

Abstract
Introduction
Mechanistic, translational and pharmacological studies led to the identification, preferred localization, binding characteristics, structure and functional properties of α1-adrenoceptor (α1-AR) subtypes in the bladder neck, bladder and prostate gland. The evidence gathered on α1-ARs, provided a molecular platform for the development of subtype selective antagonists, resulting in more effective approaches targeting those receptors for the treatment of outlet bladder obstruction and benign prostate hyperplasia.
Areas Covered
This review provides a comprehensive synopsis of advances over the last decade, in the design and optimization of Prazosin, Doxazosin, Terazosin quinazoline-based derivatives as clinically effective α1-AR antagonists. Furthermore, it discusses evidence on the metabolic and growth interference action by these agents, in addition to their smooth-muscle relaxing effects. The new action recognition emerges from compelling data on the inhibitory effect of quinazoline-based antagonists on primary tumor growth and progression to metastasis. In addition to the cellular findings in the prostate, functional validation and therapeutic impact of selected lead pharmaceutically optimized derivatives in the context of impairing vascularity and triggering tumor apoptosis, are also summarized.
Expert Opinion
The expanding knowledge on targeting intracellular signalling pathways driving the cellular response via an α1-AR dependent and independent antagonistic action, must be invested towards the optimization of new agents that while bypassing AR, exhibit improved pharmacological efficacy against human cancer.
Keywords: adrenoceptors, benign prostate hyperplasia, cancer therapy, heart failure, quinazoline based derivatives, Prazosin
2. Functional Regulation of Adrenoceptors
All classes of ARs are classical G-protein-coupled receptors (GPCRs) with seven transmembrane domains, even though they differentially activate Gα subunits; β-ARs couple predominantly to Gs, and α1-ARs to Gq, although both β2- and α1-AR subtypes can also couple to Gi [1]. Within the family of GPCRs, ARs mediate the functional signaling of catecholamines, epinephrine and norepinephrine (Figure 1) [2]. α1-ARs are comprised of multiple subtypes that can be classified by both pharmacological and binding studies into at least three subtypes, α1A-AR, α1B-AR, and α1D-AR; specifically, α1-AR subtypes are expressed in different organs of the human body including brain, heart, liver, kidney, prostate, spleen and blood vessels, in which they mediate a wide panel of functional effects such as modulation of neurotransmission, vasoconstriction, cardiac inotropy, chronotropy, and regulation of metabolism [2]. With respect to the multiplicity of α1-AR subtypes, it is of interest that all three subtypes recognize norepinephrine and epinephrine with similar affinity [3]. Functional studies on each AR subtype revealed that the α1A-AR subtypes is responsible for mediating positive inotropic responses in cardiac myocytes of rat right atrium, and it also mediates the contractions of pig uterine artery, rat tail artery, rabbit ear artery, rat renal artery, dog mesenteric artery, rat mesenteric artery, and human umbilical veins, along with contractions of rat corpus cavernosum, human vas deferens, rat vas deferens, human prostate, and human urethra [4]. The α1B-AR subtype in turn mediates contractions of cardiac myocytes of rat right atrium, human prostate (similarly with the α1A-AR subtype), rat spleen, mouse spleen, venules of rat skeletal muscle, pig uterine artery, rat thoracic aorta, rabbit corpus cavernosum, rabbit cutaneous resistance arteries, and canine aorta [4]. Furthermore, the α1D-AR subtype mediates the responses of rat iliac artery, arterioles of rat skeletal muscle, rat carotid artery, rat aorta, rat mesenteric artery, rat pulmonary artery, rabbit aorta, rabbit ventricle myocytes, rat renal artery, and canine mesenteric vein [4].
Figure 1.

Signaling pathways downstream of α1-AR activation. Activation of α1-ARs via agonistic binding of norepinephrine or mutations results in activation of Gα subunits and further activation of various effectors including PLC, PLA2, PLD, leading to induction of different transcription factors leading to heart diseases and possibly cancer [7].
Stimulation of α1-ARs results in the activation of various effectors including phospholipase C (PLC), phospholipase A2 (PLA2), and phospholipase D (PLD), as well as activation of Ca2+ channels, Na+-H+ and Na+-Ca2+ exchange, along with activation or inhibition of K+ channels [3]. Predominantly, the primary functional response after activation of all α1-ARs subtypes is a significant increase in intracellular Ca2+ [3]. Earlier studies revealed that stimulation of α-ARs and β-ARs in neonatal rat cardiomyocytes rapidly induces mRNA up-regulation of early genes c-fos, c-jun, egr-1, and additionally an increase in the assembly of the contractile protein myosin light chain-2 [5]. Furthermore α1A-ARs agonists induce stimulation of phosphoinositide hydrolysis, transcriptional induction of atrial natriuretic factor (ANF) gene expression, and an increase in myocardial cell size [6]. Significantly enough phenylephrine treatment of rat cardiomyocytes increases Raf-1 and MAPK activities with Rho, a member of the Ras superfamily of GTPases, being implicated in the phenylephrine-induced transcriptional activation of ANF and α1A-AR mediated cardiac myofibrillogenesis [7]. Ras as an important regulator of hypertrophy both in vitro and in vivo activates a kinase cascade involving Raf, the mitogen-activated protein kinase kinase (MEK), the extracellular signal-regulated protein kinase (ERK), and can also activate the c-Jun NH2-terminal kinase (JNK) in cardiomyocytes [8]. α1A-AR mediates its effect on JNK through a pathway requiring Ras and MEK kinase (MEKK). Noradrenaline stimulation of α1A-AR in human vascular smooth muscle cells was able to increase DNA synthesis and ERK activity in a PI3-K-dependent mechanism clearly showing that activation of α1A-AR outside the myocardium stimulates mitogenesis [7]. α1A-AR mediated PI3-K activation was further demonstrated to result in stimulation of the 70-kDa S6 kinase (p70S6K), which appears to play a role in the activation of protein synthesis regulation [9]. Moreover α1A-AR stimulation of adult rat hepatocytes markedly potentiated the proliferative effects of transforming growth factor alpha (TGFα) via mechanistic recruitment of PLC, protein kinase C (PKC), MAPK, PI3-K, and p70S6K [10]. In the same study, α2-AR stimulation had a similar positive effect on proliferation, while β2-AR showed antagonistic effects on TGFα mediated proliferation. Establishing the α1-AR mediated MAPK activation and signaling, it was additionally demonstrated that α1-AR interacts with β-arrestin in coordinating a different signaling networks as β-arrestins are scaffolds for components of the MAPK cascade thus mediating MAPK activation induced by various GPCRs [2]. In dissecting further signaling pathways involving α1-AR mediation, it was revealed that α1B-AR inhibits interleukin 6 (IL-6) signaling in hepatocytes after treatment with noradrenaline, by induction of MEK1/p42/44 activity resulting in attenuation of IL-6/STAT3 activation [11]. In addition to Gq coupling, α1A-ARs can signal through Gs resulting in elevation of intracellular cAMP levels, and leading to cyclic AMP-response element-binding protein (CREB) phosphorylation in a PKA dependent manner [12]. Regulation of α1A-ARs is shown to occur under a variety of conditions including hypoxia, ischemic reperfusion, catecholamine stimulation, cAMP levels, and growth factors [7].
