

Abstract
Most current diagnostic tests for transmissible spongiform encephalopathies (TSE) rely on the presence of proteinase K (PK)-resistant PrPSc (PrP-res) in postmortem tissues as an indication of TSE disease. However, a number of studies have highlighted a discrepancy between TSE infectivity and PrP-res levels in both natural and experimental cases of TSE disease. Previously, we have shown high TSE infectivity levels in the brain tissue of mice that have a clinical TSE disease with associated vacuolar pathology but little or no detectable PrP-res. Here, the levels of TSE infectivity and PrP-res within a peripheral tissue of this mouse model were investigated. Biochemical analysis showed that low levels of PrP-res were present in the spleen tissue in comparison to the levels observed in the spleen of mice infected with ME7 or 79A. However, upon subpassage of brain and spleen tissue from clinically ill mice with little or no PrP-res detectable, similar short incubation periods to disease were observed, indicating that infectivity levels were similarly high in both tissues. Thus, the discrepancy between PrP-res and TSE infectivity was also present in the peripheral tissues of this disease model. This result indicates that peripheral tissues can contain higher levels of infectivity given the correct combination of host species, PrP genotype, and TSE agent. Therefore, the assumption that the levels of peripheral infectivity are lower than those in the central nervous system is not always correct, and this could have implications for current food safety regulations.
INTRODUCTION
Transmissible spongiform encephalopathies (TSE) are a group of fatal neurodegenerative diseases that can affect both humans and animals. TSE can be sporadic, familial, or acquired diseases. The nature of the infectious agent remains unknown; however, the prion hypothesis proposes that a misfolded form (PrPSc) of the host glycoprotein, PrPC, acts as the sole or main component of the infectious agent in TSE disease (1). The abnormal, disease-associated form has increased β-sheet content and is detergent insoluble and partially resistant to proteinase K (PK) digestion. Based on the prion hypothesis, the majority of current diagnostic tests rely on the protease resistance of the disease-associated form (PrP-res) to identify cases of TSE disease postmortem. However, a number of experimental cases of TSE disease have been identified in which PrP-res was not detectable, including wild-type mice infected with bovine spongiform encephalopathy (BSE) (2) and wild-type mice infected with hamster scrapie (3). In both cases, subpassage of the tissue confirmed the presence of TSE infectivity despite the absence of PrP-res.
The highest levels of TSE infectivity are found in tissues of the central nervous system (CNS) of infected animals. However, the infectious agent has also been demonstrated to be present in peripheral tissues of some animals, specifically, the lymphoreticular system (LRS), including the spleen, with the presence of PrP-res and/or TSE infectivity during disease pathogenesis being dependent upon the host species, PrP genotype, and strain of TSE agent. In scrapie-infected sheep, VRQ/VRQ homozygotes have PrP-res deposition in the spleen, while other genotypes, ARR/ARR and VRQ/ARR, do not show any PrP-res accumulation in the spleen (4). In terminal BSE-infected cattle, no PrP-res has been detected in the spleen tissue (5, 6), indicating that the LRS is not involved in the disease pathogenesis of BSE in cattle. However, in sheep experimentally infected with BSE, TSE infectivity and PrPSc have been identified in the peripheral tissues, including the LRS and the spleen (7, 8). In murine models, the infectivity levels in the spleen tissue have been shown to have an initial increase in titer within days of inoculation that then plateaus for the remainder of the disease duration, long before infectivity is detected in the CNS (9, 10). At disease endpoint, the spleen contains lower titers of infectivity than the corresponding brain tissue, regardless of the route of infection. For example, it has been shown that wild-type/139A mouse spleens contain 2 to 3 log10 units less infectivity than the brain (11). In correlation with the infectivity data, the levels of PrP-res in the spleen of infected 139A mice were 500-fold less than those observed in the brain tissue at the terminal stage of disease (11). Further studies showed that the levels of PrP-res were 200- to 300-fold less in the spleen than in the brain of mice infected with a mouse-adapted strain of Gerstmann-Sträussler-Scheinker (GSS) originating from a Fukuoka-1 GSS case (12).
If the peripheral levels of infectivity are equally low in all cases of TSE disease, the current safety measures in place for removal of specified-risk material should prevent any infected material from entering the food chain for human consumption. However, a number of natural and experimental TSE isolates have been identified that do not have the expected association between levels of TSE infectivity and PrP-res in all tissues (2, 3, 13–15). Indeed, one study of atypical scrapie in sheep identified the presence of high levels of TSE infectivity in the spleen tissue despite the absence of abnormal PrP (13). Given that the majority of current diagnostic tests rely on the presence of PrP-res as an indication of TSE disease, disease cases and tissues where PrP-res is absent may be falsely identified as negative. Furthermore, the disparity between PrP-res and TSE infectivity levels may result in an underestimation of the presence of infectivity levels in peripheral tissues that are allowed to enter the food chain, increasing the potential for zoonotic transfer.
