Biological and molecular properties of Tomato rugose mosaic virus (ToRMV), a new tomato-infecting begomovirus from Brazil. Plant Pathol. 55:513–522 [Google Scholar]
- 22. Rochester DE, DePaulo JJ, Fauquet CM, Beachy RN. 1994. Complete nucleotide sequence of the geminivirus Tomato yellow leaf curl virus, Thailand isolate. J. Gen. Virol. 75:477–486 [DOI] [PubMed] [Google Scholar]
- 23. Guo W, Yang X, Xie Y, Cui X, Zhou X. 2009. Tomato yellow leaf curl Thailand virus-[Y72] from Yunnan is a monopartite begomovirus associated with DNAβ. Virus Genes 38:328–333 [DOI] [PubMed] [Google Scholar]
- 24. Galvão RM, Mariano AC, Luz DF, Alfenas PF, Andrade EC, Zerbini FM, Almeida MR, Fontes EPB. 2003. A naturally occurring recombinant DNA-A of a typical bipartite begomovirus does not require the cognate DNA-B to infect Nicotiana benthamiana systemically. J. Gen. Virol. 84:715–726 [DOI] [PubMed] [Google Scholar]
- 25. Hou YM, Paplomatas EJ, Gilbertson RL. 1998. Host adaptation and replication properties of two bipartite geminiviruses and their pseudorecombinants. Mol. Plant Microbe Interact. 11:208–217 [Google Scholar]
- 26. Márquez-Martín B, Aragón-Caballero L, Fiallo-Olivé E, Navas-Castillo J, Moriones E. 2011. Tomato leaf deformation virus, a novel begomovirus associated with a severe disease of tomato in Peru. Eur. J. Plant Pathol. 129:1–7 [Google Scholar]
- 27. Murayama A, Aragon L, Fernandez-Northcote NE. 2005. Nuevo begomovirus del grupo del Nuevo Mundo asociado al encrespamiento de la hoja del tomate en la costa del Peru. Fitopatologia 40:1 (In Spanish.) [Google Scholar]
- 28. Gilbertson RL, Hidayat SH, Martinez RT, Leong SA, Faria JC, Morales F, Maxwell DP. 1991. Differentiation of bean-infecting geminiviruses by nucleic acid hybridization probes and aspects of bean golden mosaic in Brazil. Plant Dis. 75:336–342 [Google Scholar]
- 29. Rojas MR, Gilbertson RL, Russell DR, Maxwell DP. 1993. Use of degenerate primers in the polymerase chain reaction to detect whitefly-transmitted geminiviruses. Plant Dis. 77:340–347 [Google Scholar]
- 30. Dellaporta SL, Wood J, Hicks JB. 1983. A plant DNA minipreparation: version II. Plant Mol. Biol. Rep. 1:19–21 [Google Scholar]
- 31. Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19:11–15 [Google Scholar]
- 32. Briddon RW, Bull SE, Mansoor S, Amin I, Markham PG. 2002. Universal primers for the PCR-mediated amplification of DNA β. Mol. Biotechnol. 20:315–318 [DOI] [PubMed] [Google Scholar]
- 33. Inoue-Nagata AK, Albuquerque LC, Rocha WB, Nagata T. 2004. A simple method for cloning the complete begomovirus genome using the bacteriophage ϕ29 DNA polymerase. J. Virol. Methods 116:209–211 [DOI] [PubMed] [Google Scholar]
- 34. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Posada D. 2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25:1253–1256 [DOI] [PubMed] [Google Scholar]
- 37. Page RD. 1996. TreeView: an application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12:357–358 [DOI] [PubMed] [Google Scholar]
- 38. Martin DP. 2009. Recombination detection and analysis using RDP3. Methods Mol. Biol. 537:185–205 [DOI] [PubMed] [Google Scholar]
- 39. Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. 1999. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Librado P, Rozas J. 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452 [DOI] [PubMed] [Google Scholar]
- 42. Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Korber B. 2002. HIV signature and sequence variation analysis, p 55–72 In Rodrigo AG, Learn GH. (ed), Computational analysis of HIV molecular sequences. Kluwer Academic Publishers, Boston, MA [Google Scholar]
- 44. Zaaijer HL, van Hemert FJ, Koppelman MH, Lukashov VV. 2007. Independent evolution of overlapping polymerase and surface protein genes of hepatitis B virus. J. Gen. Virol. 88:2137–2143 [DOI] [PubMed] [Google Scholar]
- 45. Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. (Oxf.) 41:95–98 [Google Scholar]