While prostate growth is controlled primarily by endocrine related molecules, the nervous system plays an important role in its functions, regulating the prostate via noradrenergic and cholinergic innervation [13]. Noradrenergic nerves primarily innervate the prostatic fibromuscular stroma, and in addition to noradrenaline, other neurotransmitters and neuromodulators may cause contractions and regulate the tone of prostatic smooth muscle via the activation of α1-ARs [13]. The prostate gland is a rich source of α1-ARs, and in prostate cancer epithelial cells α1-ARs are directly functionally coupled to Ca2+-permeable diacylglycerol (DAG) – gated cationic channels activating cationic membrane currents by Ca2+ influx and causing contractions [14]. Stimulation of α1-ARs promotes proliferation of primary human prostate cancer epithelial cells by inducing store-independent Ca2+ entry and subsequent activation of nuclear factor of activated T cells (NFAT) transcription factor [15]. Interestingly, it has been determined that the α1A-AR subtype is dominant in the prostate gland while in two androgen-insensitive, human metastatic cancer cell lines DU145 and PC3 as well as the mouse TRAMP cell lines the major α1-AR subtype found is the α1B-AR [16]. Recently vascular endothelial growth factor (VEGF) expression significantly correlated with activation of α1-ARs. VEGF is a critical inducer for both normal and tumor cell angiogenesis, and its over-expression is associated with progression of and poor prognosis for several tumors, including prostate, breast, as well as hepatocellular carcinoma (HCC) [17]. Norepinephrine induced VEGF expression and HIF-1α protein amount increase via the cAMP-dependent PKA/PI3K/Akt/p70S6K pathway via activation of ARs [17]. In addition, both α1-AR and β-AR are involved in norepinephrine induced VEGF expression and angiogenesis in prostate cancer and HCC cells, whereas only β-AR is required for breast cancer cells [17]. The signal transduction pathways mediated by α1-AR stimulation are summarized in Figure 1. The comparison between hyperplastic and non-hyperplastic prostate tissue revealed that the overall level of α1-AR mRNA does not significantly change, but essential differences are observed in the ratio of the α1-AR subtypes. In both glandular and stromal hyperplasia, there is a significant reduction of the α1B-AR mRNA expression while the α1A-AR is detected in the stroma and not in the glandular epithelium [18]. The α1B-AR is predominantly localized in the epithelium rather than the stroma, whereas the α1D-AR is partially detected in the stroma and is abundant in blood vessels [18]. The α1D-AR subtype is also associated with bladder muscle contraction [19].
3. Therapeutic Targeting of Adrenoceptors
Validation of agents selective for each of the three α1-AR subtypes is ongoing work for over thirty years because of the abundant possible therapeutic applications against different clinical conditions recognized by investigators around the globe in academic settings and big pharmaceutical corporations. In addition to blood pressure reduction, α1-AR antagonists offer the advantage of a favorable effect on plasma lipoproteins and a low incidence of sexual dysfunction [20]. Since the α1A-AR subtype is the predominant receptor involved in human prostate physiology, consequently α1-AR antagonists are effective drugs also for the treatment of benign prostatic hyperplasia (BPH). This is an important consideration given these two conditions occur concomitantly in about 25% of men over the age of 60, and the use of agents to treat both diseases simultaneously is convenient and preferable from the standpoint of patient acceptance, cost, compliance, and safety [21]. BPH as a condition is governed by the non-malignant proliferation of both epithelial and stromal cells mainly in the periurethral and transition zones of the prostate gland, and its incidence approaches about 90% of men by the age of 80 [19]. Prazosin was the first agent reported to block α-AR signaling, and subsequent agents were developed to maximize blocking efficacy and specificity against different AR subtypes. The nature, intensity and tolerance of the side-effects associated with Prazosin is diverse. The most frequently observed side effects observed in patients taking quinazoline α1-AR antagonists include vertigo, dizziness, malaise, headache (that can be well-tolerated), and minor gastro-intestinal disorders (nausea, gastralgia, diarrhea or vomiting). There are also less frequently reported side-effects associated with to cardiovascular toxicity such as postural hypotension, syncope, tachycardia, palpitations, fatigue, drowsiness, rash, flushes and oedema.
3.1 Prazosin and other Quinazoline-based Adrenoceptor Antagonists
As Prazosin (Figure 2) was reported to act as an oral anti-hypertensive agent, its ability to induce peripheral arterioral vasodilatation following direct vascular smooth muscle relaxation and interference with peripheral sympathetic function was established [22]. It was further shown that Prazosin exerts its antihypertensive effect by relaxation of peripheral arterioles as a consequence of post-synaptic α-AR blockade, rather than by direct relaxation of arteriolar vascular muscle, while it had no apparent central action on blood pressure and no effect on neuronal adrenergic function [23]. Prazosin differed from the previously-developed α-AR blockers, phentolamine and phenoxybenzamine, in that they do not block the pre-synaptic α-AR which modulate the release of neurotransmitter, and therefore do not cause as much reflex activation of the sympathetic nervous system [24]. The antihypertensive properties of Prazosin make it straight choice for the treatment of high blood pressure, and in addition it can be used for the treatment of lower urinary tract symptoms (LUTS) associated with BPH, therefore it is a choice for patients who suffer from both problems simultaneously.
Figure 2.

Chemical structures of Prazosin and quinazoline α1-AR antagonistc derivatives Doxazosin, Terazosin, and Tamsulosin [98]. Those agents selectively antagonize the α1-AR mediated contraction of the prostate, prostatic capsule, proximal urethra and bladder base, and urinary symptoms associated with BPH. Recently, Doxazosin, Terazosin and their derivatives have affiliated as potential anti-cancer agents.
The structure of Doxazosin, shown on Figure 2, is a readily absorbed α1-AR antagonist with high bioavailability and long plasma half-life, and this accounts for the prolonged pharmacologic activity of Doxazosin following a single oral dose [25]. Doxazosin has a positive effect on coronary heart disease by decreasing lipids like total cholesterol, total triglycerides, and low density lipoprotein cholesterol, while positive indicators high density lipoprotein and high density lipoprotein/total cholesterol ratio are increased [26]. In BPH, Doxazosin is effective of relieving bladder outflow obstruction through a reduction in prostatic tone mediated via α1-AR blockade [27]. Similar to Doxazosin, Terazosin (Figure 2) and Alfuzosin selectively antagonize the α1-AR mediated contraction of the prostate, prostatic capsule, proximal urethra and bladder base, and urinary symptoms associated with BPH, by reducing the structure tone [28–31].
More than a decade ago we demonstrated that Doxazosin induced smooth muscle cell apoptosis correlated, decreased alpha-smooth muscle actin expression, as an additional mechanism for improving symptoms associated with BPH in patients, while there were no significant changes in the kinetics of proliferation of either prostate epithelial or stromal cells [32]. Moreover, Doxazosin or Terazosin treatment correlated with a significant loss of cell viability in prostate cancer and smooth muscle cells via induction of apoptosis in a dose-dependent manner, whereas it did not induce a significant effect on the rate of cell proliferation [33]. Concurrent inhibition of α1-ARs did not abrogate the apoptotic effect of Doxazosin or Terazosin against human prostate cancer or smooth muscle cells, suggesting that the apoptotic activity of the agent is independent of its α1-AR blockade capacity [33]. Similar results were documented in experimental studies using a mouse model of prostate hyperplasia, in which Doxazosin exhibited a potent apoptotic effect against oncogene induced glandular prostate growth [34]. It was implicated that quinazoline-driven action is potentially effective against hormone-dependent tissues [35]. It was proposed that these quinazoline-based α1-AR antagonists may potentially have some tyrosine kinase activity that can potentially interfere with the signaling of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2/neu), and PI-3K pathways, all of which have previously been shown to have repressive effects on TGF-β signaling [35]. Prostate epithelial cells induce TGF-β expression to counterbalance the increased proliferation thus preventing aberrant prostate growth [36]. The mode of action of quinazoline based tyrosine kinase inhibitors (e.g. Gefitinib) that elicit apoptotic and anti-angiogenic effects against many cancer cell types involves preventing the phosphorylation of tyrosine kinase by competing with the ATP-binding site, and it is likely that the intrinsic quinazoline component confers tyrosine kinase inhibitor activity to α1-AR inhibitors [37]. The sensitivity of cells to Terazosin is shown to be independent of p53 and Rb, however Terazosin mediated apoptosis is associated with G1 phase cell cycle arrest, up-regulation of p27/KIP1, up-regulation of Bax and down-regulation of Bcl-2 [38]. Except inducing apoptosis, Doxazosin and Terazosin were additionally shown to inhibit cell adhesion to the extracellular matrix by inducing anoikis (another form of programmed cell death), and prevent cell invasion and migration of prostate cancer epithelial and vascular endothelial cells [37]. It was additionally indicated that Terazosin treatment results in down-regulation of VEGF and consequently inhibition of angiogenesis in vivo [39], while analysis of clinical BPH specimens revealed a considerable decrease of VEGF protein levels after Terazosin treatment in comparison to the untreated control [40]. Doxazosin treatment alsoinhibited proliferation of murine and human pituitary tumor cells in vitro and in vivo, induced G0-G1 cell cycle arrest, and increased apoptosis independently of its action against α1-AR [41]. Moreover, Doxazosin treatment reduced EGFR phosphorylation and activity in breast cancer cells along with NFκB signaling [42].