We have previously identified a unique mouse model of infectious TSE disease that highlights the discrepancy between PrP-res levels and the corresponding TSE infectious titers (14, 16, 17). Previous work has performed extensive characterization of the relationship between PrP-res and TSE infectivity in the brain tissue of these disease models. Here, we investigate the relationship between PrP-res and TSE infectivity within the peripheral spleen tissue of these mice using both biochemical analyses and mouse bioassay and demonstrate that extremely high levels of infectivity can be present in peripheral tissue without a corresponding increase in PrP-res. If peripheral tissues can be as infectious as brain tissue, this could represent an increased risk to food safety. Indeed, if the correct combination of host species, PrP genotype, and strain of agent that resulted in high peripheral infectivity was to occur naturally, infected peripheral tissue could have the potential to enter the food chain.
MATERIALS AND METHODS
Ethics statement.
All experimental protocols were submitted to the Local Ethical Review Committee for approval before mice were inoculated. All experiments were performed under license and in accordance with the United Kingdom Home Office Regulations [Animals (Scientific Procedures) Act of 1986].
Primary transmission to 101LL transgenic mice.
The inbred, gene-targeted transgenic mouse line 101LL and the wild-type 129/Ola control line have been previously described (16). 101LL/GSS tissues were produced by inoculation of 101LL mice with 1% (wt/vol) brain homogenate from the frontal cortex of a P102L GSS brain that had confirmed clinical GSS disease and abundant PrP-res detectable by immunoblot (provided by J. Ironside, National CJD Surveillance and Research Unit [NCJDSRU], Edinburgh, United Kingdom). 101LL/263K tissues were produced by inoculation of 101LL transgenic mice with 1% (wt/vol) brain homogenate from a 263K-infected hamster. As P102L GSS and 263K do not transmit efficiently to wild-type mice (16, 17), alternative controls were selected to provide tissue from models in which the relationship between PrPSc and infectivity in the periphery had been previously examined (ME7 and 79A mouse-adapted scrapie).
Clinical assessment and vacuolation scoring.
Mice were assessed for the presence of clinical disease as previously described (18) and were culled by cervical dislocation when either a clinical TSE disease or an intercurrent illness was observed. All experiments were performed under license and in accordance with the United Kingdom Home Office Regulations [Animals (Scientific Procedures) Act of 1986]. Brain and spleen tissue were recovered at postmortem for biochemical and immunohistochemical analysis. Half-brain sections (6 μm) were stained using hematoxylin and eosin, and the abundance of TSE-related vacuolation was assessed at nine gray matter regions (medulla, cerebellum, superior colliculus, hypothalamus, thalamus, hippocampus, septum, retrospinal cortex, cingulated, and motor cortex; see areas 1 to 9, respectively, in Fig. 4) and three regions of white matter (cerebellar white matter, midbrain white matter, and cerebral peduncle; see areas 1*, 2*, and 3*, respectively, in Fig. 4) as described previously (10). DNA was extracted from tail tissue that was taken postmortem. PCR and restriction enzyme digestion were performed as previously described (16) to confirm the presence of the P101L mutation.
Fig 4.

Similar lesion profiles, with slight variations in vacuolation levels depending on the tissue type inoculated. Lesion profiles were similar for both brain and spleen tissue inocula in 101LL/GSS(a) (A), 101LL/GSS (b) (B), 101LL/263K(c) (C), and 101LL/263K(d) (D), but a higher vacuolation level was observed after inoculation of the spleen tissue than after inoculation of the brain tissue. In comparison, the lesion profiles from inoculation of brain and spleen tissue from wild-type/79A mice (E) followed the same pattern, with inoculation of the brain tissue producing a slightly higher vacuolation level than the spleen tissue. Numbers 1 to 9 represent nine areas of grey matter scored and 1* to 3* represent three areas of white matter scored (see Materials and Methods).
Subpassage inoculations.
Inocula were prepared from the brains and spleens of two 101LL/GSS mice [designated 101LL/GSS(a) and 101LL/GSS(b)] and two 101LL/263K mice [designated 101LL/263K(c) and 101LL/263K(d)] that all had confirmed clinical TSE disease with associated TSE vacuolar pathology. Inocula were prepared from a wild-type/79A mouse brain and spleen tissue as controls [designated wild-type/79A(e)]. A 10% (wt/vol) homogenate of each tissue was prepared in sterile saline and was used to produce a dilution series of 10−2, 10−3, and 10−4 for each homogenate. Utilizing previous data (14), it was hypothesized that a 10−4 dilution of brain homogenate would produce a 100% attack rate. Given the expected 2- to 3-log10 difference in TSE infectivity titers between brain and spleen tissues in murine models (11), the inoculation of these three dilutions was predicted to allow the identification of differences in incubation period between tissues and to provide an estimation of differences in titer. Each dilution (20 μl) was inoculated intracerebrally under anesthesia into groups of 101LL mice for 101LL/GSS and 101LL/263K inocula and wild-type 129/Ola mice for wild-type/79A inocula. Dilutions of brain homogenate were inoculated into groups of 6 mice at the 10−2 and 10−3 dilutions and 8 mice at the 10−4 dilution. Dilutions of spleen homogenate were inoculated into groups of 6 mice at the 10−2 dilution and 8 mice at the 10−3 and 10−4 dilutions due to the predicted lower titers.
Immunohistochemistry to detect PrP deposition.