- 46. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hajdukiewicz P, Svab Z, Maliga P. 1994. The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol. 25:989–994 [DOI] [PubMed] [Google Scholar]
- 48. Kon T, Gilbertson RL. 2012. Two genetically related begomoviruses causing tomato leaf curl disease in Togo and Nigeria differ in virulence and host range but do not require a betasatellite for induction of disease symptoms. Arch. Virol. 157:107–120 [DOI] [PubMed] [Google Scholar]
- 49. Wise AA, Liu Z, Binns AN. 2006. Three methods for the introduction of foreign DNA into Agrobacterium. Methods Mol. Biol. 343:43–53 [DOI] [PubMed] [Google Scholar]
- 50. Kon T, Rojas MR, Abdourhamane IK, Gilbertson RL. 2009. Roles and interactions of begomoviruses and satellite DNAs associated with okra leaf curl disease in Mali, West Africa. J. Gen. Virol. 90:1001–1013 [DOI] [PubMed] [Google Scholar]
- 51. Wang HL, Gilbertson Rl, Lucas WJ. 1996. Spatial and temporal distribution of Bean dwarf mosaic geminivirus in Phaseolus vulgaris and Nicotiana benthamiana. Phytopathology 86:1204–1214 [Google Scholar]
- 52. Rojas MR, Jiang H, Salati R, Xoconostle-Cázares B, Sudarshana MR, Lucas WJ, Gilbertson RL. 2001. Functional analysis of proteins involved in movement of the monopartite begomovirus, Tomato yellow leaf curl virus. Virology 291:110–125 [DOI] [PubMed] [Google Scholar]
- 53. Chen LF, Brannigan K, Clark R, Gilbertson RL. 2010. Characterization of curtoviruses associated with curly top disease of tomato in California and monitoring for these viruses in beet leafhoppers. Plant Dis. 94:99–108 [DOI] [PubMed] [Google Scholar]
- 54. Arendt J, Reznick D. 2008. Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol. Evol. 23:26–32 [DOI] [PubMed] [Google Scholar]
- 55. Carrillo-Tripp J, Lozoya-Gloria E, Rivera-Bustamante RF. 2007. Symptom remission and specific resistance of pepper plants after infection by Pepper golden mosaic virus. Phytopathology 97:51–59 [DOI] [PubMed] [Google Scholar]
- 56. Hagen C, Rojas MR, Kon T, Gilbertson RL. 2008. Recovery from Cucurbit leaf crumple virus (family Geminiviridae, genus Begomovirus) infection is an adaptive antiviral response associated with changes in viral small RNAs. Phytopathology 98:1029–1037 [DOI] [PubMed] [Google Scholar]
- 57. Zamir D, Ekstein-Michelson I, Zakay Y, Navot N, Zeidan M, Sarfatti M, Eshed Y, Harel E, Pleban T, Van-Oss H, Kedar N, Rabinowitch HD, Czosnek H. 1994. Mapping and introgression of a tomato yellow leaf curl virus tolerance gene, TY-1. Theor. Appl. Genet. 88:141–146 [DOI] [PubMed] [Google Scholar]
- 58. Rybicki EP. 1994. A phylogenetic and evolutionary justification for three genera of Geminiviridae. Arch. Virol. 139:49–77 [DOI] [PubMed] [Google Scholar]
- 59. Silva SJC, Castillo-Urquiza GP, Hora-Júnior BT, Assunção IP, Lima GSA, Pio-Ribeiro G, Mizubuti ESG, Zerbini FM. 2012. Species diversity, phylogeny and genetic variability of begomovirus populations infecting leguminous weeds in northeastern Brazil. Plant Pathol. 61:457–467 [Google Scholar]
- 60. Gilbertson RL, Faria J, Ahlquist P, Maxwell DP. 1993. Genetic diversity in geminiviruses causing bean golden mosaic disease: the nucleotide sequence of the infectious cloned DNA components of a Brazilian isolate of bean golden mosaic geminivirus. Phytopathology 83:709–715 [Google Scholar]
- 61. Hagen C, Rojas MR, Sudarshana MR, Xoconostle-Cazares B, Natwick ET, Turini TA, Gilbertson RL. 2008. Biology and molecular characterization of Cucurbit leaf crumple virus, an emergent cucurbit-infecting begomovirus in the Imperial Valley of California. Plant Dis. 92:781–793 [DOI] [PubMed] [Google Scholar]
- 62. Qazi J, Ilyas M, Mansoor S, Briddon RW. 2007. Legume yellow mosaic viruses: genetically isolated begomoviruses. Mol. Plant Pathol. 8:343–348 [DOI] [PubMed] [Google Scholar]
- 63. Gutiérrez S, Michalakis Y, Blanc S. 2012. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2:546–555 [DOI] [PubMed] [Google Scholar]