Doxazosin, Terazosin, and Alfuzosin are adrenoceptor antagonists that show equal affinity for all α1-AR subtypes and are therefore categorized as non subtype selective [43]. A fourth adrenoceptor antagonist currently in use is Tamsulosin, which is a sulphonamide chemical structure (Fig. 2), with a higher affinity for the α1A-AR and α1D-AR subtypes, that quickly became known as a subtype selective or super-selective antagonist [19]. Because of its specific interaction with α1A-AR, Tamsulosin is effective in patients with mild to severe LUTS associated with BPH, in patients with diabetes mellitus and in the elderly, does not interfere with concomitant antihypertensive therapy, has not been associated with clinically significant changes in blood pressure [44]. Several clinical trials have established that Tamsulosin has an improved safety profile relative to the quinazoline-based α1-adrenoceptor antagonists [45].
3.2 Recent Advances
Quinazoline bearing compounds are in general typified by Prazosin, and the 2,4-diamino-6,7-dimethoxyquinazoline moiety is considered as analogue of noradrenaline, where a dominant role in the AR recognition process is played by the protonated N1 of the moiety which mimics the amine function of the neurotransmitter, protonated at physiological pH [46]. The different localization of AR subtypes accelerated the possibility of designing drugs that selectively interact with distinct AR subtypes, thus avoiding the occurrence of possible side effects in other organs. Silodosin is the most recently developed, highly selective α1A-AR antagonist, and its selectivity towards α1A-AR blockade was reported to be 38 times higher than Tamsulosin hydrochloride, in Chinese hamster ovary cells expressing three human α1A-AR subtypes, exhibit a high selectivity of Silodosin for the lower urinary tract where α1A-AR is the predominant subtype [47]. Tamsulosin preferentially blocks α1A-AR and α1D-AR, with a 10-fold greater affinity than for α1B-AR, whereas Silodosin is highly selective for α1A-AR, with a 162-fold greater affinity than α1B-AR and about a 50-fold greater affinity than for α1D-AR [48]. In comparison with Tamsulosin and Prazosin, Silodosin showed favourable α1A-AR selectivity, as determined by the ratio between the dose required to inhibit intra-urethral pressure and that to decrease blood pressure in rat and dog models [47]. Except blocking α1A-AR signaling and inducing smooth muscle relaxation, Silodosin also targets afferent nerves in the bladder, thus acting on bladder overactivity and storage symptoms [49]. In different randomized clinical trials, Silodosin showed significant improvements in the International Prostate Symptom Score (IPSS), maximum urinary flow rate, and favourable blood pressure related tolerability [47,48,50,51].
The use of monoclonal antibodies prevents ligand binding on receptor tyrosine kinases and small molecule tyrosine kinase inhibitors (TKIs) repressing their enzymatic activity of autophosphorylation and downstream intracellular signaling. EGFR is an excellent therapeutic target for cancer treatment that delivered much promise and hope. The most promising small molecule selective EGFR-TKIs are currently Erlotinib (6,7-bis(2-methoxy-ethoxy)-quinazolin-4-yl-(3-ethynylphenyl)amine) (OSI-774) and Gefitinib (4-(3-chloro-4-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy) quinazoline) (ZD1839), both containing the quinazoline nucleus (Figure 3) [52]. Both TKIs showed promising results in preclinical and clinical studies in many human epithelial cancers, including head and neck squamous-cell carcinoma (HNSCC), non small cell lung cancer (NSCLC), colorectal cancer (CRC), breast cancer, pancreatic cancer and brain cancer, where aberrant expression or activity of EGFR has been identified as an important factor of cancer progression [53]. Vandetanib (ZD6474), another orally bioavailable quinazoline substituted with a halogen at the 2- and 4-positions on the phenyl group in phase III clinical trials, is considered to be a dual tyrosine kinase inhibitor targeting EGFR and VEGFR-2 [54].
Figure 3.
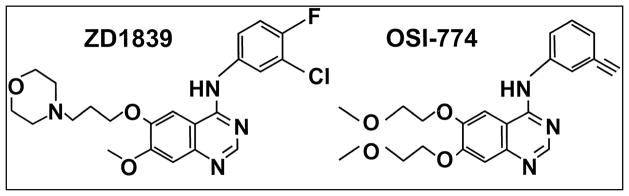
Chemical structures of the tyrosine kinase inhibitors Gefitinib (ZD1839) and Erlotinib (OSI-774) containing the quinazoline core. Both TKIs show promising results in preclinical and clinical studies in many human epithelial cancers, including HNSCC, NSCLC, CRC, breast cancer, pancreatic cancer and brain cancer, where aberrant expression or activity of EGFR has been identified as an important factor of cancer progression.
New quinazoline derivatives were very recently synthesized from 4-chloro-6,7-dimethoxyquinazoline 3, 4-chloro-6,7-methylenedioxyquinazoline, and commercially available anilines, and these molecules were evaluated as potential DNA intercalating agents exhibiting cytotoxic activity via DNA binding [55]. Other compounds also recently synthesized, successfully differentiated the ether linker (methoxy or diethylaminoethoxy) at the C-6 and C-7 positions of the quinazoline core with expected improved water solubility, bioavailability, and cell penetration. These compounds were tested in the androgen-independent prostate cancer cells, PC-3 and the inhibition of various kinase activities; suppression of the basic side chain on the quinazoline core confers an increase of cellular and enzymatic inhibitory activity with high selectivity on EGFR and VEGFR [54]. In addition, replacement of the urea entity by a carbamic acid methyl ester group presented a dual EGFR/VEGFR-2 activity, and it was suggested that this type of compound could bind in the ATP pocket of the receptors with better affinity leading to a more efficient growth inhibition of the tumors [54]. AZD2171 (4-{(4-Fluoro-2-methyl-1H-indol-5-yl)oxy}-6-methoxy-7-{3-(pyrrolidin-1-yl)propoxy}quinazoline) is another orally bioavailable prazosin derivative bearing the quinazoline core that shows strong selectivity and inhibition of VEGF signaling and angiogenesis by targeting VEGFR2 [56]. With its current name Cediranib in new clinical trials, this compound is suggested against a wide array of solid cancers including NSCLC, HCC, RCC, glioblastomas, ovarian, brain, lung, liver, and prostate cancer with the aim of inhibiting angiogenesis and subsequently regressing tumor progression [57–66].