Immunohistochemistry was performed to detect PrP deposition in the brain tissue. Briefly, following fixation in 10% formal saline (10% formaldehyde in phosphate-buffered saline [PBS]), brains were treated for 1.5 h in 98% formic acid, dissected, and embedded in paraffin. Sections (6 μm) of brain tissue were hydrated, autoclaved for 15 min at 121°C, and incubated in 98% formic acid for 5 min to expose the PrP epitopes. Sections were incubated in 1% hydrogen peroxide-methanol and washed in 0.2% bovine serum albumin (BSA)-PBS. Sections were blocked with normal goat serum and incubated overnight in anti-PrP monoclonal antibody 6H4 (Prionics) (0.5 μg/ml) and then with secondary anti-mouse biotinylated antibody (Jackson ImmunoResearch Laboratories) (2 μg/ml) for 1 h. Sections were processed using the ABC elite kit (Vector Laboratories), and the signal was visualized by a reaction with hydrogen peroxidase-activated diaminobenzidine.
Precipitation of PrP-res from primary spleen tissue using NaPTA.
Sodium phosphotungstic acid (NaPTA) precipitation has previously been shown to increase the sensitivity of immunoblotting for the detection of PrPSc (19). All incubations were performed at 37°C with agitation. Briefly, spleen tissue from terminal animals was homogenized in 0.1 M Tris-HCl (pH 7.4) to a 10% (wt/vol) homogenate with a glass Dounce homogenizer. Cellular debris was removed through centrifugation at 1,000 rpm for 5 min. An equal volume of 2% Sarkosyl-0.1 M Tris-HCl was added to the supernatant and incubated for 10 min. Homogenates were digested with 50 μg/ml PK for 1 h. Digestion was stopped by the addition of phenylmethylsulfonyl fluoride to 2 mM. PrP-res was selectively precipitated with 0.3% (wt/vol) NaPTA for 20 min. Homogenates were centrifuged at 15,400 × g for 30 min. Pellets were washed with 83 mM EDTA, 0.1% Sarkosyl-0.1 M Tris-HCl, and a further 30 min of centrifugation (15,400 × g) produced the PrP-res pellet, which was resuspended in the appropriate volume of Tris-glycine sample buffer (Invitrogen).
Precipitation of PrP-res with NaPTA from residual inocula and brain tissue of recipient mice.
All incubations were performed at 37°C with agitation to allow precipitation of PrP-res from residual inocula. Briefly, to a 50-μl aliquot of each 10% (wt/vol) homogenate, 0.1 M magnesium chloride was added, and the mixture incubated for 15 min. An equal volume of 4% Sarkosyl-0.1 M Tris-HCl was added to the supernatant and incubated for 5 min. Homogenates were digested with 50 μg/ml PK for 1 h, and precipitation of PrP-res with NaPTA performed as described for spleen tissues. The PrP-res pellet was resuspended in 15 μl of Tris-glycine sample buffer (Invitrogen).
Immunoblotting of PrP-res.
PrP-res pellets produced from NaPTA precipitation were diluted to produce samples from specific tissue equivalents. Samples were loaded on 12% Tris-glycine polyacrylamide gels (Invitrogen) and separated by SDS-PAGE. Separated proteins were transferred onto a polyvinylidene difluoride membrane by the semidry transfer system (Bio-Rad). PrP was detected with monoclonal antibody (MAb) 8H4 (Sigma) using chemiluminescent solution (West Dura ECL substrate, Pierce), with images captured on X-ray film.
RESULTS
Low levels of PrP-res in the spleen tissue of 101LL/GSS and 101LL/263K mice.
Spleens were harvested at cull from several 101LL transgenic mice that had been inoculated with P102L GSS or hamster 263K scrapie. All spleens selected for analysis were from mice which showed clinical signs of TSE disease and confirmed vacuolar pathology but had low levels of PrP deposition in the brain tissue as shown by immunohistochemical analysis. The levels of PrP-res in the spleens of infected mice were investigated to determine if PrP conversion and, therefore, replication of infectivity was occurring in the spleen tissue of these unique disease models. It has previously been shown that it is necessary to concentrate the spleen material in order to detect disease-associated forms of PrP (20) and to minimize IgG cross-reactivity during the immunoblot procedure. Therefore, in these studies, PrP-res in spleen tissue was precipitated using sodium phosphotungstic acid (NaPTA) (21) to concentrate the PrP-res present for detection by immunoblotting. Analysis of the PK-digested, NaPTA-precipitated material identified variations in the levels of PrP-res between individual spleens (Fig. 1A). However, the levels of PrP-res in some 101LL/GSS and 101LL/263K spleens appeared to be lower than the levels of PrP-res in the wild-type/ME7 spleens from which PrP-res was isolated as a control (Fig. 1A). In order to confirm the differences in levels, doubling dilutions of the PK-digested, NaPTA-precipitated material from 101LL/GSS and 101LL/263K spleens and a wild-type/ME7 spleen were analyzed to determine the minimum tissue equivalent at which PrP-res could be detected with a semiquantitative immunoblot (Fig. 1B). 101LL/GSS spleens were shown to contain PrP-res at levels similar to the PrP-res levels in wild-type/ME7 spleens, albeit with lighter band intensities. 101LL/263K spleens contained PrP-res at levels 2-fold lower than the levels observed in the wild-type/ME7 spleens. Importantly, of all the spleen tissue analyzed, that of 3 of 20 101LL/GSS mice and 1 of 12 101LL/263K mice analyzed contained no detectable PrP-res even with NaPTA precipitation.