- 64. Qin S, Ward BM, Lazarowitz SG. 1998. The bipartite geminivirus coat protein aids BR1 function in viral movement by affecting the accumulation of viral single-stranded DNA. J. Virol. 72:9247–9256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou YC, Garrido-Ramirez ER, Sudarshana MR, Yendluri S, Gilbertson RL. 2007. The N-terminus of the begomovirus nuclear shuttle protein (BV1) determines virulence or avirulence in Phaseolus vulgaris. Mol. Plant Microbe Interact. 20:1523–1534 [DOI] [PubMed] [Google Scholar]
- 66. Jupin I, De Kouchkovsky F, Jouanneau F, Gronenborn B. 1994. Movement of Tomato yellow leaf curl geminivirus (TYLCV): involvement of the protein encoded by ORF C4. Virology 204:82–90 [DOI] [PubMed] [Google Scholar]
- 67. Amin I, Patil BL, Briddon RW, Mansoor S, Fauquet CM. 2011. Comparison of phenotypes produced in response to transient expression of genes encoded by four distinct begomoviruses in Nicotiana benthamiana and their correlation with the levels of developmental miRNAs. Virol. J. 8:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dogra SC, Eini O, Rezaian MA, Randles JW. 2009. A novel shaggy-like kinase interacts with the Tomato leaf curl virus pathogenicity determinant C4 protein. Plant Mol. Biol. 71:25–38 [DOI] [PubMed] [Google Scholar]
- 69. Raja P, Wolf JN, Bisaro DM. 2010. RNA silencing directed against geminiviruses: Post-transcriptional and epigenetic components. BBA Gen. Regul. Mech. 1799:337–351 [DOI] [PubMed] [Google Scholar]
- 70. Tomás DM, Cañizares MC, Abad J, Fernández-Muñoz R, Moriones E. 2011. Resistance to Tomato yellow leaf curl virus accumulation in the tomato wild relative Solanum habrochaites associated with the C4 viral protein. Mol. Plant Microbe Interact. 24:849–861 [DOI] [PubMed] [Google Scholar]
- 71. Vanitharani R, Chellappan P, Pita JS, Fauquet CM. 2004. Differential roles of AC2 and AC4 of cassava geminiviruses in mediating synergism and suppression of posttranscriptional gene silencing. J. Virol. 78:9487–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rodríguez-Negrete EA, Carrillo-Tripp J, Rivera-Bustamante RF. 2009. RNA silencing against geminivirus: complementary action of posttranscriptional gene silencing and transcriptional gene silencing in host recovery. J. Virol. 83:1332–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen L-F, Rojas M, Kon T, Gamby K, Xoconostle-Cazares B, Gilbertson RL. 2009. A severe symptom phenotype in tomato in Mali is caused by a reassortant between a novel recombinant begomovirus (Tomato yellow leaf curl Mali virus) and a betasatellite. Mol. Plant Pathol. 10:415–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hou YM, Gilbertson RL. 1996. Increased pathogenicity in a pseudorecombinant bipartite geminivirus correlates with intermolecular recombination. J. Virol. 70:5430–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lefeuvre P, Lett JM, Varsani A, Martin DP. 2009. Widely conserved recombination patterns among single-stranded DNA viruses. J. Virol. 83:2697–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. García-Andrés S, Accotto GP, Navas-Castillo J, Moriones E. 2007. Founder effect, plant host, and recombination shape the emergent population of begomoviruses that cause the tomato yellow leaf curl disease in the Mediterranean basin. Virology 359:302–312 [DOI] [PubMed] [Google Scholar]
- 77. Sanz AI, Fraile A, Gallego JM, Malpica JM, García-Arenal F. 1999. Genetic variability of natural populations of Cotton leaf curl geminivirus, a single-stranded DNA virus. J. Mol. Evol. 49:672–681 [DOI] [PubMed] [Google Scholar]
- 78. Duffy S, Holmes EC. 2008. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus Tomato yellow leaf curl virus. J. Virol. 82:957–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Duffy S, Shackelton LA, Holmes EC. 2008. Rates of evolutionary change in viruses: patterns and determinants. Nat. Rev. Genet. 9:267–276 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.