Quinazolines were used as carriers to prepare a series of N-mustard-quinazoline conjugates having a urea or hydrazinecarboxamide linker [67]. These conjugates possess antitumor potential against different human tumor xenografts, and both urea and hydrazinecarboxamide linkers attached to the C-4 position of the 4-aminoquinazolines are able to lower the reactivity of the N-mustard moiety resulting in a longer half-life in rat plasma; in addition the quinazoline core is proved to be valuable carriers for building DNA-directed alkylating agents [67–69]. Doxazosin induces apoptosis in androgen independent human prostate cancer cells by enhancing Fas-associated death domain (FADD) recruitment and caspase-8 activation, indicating Fas-mediated apoptosis as the underlying mechanism, but also including up-regulation of TGFβ signaling and engaging the inhibitor of NFκBα (IκBα) [70,71]. Microarray-based examination of Doxazosin mediated gene expression in prostate cancer cells revealed the rapid up-regulation of two TGF-β1-modulated genes, IκBα and p21WAF-1 via induction of TIEG1 and Smad4 mRNA levels [70], while Doxazosin can also potentially induce increase of Bax protein levels and caspase-8 activation along with caspase-3 activation via FADD recruitment and formation of the death-inducing signaling complex (DISC) [71].
Subsequent structural optimization studies to enhance the apoptotic action of Doxazosin led to the generation of compounds with significantly increased antitumor efficacy. The main modification strategies are illustrated on Figure 4; the aryl carboxamide function of Doxazosin was substituded with aryl sulfonamides to generate intermediate compounds, and then the piperazine moiety of the optimal compounds was replaced by an ethylenediamine linker, while in another strategy the methoxy side chains on the quinazoline ring of the intermediate compounds were replaced [72]. The apoptotic action of Doxazosin was correlated with its efficacy in inhibiting intracellular levels of the survival pathway driven by protein kinase B (PKB)/Akt phosphorylation/activation. Among the new quinazoline compounds, structures with the side chains of tert-butylphenyl, biphenyl, and phenanthren-9-yl-phenyl, represented the optimal compounds, with IC50 values in the range of 5-fold less than Doxazosin resulting in apoptosis induction attributable in part, to the inhibition of Akt activation [72]. Structure-activity studies identify that the lead compound, named DZ-50, significantly reduced the ability of prostate cancer epithelial cells to attach to extracellular matrix and migrate through endothelial cells, while in vivo studies showed that DZ-50 treatment led to significant suppression of tumor growth as well as prevented prostate cancer initiation by targeting tissue vascularity [73]. Furthermore, prostate tumor cell metastatic lung colonization was inhibited by DZ-50, further evidence confirming that the development of this class of lead quinazoline-based compounds generated agents with higher potency and stronger efficancy than Doxazosin, in suppressing prostate growth at lower concentrations, thus potentially minimizing toxicity [73]. This profound anti-angiogenesis action has also been manifested in human renal tumors: DZ-50 was recently demonstrated to significantly inhibit tumor cell adhesion, migration, and invasion at lower doses than Doxazosin in renal cancer cell lines, by repressing the focal adhesion complex signaling and downstream the Akt survival pathway [74]. Additional novel Prazosin related compounds inducing apoptosis were synthesized by an independent group of investigators where 2-Chloro-N-(4-methoxyphenyl)-N-methylquinazolin-4-amine was prepared from reaction of 2,4-dichloroquinazoline with N-methyl-4-methoxyaniline [75]. This compound inhibited tubulin polymerization in cells over-expressing Pgp1, and exerted antitumor action against in the human MX-1 breast and PC-3 prostate cancer mouse models; moreover the methyl group on the nitrogen linker of the compound was essential its the apoptosis-inducing activity and substitution in the 6- and 7-positions of the quinazoline core structure decreased potency against EGFR [75]. Terazosin exists in four solvent-free forms that can be prepared directly to the forms of dehydrate or methanolate, but also an isomorphic solvent free form can be prepared by desolvation of the methanolate, thus showing improved stability than the commercially available Terazosin monohydrochloride [76].
Figure 4.
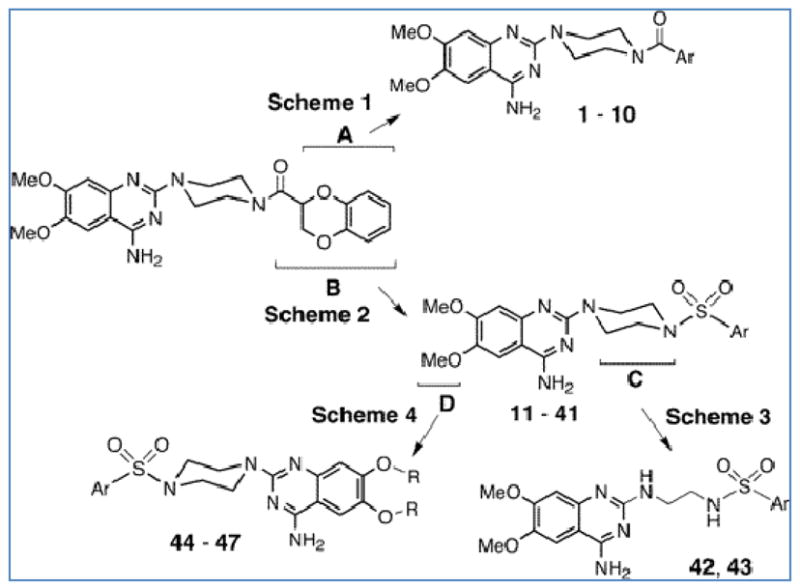
Strategies for structural modifications of doxazosin. A, B, C, and D indicate four modification strategies that target the 2,3-dihydro-benzo{1,4} dioxane moiety, the terminal acyl function, the piperazine linker, and the methoxy side chain ofthe quinazoline base, respectively leading to the synthesis of new Doxazosin derivatives with different properties [72].
Other Prazosin derivatives were also designed by transforming the piperazinyl-quinazoline moiety into an amino-methyl-tetrahydroacridine system [77]. The tetrahydroacridine moiety was shown to be the most promising skeleton for α1-AR antagonism and the derivatives displayed a tendency to selectively target α1B-AR, whereas the pharmacological profile of these compounds at α1A-AR and α1D-AR was significantly negatively affected [77]. In another study, hybrid tetra-amine disulfides were synthesized by combining the structural features of Prazosin, and Benextramine, an irreversible α1/α2-adrenoreceptor antagonist, and their biological profiles were assessed in isolated rat vas deferens (α1A), spleen (α1B), and aorta (α1D), revealing that the disulfide bridge of these molecules was responsible for their binding potential to α1A-AR and α1B-AR but not in α1D-AR; it was postulated that α1A-AR and α1B-AR subtypes but not α1D-AR probably include in their binding pocket a suitable thiol function that would suffer an interchange reaction due to the disulfide moiety of the agents [78]. A critical structural feature, necessary for the α1-AR affinity is a phenyl ring on the nitrogen atom of the piperazine moiety opposite to the protonatable nitrogen atom, thus the phenylpiperazine scaffold is crucial for binding; consequently compounds possessing the positively ionizable nitrogen atom but lacking the integrity of the phenylpiperazine system are inactive [79]. Furthermore, the length of the alkyl chain acting as a spacer between the phenylpiperazine and the terminal moiety has been demonstrated to influence affinity toward a1-AR, and a three or four-carbon atom spacer represents the optimal distance between the two major molecular portions [79]. New compounds were designed and characterized by a flavone system linked through an ethoxy or propoxy spacer to a phenyl- or pyridazinone-piperazine moiety resulting in a nanomolar affinity potential toward α1-AR, whereas affinity was less pronounced for α2-AR [80]. Efforts to enhance the biological action of Prazosin related compounds, aimed at different modifications of the Prazosin structure. Replacing the furoyl moiety of prazosin with the lipoyl fragment of lipoic acid or of its lower homologues or with 1,4-naphthoquinone, new prazosin derivatives were synthesized including both α1-AR antagonist and antioxidant properties, but also anti-proliferative potential against prostate cancer cells [20]. The use of lipoic acid was based on the potential of the molecule to scavenge a number of free radicals in both membrane and aqueous domains and act as a protective anti-oxidant against pathological conditions associated with oxidative stress [20].