Fig 1.
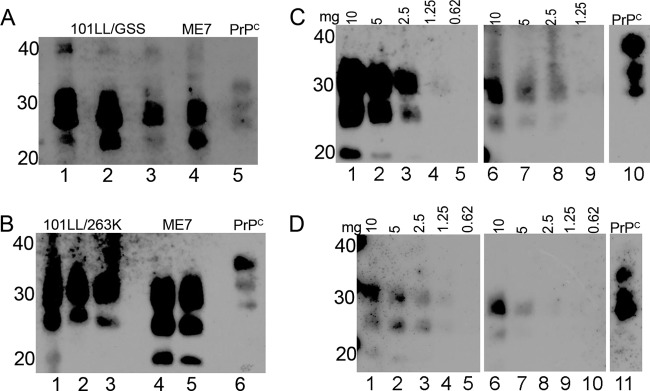
PrP-res levels in 101LL/GSS and 101LL/263K spleens compared to PrP-res levels in wild-type/ME7 spleens. Spleen homogenates were PK digested and NaPTA precipitated, and the samples loaded at a tissue equivalent of 15 mg (A and B) or at different tissue weights (mg) as indicated (C and D). PrP-res was present in 101LL/GSS (A) and 101LL/263K (B) spleens at levels that varied between three individual spleens (lanes 1 to 3). ME7 spleen samples were loaded as a control (A, lane 4; B, lanes 4 and 5). The dilution series (C and D) allowed estimation of the difference in PrP-res levels between 101LL/GSS (C, lanes 6 to 9) or 101LL/263K (D, lanes 6 to 10) spleens and wild-type/ME7 spleens (C and D, lanes 1 to 5). Normal brain homogenate was loaded on each blot to provide a PrPC band as an internal control. Blots were probed with MAb 8H4 and exposed for 5 min. Numbers to the left of the panels indicate molecular weight markers (in kilodaltons).
Brain and spleen tissues show equivalent high levels of infectivity in 101LL/GSS and 101LL/263K mice.
In order to confirm the presence of infectivity in spleen tissue and compare the levels of infectivity present in brain and spleen from the same mouse, 10% (wt/vol) homogenates of brain and spleen from two 101LL/GSS and two 101LL/263K mice were prepared and used to produce dilution series (10−2, 10−3, and 10−4) for inoculation. 101LL/GSS and 101LL/263K brain and spleen homogenates were inoculated into groups of 101LL transgenic mice. As both P102L GSS and hamster 263K scrapie had been shown previously to be poorly transmissible to wild-type mice (16, 17), brain and spleen homogenates from murine 79A scrapie were inoculated as controls. The average incubation periods to disease were similar for each brain-and-spleen-inoculum pair from 101LL/GSS and 101LL/263K mice (Fig. 2). No statistical differences (Student's t test) were observed between the average incubation periods to disease at each dilution for 101LL/263K. Although no statistical differences between incubation periods were observed for the majority of dilutions of 101LL/GSS, the two exceptions were the 101LL/GSS(a) 10−3 dilution of brain homogenate, which had a significantly (P value, 8.3 × 10−5) shorter incubation period to disease than the spleen homogenate at the same dilution, and the 101LL/GSS(b) 10−2 dilution, which had a significantly (P value, 5.6 × 10−4) shorter incubation period to disease from the brain homogenate than from the spleen homogenate. However, these significant differences were not consistent and were not observed for all dilutions of the same inoculum. Overall, the data (Fig. 2) show that the brain and spleen incubation periods were similar in 101LL/GSS and 101LL/263K tissues. In comparison, significantly different incubation periods to disease were observed for brain and spleen homogenates from wild-type/79A mice at each dilution. The average incubation period to disease for the wild-type/79A spleen homogenates was 30 to 40 days longer than the average incubation period to disease with the corresponding brain homogenates.
Fig 2.

Similar average incubation periods to disease from brain and spleen transmissions from 101LL/GSS and 101LL/263K. In comparison, the average incubation periods for transmissions of brain and spleen tissue from wild-type mice were statistically different, with longer incubation periods produced from inoculation with spleen tissue (blue bars). Statistical calculations with Student's two-tailed t test determined P values of 2.88 × 10−5, 3.66 × 10−6, and 1.08 × 10−8 for the 10−2, 10−3, and 10−4 dilution group, respectively, for wild-type/79A transmission. One dilution of 101LL/GSS(a), 10−3, produced a significant difference (P value, 8.3 × 10−5), and one dilution of 101LL/GSS(b), 10−2, produced a significant difference (P value, 5.6 × 10−4).
Analysis of PrP-res levels in residual inocula.