The finding that affinity profiles of Prazosin derivatives depends on the type of moiety linking the two nitrogen atoms of the piperazine ring of Prazosin, led to the synthesis of Cystazosin. The structural design of Cystazosin involved replacement of the piperazine ring with a cystamine moiety, which is a structural feature of benextramine, an irreversible α-AR antagonist [81]. Significantly enough, Cystazosin displayed a higher affinity profile for α1D-AR subtype and a significantly lower selectivity for all other α1-AR subtypes when compared with Prazosin [81]. Cyclazosin was another potent molecule synthesized as a Prazosin derivative with imporoved selectivity. Cyclazosin (+)-2-{(4aS,8aR)-4-(2-furoyl)octahydroquinoxalin-1(2H)-yl}-6,7-dimethoxyquinazolin-4-amine-hydrochloride(+)-1} was designed as a cis-octahydroquinoxaline analogue of Prazosin with further insertion of an alkane chain in the octahydroquinoxaline core, and showed very strong selectivity on α1A-ARs [82,83]. Additional Cyclazosin derivatives were generated by introducing structural modifications on its furan ring by integration of selected substituents at position 5 of the furan ring, and replacement of the furan moiety with classical isosteric ring [84]. These derivatives exhibited higher selectivity than Cyclazosin in targeting and binding α1B-AR subtype, while keeping similar selectivity for the α1B-AR over the α1D-AR subtype [84]. Further exploitation of the cyclazosin structure gave rise to two novel analogues , 2-{4-(2,3-dihydro-1,4-benzodioxin-2-ylcarbonyl)-cis-octahydroquinoxalin-1(2H)-yl}-6,7 dimethoxyquinazolin-4-amine (or also called cyclodoxazosin), and 2-{4-(2,3-dihydro-1,4-benzodioxin2-ylcarbonyl)-trans-octahydroquinoxalin-1(2H)-yl}-6,7-dimethoxyquinazolin-4-amine, incorporating a 2,3-dihydro-1,4-benzodioxine-2-carbonyl moiety instead of the 2-furoyl group [85]. These compounds showed enhanced anti-proliferative and apoptotic properties against three different prostate cancer cell lines, while their anti-angiogenic potential was significantly higher than Doxazosin [85]. Several new octahydroquinazoline derivatives structurally similar to Prazosin were generated using Mannich reaction of 3-(4-chlorophenylamino)-5,5-dimethyl-2-cyclohexenone with different aromatic amines in the presence of formaline [86]. The newly synthesized compounds showed high hypotensive effect through α1-AR blockade similar to Prazosin, without causing reflex tachycardia; interestingly there was prolonged duration of action when tested in adrenaline induced hypertension in anaesthetized rats [86].
Computational methods have also been used for designing pharmacologic targeting of ARs with evolving degrees of success. Development of pharmacophores to study the specific structural requirements for antagonistic activity to the various α1-AR subtypes [87], revealed that the most important structural feature of α1-AR antagonists concerning their affinity is the distance between the basic nitrogen the aromatic ring [87]. Recent computational methods have been applied for the development of specific pharmacophores for each α1-AR. Those pharmacophores were generated using the HypoGen algorithm in Catalyst software with the feature options of H-bond acceptor (HBA), H-bond donor (HBD), hydrophobic (aromatic) (Har), hydrophobic (aliphatic) (Hal) and positive ionisable (PI) [88]. The PI feature represents atoms, with the ability to be protonated and thus become positively charged, Har and Hal features were chosen to allow differentiation between aromatic (π-π) interactions and more generic hydrophobic interactions, and uncertainty values were calculated as the ratio between the maximum and minimum affinity values [88]. Additional docking studies with Prazosin (as control compound), revealed that the α1D-AR pocket is the smallest, the α1A-AR pocket the largest and the α1B-AR pocket in between [88].
A major challenge of successful outcomes of cancer chemotherapy is the emergence of drug resistance due to increased cellular expression of P-glycoprotein (Pgp1) and the multidrug resistance protein (MRP1), which act as efflux pumps for various anticancer drugs [89]. Iodoazidoaryl prazosin (IAAP), a photoactive analog of prazosin, has been previously reported to act as a multidrug resistance reversal agent by binding and inhibiting Pgp1 function [90]. As the synthesis of IAAP following the earlier route [91] proved to be dangerously unreliable, a convergent route involving direct addition of an acylated piperazine to 2-chloroquinazoline intermediate was established [92]. Erlotinib can also inhibit multidrug resistance by reversing ABC subfamily B member 1 (ABCB1; P-glycoprotein) and ABC subfamily G member 2 (ABCG2; breast cancer resistance protein/mitoxantrone resistance protein) functions in cancer cells through direct inhibition of the drug efflux function of ABCB1 and ABCG2 [93]. Other TKIs like Imatinib, Nilotinib, and Dasatinib were shown to compete labeling of ABCB1 and ABCG2, pointing to interactions at the Prazosin binding site of both proteins [94].
4. Conclusion
The aggressive pursuit of the expression profile, subtype characterization, cellular localization, pharmacological characteristics, structure and function of the α1-AR subtypes, established an ideal molecular platform for the discovery and development of a large number of subtype selective antagonists that impacted clinical care and therapeutic approaches of various conditions, primarily, benign prostate hyperplasia/bladder outlet obstruction and hypertension [46]. The existence of multiple α1-AR subtypes points out the need of developing new molecules, which target only one receptor while not affecting others localized at different sites. Prazosin still represents a valid choice of treatment for pathologies leading to hypertension and its early developed derivatives have shown clinical improvement in the treatment of LUTS symptoms associated with BPH, as well as hypertension. In this review we summarized the recent advances in design and synthesis of Prazosin derivatives and pharmacologic optimization of new compounds. While the evidence drove patients to embrace the potential use of these agents as potential prevention strategies, their scientific “acceptance” as anti-cancer agents in the pharmacological and clinical arena awaits confirmation.