Given the similar infectivity levels of the brain and spleen homogenates, we analyzed the levels of PrP-res present in the residual inocula. Little or no PrP-res was present in the residual brain homogenates from 101LL/GSS and 101LL/263K inocula samples when standard PK digestion and immunoblotting were performed on 10% homogenates (data not shown). NaPTA precipitation was required in order to detect PrP-res levels in the residual spleen homogenate by increasing the sensitivity of detection (19, 20). Low levels of PrP-res were present in the 101LL/GSS(a) and (b) residual spleen homogenates (Fig. 3B). NaPTA precipitation of the residual brain tissue homogenates (Fig. 3A) indicated that similarly low levels of PrP-res were present in 101LL/GSS(a) and (b) and 101LL/263K(c). However, it is important to highlight that no PrP-res was present following NaPTA precipitation of residual brain or spleen homogenate from 101LL/263K(d) despite this inoculum causing infectious TSE disease in the mice that received the inocula. Indeed, no difference was observed in the incubation periods for 101LL/263K(c), which contained low levels of PrP-res, and 101LL/263K(d), which did not contain detectable PrP-res.
Fig 3.
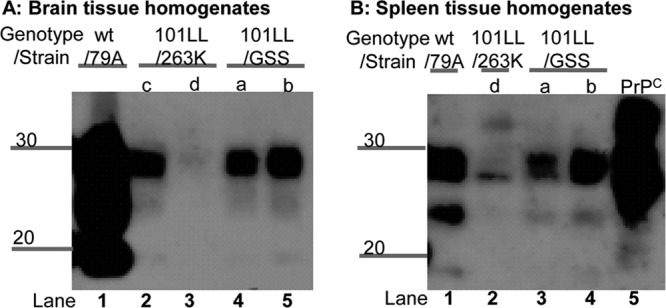
Low PrP-res levels were present in brain (A) and spleen (B) residual inocula as identified by NaPTA precipitation. In contrast, PrP-res was present in wild-type/79A brain and spleen residual homogenates. Residual homogenates were PK digested and NaPTA precipitated. Samples were loaded at a wet weight tissue equivalent of 10 mg, and proteins separated by SDS-PAGE. Immunoblots were probed with MAb 8H4 and exposed for 10 min. Numbers to the left of the panels indicate molecular weight markers (in kilodaltons).
Strain properties are maintained on subpassage, with different levels of vacuolation and PrP deposition.
The lesion profiles produced from 101LL/GSS and 101LL/263K brain and spleen inoculations (Fig. 4) followed a similar pattern, varying only in the level of vacuolation, with higher levels of vacuolation observed from inoculation of the spleen tissue than of the brain tissue. Immunohistochemical analysis of the PrP deposition patterns (Fig. 5) indicated that PrP deposition targeted similar areas in the recipient brains irrespective of the tissue source of the inoculum. In 101LL/GSS mice, PrP deposition was targeted to the hippocampus, thalamus, and midbrain. In cases with heavier deposition, PrP was also observed in the medulla and, occasionally, the septum. However, the PrP deposition observed was lighter in those mice inoculated with 101LL/GSS spleen homogenate than in those inoculated with 101LL/GSS brain homogenate from the same mouse. In 101LL/263K mice, PrP deposition was targeted to the hippocampus, with PrP aggregates present in the corpus callosum, midbrain, and medulla at similar deposition levels irrespective of the tissue source of the inocula.
Fig 5.

Lighter PrPSc deposition was observed from inoculation of spleen homogenates than from inoculation of brain homogenates from 101LL/GSS. Similar levels of PrPSc deposition from inoculation of 101LL/263K homogenates were observed irrespective of the tissue type. Tissues were taken from mice inoculated with the 10−2 dilution of inocula. Immunohistochemistry performed with MAb 6H4. Hippocampus regions are shown at ×2 magnification in the left-hand column. Brain areas containing PrPSc deposition are shown at ×20 magnification.
Various levels of PrP-res in the 101LL/GSS recipient brains depending on tissue type inoculated.
Given the different levels of PrP deposition for 101LL/GSS depending on the tissue source, as observed by immunohistochemistry, recipient brain tissue from animals receiving brain and spleen inocula was analyzed through PK digestion, NaPTA precipitation, and immunoblotting. The results (Fig. 6) confirm that 101LL/GSS mice that received brain homogenate had a greater level of PrP-res than mice that received spleen homogenate. The differences in the levels of PrP-res in 101LL/GSS mice were shown to be present in each dilution group inoculated. In contrast, the levels of PrP-res in 101LL/263K tissues varied independent of the tissue type inoculated, while the levels of PrP-res in wild-type/79A mice were similar irrespective of the tissue type inoculated. These results were confirmed through a number of different techniques, including standard PK digestion (performed at 37°C without the use of NaPTA) and isolation of scrapie-associated fibrils (data not shown).
Fig 6.