5. Expert Opinion
Medicinal chemistry attempts to improve the curative properties of the newly synthesized agents and points out the need for compounds that target specifically each α1-AR subtype, while they are also more stable than their parental compounds and can also bear additional properties for activity against other conditions simultaneously, thus leading to monotherapy. In the early 90s when the clinical use of AR antagonists such as Doxazosin, Prazosin and Terazosin reached an exciting peak and the big pharma had secured their place in the clinical area with a great deal of promise, it would be safe to assume that a potential anticancer action was not even philosophically considered. Just over a decade later the scenario has dramatically changed although the acceptance as anti-tumor agents is still challenged. Indeed a new series of compounds that derive from Prazosin, Doxazosin, Terazosin, and Cyclazosin, bearing the quinazoline core, are tested for their efficacy in inducing apoptotic and anti-angiogenic effects predominantly in prostate but also in other cancer models both in vitro and in vivo. Recently, novel quinazoline compounds were further developed with Erlotinib and Lapatinib as templates. The substitution pattern at the 4-substituted quinazoline pharmacophores was selected in order to confer the electronic environment that would affect the lipophilicity and hence the activity of the target molecules, towards the objective of forming these hybrids was an additional attempt to create a potent antitumor agent with enhanced activity and selectivity toward cancerous cells [95]. Novel AR antagonists harbouring the quinazoline nucleus effectively impair tumor growth and progression to metastasis by targeting vascularity of solid tumors via anoikis induction, as shown by in vitro and in vivo studies; moreover such novel compounds can potentially prevent the onset of cancer [73,96]. Ongoing efforts by independent groups are directed at molecular docking studies, pharmacophores, and utilization of software aiming at visualizing the binding sites of α1-ARs and other membrane receptors towards designing new agents with higher affinity and selectivity. Identification of the transmebrane receptors and their downstream signalling pathways, targeting of which impairs tumor growth and progression, is a primary task and a most challenging. Since Doxazosin and its derivative new compound DZ-50, significantly inhibit tumor cell adhesion, migration, and invasion, via disruption of focal adhesions, key regulators of the focal adhesion complex, including focal adhesion kinase (FAK), integrin-linked kinase (ILK), and Talin, were assigned roles as regulators determining the cellular response to these drugs [74,96]. Cell-matrix interactions mediated mainly by integrins and the focal adhesion complex, as well as cell-cell interactions mediated by cadherins, provide not only solid structural support, but also mediate survival of tumor cells via anoikis inhibition [97]. The work demonstrating that a subclass of α1-AR antagonists, (the quinazoline-based), disrupts cancer cell survival and induce apoptosis or anoikis, warrants further efforts to enhance our understanding of mechanisms of apoptotic action and identification of critical anti-apoptotic regulators to be functionally interrupted. Structural exploitation of the different chemical components of Prazosin and related derivatives is also required in understanding the acquisition of anti-tumor action and the molecular targets downstream of α1-AR signaling. The knowledge gathered so far must be processed with careful consideration of the multiple signalling pathways navigating the cellular response to α1-adrenoceptor-dependent and independent antagonistic inhibition towards the optimization of existing and newly synthesized compounds with minimized toxicity and improved pharmacological profiles for the treatment of human cancer.
Highlights.
α1-ARs are comprised of multiple subtypes that can be classified by both pharmacological and binding studies into at least three subtypes, α1A-AR, α1B-AR, and α1D-AR.
Prazosin was the first agent reported to block α-AR activation and signaling, and subsequently further agents were developed to maximize blocking efficacy and specificity against different AR subtypes.
Quinazoline α1-AR antagonistc derivatives Doxazosin, Terazosin, and Tamsulosin selectively antagonize the α1-AR mediated contraction of the prostate, prostatic capsule, proximal urethra and bladder base, and urinary symptoms associated with BPH.
Tyrosine kinase inhibitors Gefitinib and Erlotinib containing the quinazoline core along with Doxazosin, Terazosin and other derivatives show promising results in preclinical and clinical studies as anti-cancer agents.
Ongoing efforts are directed at molecular docking studies, pharmacophores, and utilization of software aiming at visualizing and describing the binding sites of α1-ARs and other receptors in the process of designing new agents with better affinity and selectivity.
Abbreviations
- AR
Adrenoceptor
- GPCR
G-protein-coupled receptor
- PLC
phospholipase C
- PLA2
phospholipase A2
- PLD
phospholipase D
- ANF
atrial natriuretic factor
- MEK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated protein kinase
- JNK
c-Jun NH2-terminal kinase
- MEKK
MEK kinase
- PI3K
phosphoinositol–3 kinase
- p70S6K
70-kDa S6 kinase
- TGFα
transforming growth factor alpha
- PKC
protein kinase C
- IL-6
interleukin 6
- CREB
cyclic AMP-response element-binding protein
- DAG
diacylglycerol
- NFAT
nuclear factor of activated T cells transcription factor
- VEGF
vascular endothelial growth factor
- HCC
hepatocellular carcinoma
- BPH
benign prostatic hyperplasia
- LUTS
lower urinary tract symptoms
- EGFR
epidermal growth factor receptor
- HER2/neu
human epidermal growth factor receptor 2
- IPSS
International Prostate Symptom Score
- TKI
tyrosine kinase inhibitor
- HNSCC
head and neck squamous-cell carcinoma
- NSCLC
non small cell lung cancer
- CRC
colorectal cancer
- IκBα
inhibitor of NFκBα
- PKB
protein kinase B
- HBA
H-bond acceptor
- HBD
H-bond donor
- Har
hydrophobic (aromatic)
- Hal
hydrophobic (aliphatic)
- PI
positive ionisable
- Pgp1
P-glycoprotein
- MRP1
multidrug resistance protein 1
- IAAP
Iodoazidoaryl Prazosin
- ABCB1
ABC subfamily B member 1
- ABCG2
ABC subfamily G member 2
- FADD
Fas-associated death domain
- DISC
death-inducing signaling complex
- FAK
focal adhesion kinase
- ILK
integrin-linked kinase
Footnotes
Declaration of Interest
The authors were supported by the National Institute of Health, grant.
References
- 1.Jensen BC, O'Connell TD, Simpson PC. Alpha-1-adrenergic receptors: Targets for agonist drugs to treat heart failure. J Mol Cell Cardiol. 2010 doi: 10.1016/j.yjmcc.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cotecchia S. The alpha1-adrenergic receptors: Diversity of signaling networks and regulation. J Recept Signal Transduct Res. 2010;30:410–9. doi: 10.3109/10799893.2010.518152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwa J, Graham RM, Perez DM. Identification of critical determinants of alpha 1-adrenergic receptor subtype selective agonist binding. J Biol Chem. 1995;270:23189–95. doi: 10.1074/jbc.270.39.23189. [DOI] [PubMed] [Google Scholar]
- 4.Civantos Calzada B, Aleixandre de Artinano A. Alpha-adrenoceptor subtypes. Pharmacol Res. 2001;44:195–208. doi: 10.1006/phrs.2001.0857. [DOI] [PubMed] [Google Scholar]
- 5.Iwaki K, Sukhatme VP, Shubeita HE, Chien KR. Alpha- and beta-adrenergic stimulation induces distinct patterns of immediate early gene expression in neonatal rat myocardial cells. Fos/jun expression is associated with sarcomere assembly; egr-1 induction is primarily an alpha 1-mediated response. J Biol Chem. 1990;265:13809–17. [PubMed] [Google Scholar]
- 6.Knowlton KU, Michel MC, Itani M, et al. The alpha 1a-adrenergic receptor subtype mediates biochemical, molecular, and morphologic features of cultured myocardial cell hypertrophy. J Biol Chem. 1993;268:15374–80. [PubMed] [Google Scholar]
- 7**.Michelotti GA, Price DT, Schwinn DA. Alpha 1-adrenergic receptor regulation: Basic science and clinical implications. Pharmacol Ther. 2000;88:281–309. doi: 10.1016/s0163-7258(00)00092-9. A compehensive review on α1-AR signaling. [DOI] [PubMed] [Google Scholar]
- 8.Ramirez MT, Sah VP, Zhao XL, et al. The mekk-jnk pathway is stimulated by alpha1-adrenergic receptor and ras activation and is associated with in vitro and in vivo cardiac hypertrophy. J Biol Chem. 1997;272:14057–61. doi: 10.1074/jbc.272.22.14057. [DOI] [PubMed] [Google Scholar]
- 9.Boluyt MO, Zheng JS, Younes A, et al. Rapamycin inhibits alpha 1-adrenergic receptor-stimulated cardiac myocyte hypertrophy but not activation of hypertrophy-associated genes. Evidence for involvement of p70 s6 kinase. Circ Res. 1997;81:176–86. doi: 10.1161/01.res.81.2.176. [DOI] [PubMed] [Google Scholar]
- 10.Kimura M, Ogihara M. Stimulation by transforming growth factor-alpha of DNA synthesis and proliferation of adult rat hepatocytes in primary cultures: Modulation by alpha- and beta-adrenoceptor agonists. J Pharmacol Exp Ther. 1999;291:171–80. [PubMed] [Google Scholar]
- 11.Nguyen VA, Gao B. Cross-talk between alpha(1b)-adrenergic receptor (alpha(1b)ar) and interleukin-6 (il-6) signaling pathways. Activation of alpha(1b)ar inhibits il-6-activated stat3 in hepatic cells by a p42/44 mitogen-activated protein kinase-dependent mechanism. J Biol Chem. 1999;274:35492–8. doi: 10.1074/jbc.274.50.35492. [DOI] [PubMed] [Google Scholar]
- 12.Lin RZ, Chen J, Hu ZW, Hoffman BB. Phosphorylation of the camp response element-binding protein and activation of transcription by alpha1 adrenergic receptors. J Biol Chem. 1998;273:30033–8. doi: 10.1074/jbc.273.45.30033. [DOI] [PubMed] [Google Scholar]
- 13.Pennefather JN, Lau WA, Mitchelson F, Ventura S. The autonomic and sensory innervation of the smooth muscle of the prostate gland: A review of pharmacological and histological studies. J Auton Pharmacol. 2000;20:193–206. doi: 10.1046/j.1365-2680.2000.00195.x. [DOI] [PubMed] [Google Scholar]
- 14.Sydorenko V, Shuba Y, Thebault S, et al. Receptor-coupled, dag-gated ca2+-permeable cationic channels in lncap human prostate cancer epithelial cells. J Physiol. 2003;548:823–36. doi: 10.1113/jphysiol.2002.036772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thebault S, Flourakis M, Vanoverberghe K, et al. Differential role of transient receptor potential channels in ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006;66:2038–47. doi: 10.1158/0008-5472.CAN-05-0376. [DOI] [PubMed] [Google Scholar]
- 16.Shi T, Gaivin RJ, McCune DF, et al. Dominance of the alpha1b-adrenergic receptor and its subcellular localization in human and tramp prostate cancer cell lines. J Recept Signal Transduct Res. 2007;27:27–45. doi: 10.1080/10799890601087487. [DOI] [PubMed] [Google Scholar]
- 17*.Park SY, Kang JH, Jeong KJ, et al. Norepinephrine induces vegf expression and angiogenesis by a hypoxia-inducible factor-1alpha protein-dependent mechanism. Int J Cancer. 2011;128:2306–16. doi: 10.1002/ijc.25589. Activation of α1-ARs correlates with VEGF expression. [DOI] [PubMed] [Google Scholar]
- 18.Walden PD, Gerardi C, Lepor H. Localization and expression of the alpha1a-1, alpha1b and alpha1d-adrenoceptors in hyperplastic and non-hyperplastic human prostate. J Urol. 1999;161:635–40. [PubMed] [Google Scholar]
- 19.Kaplan SA. Current role of alpha-blockers in the treatment of benign prostatic hyperplasia. BJU Int. 2008;102 (Suppl 2):3–7. doi: 10.1111/j.1464-410X.2008.08086.x. [DOI] [PubMed] [Google Scholar]
- 20.Antonello A, Hrelia P, Leonardi A, et al. Design, synthesis, and biological evaluation of prazosin-related derivatives as multipotent compounds. J Med Chem. 2005;48:28–31. doi: 10.1021/jm049153d. [DOI] [PubMed] [Google Scholar]
- 21*.Boyle P, Napalkov P. The epidemiology of benign prostatic hyperplasia and observations on concomitant hypertension. Scand J Urol Nephrol Suppl. 1995;168:7–12. Hypertension and BPH were shown to be often simultaneously present. [PubMed] [Google Scholar]
- 22.Cohen BM. Prazosin hydrochloride (cp-12,299-1), an oral anti-hypertensive agent: Preliminary clinical observations in ambulatory patients. J Clin Pharmacol J New Drugs. 1970;10:408–17. [PubMed] [Google Scholar]
- 23.Brogden RN, Heel RC, Speight TM, Avery GS. Prazosin: A review of its pharmacological properties and therapeutic efficacy in hypertension. Drugs. 1977;14 :163–97. doi: 10.2165/00003495-197714030-00002. [DOI] [PubMed] [Google Scholar]
- 24.Stokes GS, Marwood JF. Review of the use of alpha-adrenoceptor antagonists in hypertension. Methods Find Exp Clin Pharmacol. 1984;6:197–204. [PubMed] [Google Scholar]
- 25.Taylor SH. Clinical pharmacotherapeutics of doxazosin. Am J Med. 1989;87:2S–11S. doi: 10.1016/0002-9343(89)90107-1. [DOI] [PubMed] [Google Scholar]
- 26.Pool JL, Taylor AA, Nelson EB. Review of the effects of doxazosin, a new selective alpha 1-adrenergic inhibitor, on lipoproteins in patients with essential hypertension. Am J Med. 1989;87:57S–61S. doi: 10.1016/0002-9343(89)90115-0. [DOI] [PubMed] [Google Scholar]
- 27.Fulton B, Wagstaff AJ, Sorkin EM. Doxazosin. An update of its clinical pharmacology and therapeutic applications in hypertension and benign prostatic hyperplasia. Drugs. 1995;49:295–320. doi: 10.2165/00003495-199549020-00011. [DOI] [PubMed] [Google Scholar]
- 28.Wilde MI, Fitton A, Sorkin EM. Terazosin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in benign prostatic hyperplasia. Drugs Aging. 1993;3:258–77. doi: 10.2165/00002512-199303030-00007. [DOI] [PubMed] [Google Scholar]
- 29.Wilde MI, Fitton A, McTavish D. Alfuzosin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in benign prostatic hyperplasia. Drugs. 1993;45:410–29. doi: 10.2165/00003495-199345030-00008. [DOI] [PubMed] [Google Scholar]
- 30.McKeage K, Plosker GL. Alfuzosin. A review of the therapeutic use of the prolonged-release formulation given once daily in the management of benign prostatic hyperplasia. Drugs. 2002;62:633–53. doi: 10.2165/00003495-200262040-00009. [DOI] [PubMed] [Google Scholar]
- 31.Nickel JC. Role of alpha1-blockers in chronic prostatitis syndromes. BJU Int. 2008;101(Suppl 3):11–6. doi: 10.1111/j.1464-410X.2008.07496.x. [DOI] [PubMed] [Google Scholar]
- 32.Kyprianou N, Litvak JP, Borkowski A, Alexander R, Jacobs SC. Induction of prostate apoptosis by doxazosin in benign prostatic hyperplasia. J Urol. 1998;159:1810–5. doi: 10.1016/S0022-5347(01)63162-8. [DOI] [PubMed] [Google Scholar]