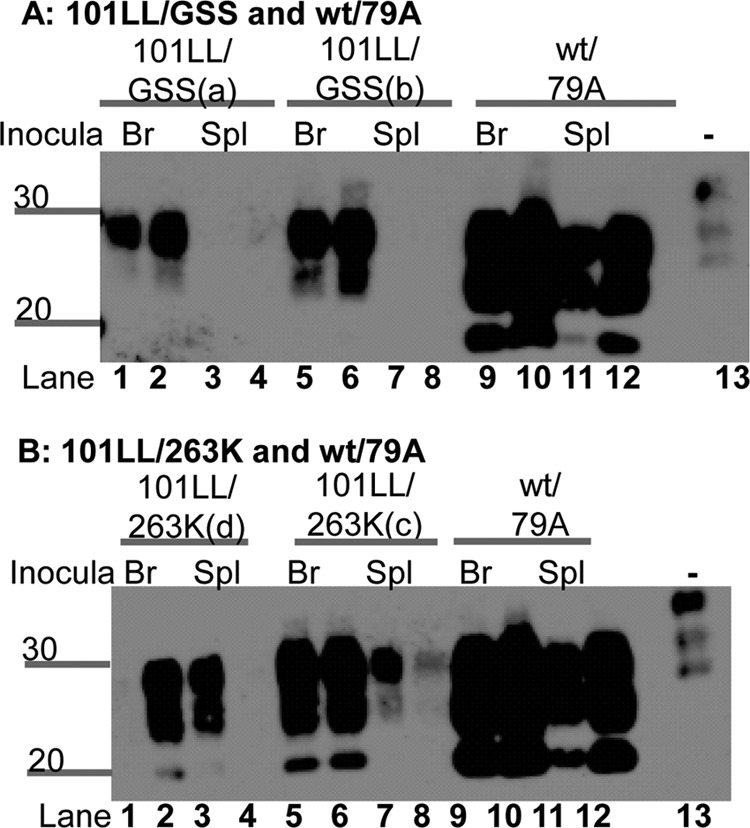
PrP-res levels in recipient mouse brains varied depending upon the tissue type inoculated from 101LL/GSS mice. This phenomenon was observed from both 101LL/GSS(a) and 101LL/GSS(b) mice (A). In contrast, 101LL/263K(c) and 101LL/263K(d) had varvarious levels of PrP-res independent of the tissue type inoculated (B), while wild-type/79A recipient brains had the same level of PrP-res independent of the tissue type inoculated (A and B). Tissue homogenates were PK digested and NaPTA precipitated. Lanes 1, 2, 5, 6, 9, and 10, brain tissue; lanes 3, 4, 7, 8, 11, and 12, spleen tissue. Samples were loaded at the equivalent of a wet tissue weight of 33 mg. An uninfected brain homogenate was loaded at 10 mg/ml (wt/vol) wet weight tissue in lane 13 as an internal control. Blots were probed with MAb 8H4. Numbers to the left of the panels indicate molecular weight markers (in kilodaltons).
DISCUSSION
The prion hypothesis proposes that PrPSc is the sole or main component of the infectious agent in TSE diseases. Based on the prion hypothesis, the majority of TSE diagnostic tests rely on the detection of PrP-res as an indication of TSE disease. However, recently data have shown that PrP-res and TSE infectivity levels do not always correlate, with infectivity being identified in tissues in which PrP-res was not detectable (2, 3, 13, 14). The involvement of the lymphoreticular system in TSE diseases is dependent upon the combination of host species, genotype, and TSE agent. In the majority of murine TSE disease models, the levels of PrP-res present in the spleen are lower than those present in the brain tissue (11, 12). However, in the disease models investigated here, we have shown equally short incubation periods following subpassage of either brain or spleen homogenate from the same mouse, indicating the presence of similar levels of infectivity in both tissues. This was in contrast to the wild-type 79A control, which showed consistently extended incubation times with spleen homogenate (30 to 40 days) when compared to the incubation times with brain homogenate. Based on previous data (14), we can therefore hypothesize that, despite the extremely low levels of PrP-res identified in both brain and spleen of these mice following NaPTA precipitation, the titer of infectivity in each tissue is similar, at approximately 107 to 109 IU/g. Thus, the spleen tissue maintains the discrepancy observed in the brain tissue of these unique disease models, with high infectivity levels being observed despite the presence of low levels or an absence of PrP-res. Full titration studies are currently planned to accurately establish the infectious titer in the spleen tissues of the 101LL/GSS and 101LL/263K mice. However, the initial data from this disease model prove that TSE agents can have the potential to replicate to high infectivity levels in peripheral tissues when the combination of host species, PrP genotype, and TSE agent provides the correct conditions for replication of the infectious agent.
Previous studies of these disease models have shown the presence of little or no PrP-res in brain tissue, despite relatively high levels of infectivity (14, 16, 17). Further investigation here has now demonstrated the presence of low levels of PrP-res in these tissues, as determined after the concentration of approximately 10 mg of tissue homogenate by NaPTA precipitation. Interestingly, the levels of PrP-res detected in brain and spleen of each mouse were similar, and experiments to determine whether this is the truly infectious subpopulation of PrPSc are ongoing in our laboratory. However, despite the identification of similar levels of PrP-res in spleens of 101LL/GSS and 79A mice (Fig. 3), the estimated levels of infectivity, based on incubation times, were significantly higher in 101LL/GSS spleen homogenates. Moreover, 101LL/263K(d) was able to transmit disease from both brain and spleen tissue despite the absence of PrP-res in the residual inocula of both tissues, even with the inclusion of NaPTA precipitation as a concentration step to increase the sensitivity levels. Together, these data support the continued lack of correlation between PrP-res levels and infectivity in these mouse models of disease.
Given that both 101LL/GSS and 101LL/263K had similar incubation periods to disease from both brain and spleen tissue, it can be presumed that this unexpected result was not solely due to the strain of TSE agent or compatibility at codon 101/102. Indeed, the hamster 263K scrapie strain is propagated in animals with proline at PrP codon 102 (equivalent to codon 101 in mice) and does not produce the same disease phenotype when passaged in hamsters. Therefore, the disease phenotype in 101LL mice is not an intrinsic characteristic of the strain but, rather, is due to the specific combination of host species, PrP genotype, and strain of agent that allows this disease phenotype of high infectivity levels but extremely low levels of PrP-res to manifest. It is possible that this phenotype is uniquely due to the 101L mutation in the recipient mice altering the disease pathogenesis or selecting a different isolate from the heterogeneous population of the infectious agent. Further analyses of other TSE strains (e.g., ME7, 79A, and 301V) in these mice (which show PrP-res levels similar to those in wild-type mice [22]) are planned to address this issue.
Several recent studies have identified the presence of quasispecies within individual cases of TSE disease in humans (23, 24) and animals (25) and in cell-culture models (26). Indeed, it has been hypothesized that subvariants of disease-associated PrP replicate preferentially in specific tissue types, with a dependence on tissue-specific host factors (25, 27). The biochemical and immunohistochemical analyses of the recipient mice from the 101LL/GSS subpassage demonstrated that PrP-res deposition was lower in the brain tissue of mice that received the spleen homogenate inocula than in those that received the brain homogenate inocula (Fig. 6), while in contrast, the vacuolation score in mice was greater in those that received the spleen homogenate than in those that received the brain homogenate (Fig. 4). These results indicate that a potentially heterogeneous population of PrP-res was present. It is hypothesized that tissue-specific conditions supported the replication of different subvariants that showed different replication efficiencies upon subpassage. If heterogeneous populations of PrP-res exist that include variants that have different replication capabilities, a variant present in peripheral tissues may have a higher level of infectivity than the corresponding brain-derived variant. Therefore, assessment of the peripheral infectivity levels from novel and emerging isolates is urgently required to ensure that an accurate titer is established to maintain food safety.
Together, these results indicate that a form of the infectious agent may be present in this disease model that remains undetectable by current standard analysis. Given the increasing costs of bioassay to identify the presence of TSE infectivity, the majority of disease cases are being confirmed by biochemical techniques specific for the presence of PrP-res, without confirmation of the presence of infectivity. This current reliance on PrP-res as indicative of TSE disease may not detect all cases of TSE disease, with the possible emergence of cases with high infectivity levels associated with low levels or an absence of PrP-res. Indeed, the discovery of significant levels of TSE infectivity despite the absence of PrP-res in spleens from sheep infected with atypical scrapie (13) indicates that this disease phenomenon can occur in natural cases of TSE disease present in the environment. Furthermore, a recent study by Gonzalez and colleagues highlighted the discrepancy between levels of PrP-res and TSE infectivity in sheep scrapie and sheep BSE and indicated that quantitative laboratory tests to detect disease-associated PrP could not be used to accurately predict infectious titers (28). While TSEs remain in the environment, the emergence of novel isolates or the possibility that a known isolate could infect a different host species remains. Our data show that a combination of host species, PrP genotype, and TSE isolate that could produce a novel disease phenotype with high levels of TSE infectivity in the absence of PrP-res has the potential to occur. Therefore, if the infectivity levels in the peripheral tissues of disease cases with low levels of PrP-res are higher than originally hypothesized from previous research into classical isolates and current biochemical tests, the emergence of a novel isolate could pose a major risk to food safety if tissues were able to enter the food chain. Together with the discrepancy between PrP-res and TSE infectivity levels presented here, the estimation of infectious titer should not rely on the detection of PrP-res as the sole indicator of TSE disease.
ACKNOWLEDGMENTS
We acknowledge J. Manson for providing the 101LL transgenic line, M.W. Head for supervision, advice, and guidance of the Ph.D. studies, S. Cumming, K. Hogan, and R. Greenan for experimental setup and care and scoring of the animals, S. Mack and G. McGregor for histology processing and sectioning, and A. Boyle for vacuolar profiling.
This work was funded by Biotechnology and Biological Sciences Research Council (BBSRC) DTG Studentship BB/D526429/1 and BBSRC Institute Strategic Grant funding BB/J004332/1.
Footnotes
Published ahead of print 13 March 2013
REFERENCES
- 1. Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216:136–144 [DOI] [PubMed] [Google Scholar]
- 2. Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, Fournier JG, Hauw JJ, Rossier J, Dormont D. 1997. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science 275:402–405 [DOI] [PubMed] [Google Scholar]
- 3. Race R, Meade-White K, Raines A, Raymond GJ, Caughey B, Chesebro B. 2002. Subclinical scrapie infection in a resistant species: persistence, replication, and adaptation of infectivity during four passages. J. Infect. Dis. 186(Suppl 2):S166–S170 [DOI] [PubMed] [Google Scholar]
- 4. Andréoletti O, Berthon P, Marc D, Sarradin P, Grosclaude J, van Keulen L, Schelcher F, Elsen JM, Lantier F. 2000. Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 81:3115–3126 [DOI] [PubMed] [Google Scholar]
- 5. Mohri S, Farquhar CF, Somerville RA, Jeffrey M, Foster J, Hope J. 1992. Immunodetection of a disease specific PrP fraction in scrapie-affected sheep and BSE-affected cattle. Vet. Rec. 131:537–539 [PubMed] [Google Scholar]
- 6. Somerville RA, Birkett CR, Farquhar CF, Hunter N, Goldmann W, Dornan J, Grover D, Hennion RM, Percy C, Foster J, Jeffrey M. 1997. Immunodetection of PrPSc in spleens of some scrapie-infected sheep but not BSE-infected cows. J. Gen. Virol. 78(Pt 9):2389–2396 [DOI] [PubMed] [Google Scholar]
- 7. Foster JD, Bruce M, McConnell I, Chree A, Fraser H. 1996. Detection of BSE infectivity in brain and spleen of experimentally infected sheep. Vet. Rec. 138:546–548 [DOI] [PubMed] [Google Scholar]
- 8. van Keulen LJ, Vromans ME, Dolstra CH, Bossers A, van Zijderveld FG. 2008. Pathogenesis of bovine spongiform encephalopathy in sheep. Arch. Virol. 153:445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dickinson AG, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J. Comp. Pathol. 79:363–366 [DOI] [PubMed] [Google Scholar]
- 10. Dickinson AG, Meikle VM, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in the brain of mice. J. Comp. Pathol. 79:15–22 [DOI] [PubMed] [Google Scholar]
- 11. Rubenstein R, Merz PA, Kascsak RJ, Scalici CL, Papini MC, Carp RI, Kimberlin RH. 1991. Scrapie-infected spleens: analysis of infectivity, scrapie-associated fibrils, and protease-resistant proteins. J. Infect. Dis. 164:29–35 [DOI] [PubMed] [Google Scholar]
- 12. Cervenakova L, Yakovleva O, McKenzie C, Kolchinsky S, McShane L, Drohan WN, Brown P. 2003. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43:1687–1694 [DOI] [PubMed] [Google Scholar]
- 13. Andreoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S, Le Dur A, Laude H, Simmons H, Lugan S, Corbiere F, Costes P, Morel N, Schelcher F, Lacroux C. 2011. Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog. 7:e1001285 doi:10.1371/journal.ppat.1001285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. 2007. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J. Biol. Chem. 282:35878–35886 [DOI] [PubMed] [Google Scholar]
- 15. Balkema-Buschmann A, Eiden M, Hoffmann C, Kaatz M, Ziegler U, Keller M, Groschup MH. 2011. BSE infectivity in the absence of detectable PrP(Sc) accumulation in the tongue and nasal mucosa of terminally diseased cattle. J. Gen. Virol. 92:467–476 [DOI] [PubMed] [Google Scholar]
- 16. Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C. 1999. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 18:6855–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barron RM, Thomson V, Jamieson E, Melton DW, Ironside J, Will R, Manson JC. 2001. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO J. 20:5070–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fraser H, Dickinson AG. 1968. The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 78:301–311 [DOI] [PubMed] [Google Scholar]
- 19. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. 2001. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180 [DOI] [PubMed] [Google Scholar]
- 20. Rubenstein R, Kascsak RJ, Merz PA, Papini MC, Carp RI, Robakis NK, Wisniewski HM. 1986. Detection of scrapie-associated fibril (SAF) proteins using anti-SAF antibody in non-purified tissue preparations. J. Gen. Virol. 67(Pt 4):671–681 [DOI] [PubMed] [Google Scholar]
- 21. Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. 1998. Eight prion strains have PrP(Sc) molecules with different conformations. Nat. Med. 4:1157–1165 [DOI] [PubMed] [Google Scholar]
- 22. Barron RM, Thomson V, King D, Shaw J, Melton DW, Manson JC. 2003. Transmission of murine scrapie to P101L transgenic mice. J. Gen. Virol. 84:3165–3172 [DOI] [PubMed] [Google Scholar]
- 23. Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS, Williams MC, McCardle L, MacKenzie J, Knight R, Will RG, Ironside JW. 2004. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991-2002. Ann. Neurol. 55:851–859 [DOI] [PubMed] [Google Scholar]
- 24. Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. 2006. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am. J. Pathol. 168:151–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. 2012. Facilitated cross-species transmission of prions in extraneural tissue. Science 335:472–475 [DOI] [PubMed] [Google Scholar]
- 26. Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. 2010. Darwinian evolution of prions in cell culture. Science 327:869–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Collinge J, Clarke AR. 2007. A general model of prion strains and their pathogenicity. Science 318:930–936 [DOI] [PubMed] [Google Scholar]
- 28. Gonzalez L, Thorne L, Jeffrey M, Martin S, Spiropoulos J, Beck KE, Lockey RW, Vickery CM, Holder T, Terry L. 2012. Infectious titres of sheep scrapie and bovine spongiform encephalopathy agents cannot be accurately predicted from quantitative laboratory test results. J. Gen. Virol. 93:2518–2527 [DOI] [PubMed] [Google Scholar]