- 33**.Kyprianou N, Benning CM. Suppression of human prostate cancer cell growth by alpha1-adrenoceptor antagonists doxazosin and terazosin via induction of apoptosis. Cancer Res. 2000;60:4550–5. Potential of Doxazosin and Terazosin as anti-cancer agents was demonstrated. [PubMed] [Google Scholar]
- 34.Yang G, Timme TL, Park SH, et al. Transforming growth factor beta 1 transduced mouse prostate reconstitutions: Ii. Induction of apoptosis by doxazosin. Prostate. 1997;33:157–63. doi: 10.1002/(sici)1097-0045(19971101)33:3<157::aid-pros2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 35.Anglin IE, Glassman DT, Kyprianou N. Induction of prostate apoptosis by alpha1-adrenoceptor antagonists: Mechanistic significance of the quinazoline component. Prostate Cancer Prostatic Dis. 2002;5:88–95. doi: 10.1038/sj.pcan.4500561. [DOI] [PubMed] [Google Scholar]
- 36.Kyprianou N, Tu H, Jacobs SC. Apoptotic versus proliferative activities in human benign prostatic hyperplasia. Hum Pathol. 1996;27:668–75. doi: 10.1016/s0046-8177(96)90396-2. [DOI] [PubMed] [Google Scholar]
- 37.Tahmatzopoulos A, Kyprianou N. Apoptotic impact of alpha1-blockers on prostate cancer growth: A myth or an inviting reality? Prostate. 2004;59:91–100. doi: 10.1002/pros.10357. [DOI] [PubMed] [Google Scholar]
- 38.Xu K, Wang X, Ling PM, Tsao SW, Wong YC. The alpha1-adrenoceptor antagonist terazosin induces prostate cancer cell death through a p53 and rb independent pathway. Oncol Rep. 2003;10:1555–60. doi: 10.3892/or.10.5.1555. [DOI] [PubMed] [Google Scholar]
- 39.Pan SL, Guh JH, Huang YW, et al. Identification of apoptotic and antiangiogenic activities of terazosin in human prostate cancer and endothelial cells. J Urol. 2003;169:724–9. doi: 10.1097/01.ju.0000037731.83941.db. [DOI] [PubMed] [Google Scholar]
- 40*.Keledjian K, Borkowski A, Kim G, et al. Reduction of human prostate tumor vascularity by the alpha1-adrenoceptor antagonist terazosin. Prostate. 2001;48:71–8. doi: 10.1002/pros.1083. Decrease of VEGF protein levels after Terazosin treatment in clinical BPH specimens was revealed. [DOI] [PubMed] [Google Scholar]
- 41.Fernando MA, Heaney AP. Alpha1-adrenergic receptor antagonists: Novel therapy for pituitary adenomas. Mol Endocrinol. 2005;19:3085–96. doi: 10.1210/me.2004-0471. [DOI] [PubMed] [Google Scholar]
- 42.Hui H, Fernando MA, Heaney AP. The alpha1-adrenergic receptor antagonist doxazosin inhibits egfr and nf-kappab signalling to induce breast cancer cell apoptosis. Eur J Cancer. 2008;44:160–6. doi: 10.1016/j.ejca.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 43.Beduschi MC, Beduschi R, Oesterling JE. Alpha-blockade therapy for benign prostatic hyperplasia: From a nonselective to a more selective alpha1a-adrenergic antagonist. Urology. 1998;51:861–72. doi: 10.1016/s0090-4295(98)00140-x. [DOI] [PubMed] [Google Scholar]
- 44.Dunn CJ, Matheson A, Faulds DM. Tamsulosin: A review of its pharmacology and therapeutic efficacy in the management of lower urinary tract symptoms. Drugs Aging. 2002;19:135–61. doi: 10.2165/00002512-200219020-00004. [DOI] [PubMed] [Google Scholar]
- 45.Michel MC, Flannery MT, Narayan P. Worldwide experience with alfuzosin and tamsulosin. Urology. 2001;58:508–16. doi: 10.1016/s0090-4295(01)01335-8. [DOI] [PubMed] [Google Scholar]
- 46.Rosini M, Bolognesi ML, Giardina D, et al. Recent advances in alpha1-adrenoreceptor antagonists as pharmacological tools and therapeutic agents. Curr Top Med Chem. 2007;7:147–62. doi: 10.2174/156802607779318244. [DOI] [PubMed] [Google Scholar]
- 47.Kawabe K, Yoshida M, Homma Y. Silodosin, a new alpha1a-adrenoceptor-selective antagonist for treating benign prostatic hyperplasia: Results of a phase iii randomized, placebo-controlled, double-blind study in japanese men. BJU Int. 2006;98 :1019–24. doi: 10.1111/j.1464-410X.2006.06448.x. [DOI] [PubMed] [Google Scholar]
- 48.Rossi M, Roumeguere T. Silodosin in the treatment of benign prostatic hyperplasia. Drug Des Devel Ther. 2010;4:291–7. doi: 10.2147/DDDT.S10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yokoyama O, Ito H, Aoki Y, et al. Selective alpha1a-blocker improves bladder storage function in rats via suppression of c-fiber afferent activity. World J Urol. 2010;28:609–14. doi: 10.1007/s00345-009-0481-2. [DOI] [PubMed] [Google Scholar]
- 50.Chapple CR, Montorsi F, Tammela TL, et al. Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: Results of an international, randomized, double-blind, placebo- and active-controlled clinical trial performed in europe. Eur Urol. 2011;59:342–52. doi: 10.1016/j.eururo.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 51.Lepor H, Hill LA. Silodosin for the treatment of benign prostatic hyperplasia: Pharmacology and cardiovascular tolerability. Pharmacotherapy. 2010;30:1303–12. doi: 10.1592/phco.30.12.1303. [DOI] [PubMed] [Google Scholar]
- 52.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7:2958–70. [PubMed] [Google Scholar]
- 53**.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to egfr inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7:493–507. doi: 10.1038/nrclinonc.2010.97. A review on EGFR inhibitors and treatment strategies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garofalo A, Goossens L, Lemoine A, Farce A, Arlot Y, Depreux P. Quinazoline-urea, new protein kinase inhibitors in treatment of prostate cancer. J Enzyme Inhib Med Chem. 2010;25:158–71. doi: 10.3109/14756360903169485. [DOI] [PubMed] [Google Scholar]
- 55.Garofalo A, Goossens L, Baldeyrou B, et al. Design, synthesis, and DNA-binding of n-alkyl(anilino)quinazoline derivatives. J Med Chem. 2010;53:8089–103. doi: 10.1021/jm1009605. [DOI] [PubMed] [Google Scholar]
- 56.Wedge SR, Kendrew J, Hennequin LF, et al. Azd2171: A highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 57.Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol Oncol. 2011;121:230–38. doi: 10.1016/j.ygyno.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 58.Lindsay CR, MacPherson IR, Cassidy J. Current status of cediranib: The rapid development of a novel anti-angiogenic therapy. Future Oncol. 2009;5:421–32. doi: 10.2217/fon.09.18. [DOI] [PubMed] [Google Scholar]
- 59.Ulahannan SV, Brahmer JR. Antiangiogenic agents in combination with chemotherapy in patients with advanced non-small cell lung cancer. Cancer Invest. 2011;29:325–37. doi: 10.3109/07357907.2011.554476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huynh H. Molecularly targeted therapy in hepatocellular carcinoma. Biochem Pharmacol. 2010;80:550–60. doi: 10.1016/j.bcp.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 61.Ansari J, Glaholm J, McMenemin R, et al. Recent advances and future directions in the management of metastatic renal cell carcinoma. Anticancer Agents Med Chem. 2010;10:225–35. doi: 10.2174/1871520611009030225. [DOI] [PubMed] [Google Scholar]
- 62.Huynh H. Tyrosine kinase inhibitors to treat liver cancer. Expert Opin Emerg Drugs. 2010;15:13–26. doi: 10.1517/14728210903571659. [DOI] [PubMed] [Google Scholar]
- 63.Aragon-Ching JB, Dahut WL. Vegf inhibitors and prostate cancer therapy. Curr Mol Pharmacol. 2009;2:161–8. doi: 10.2174/1874467210902020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerstner ER, Duda DG, di Tomaso E, et al. VEGf inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6:229–36. doi: 10.1038/nrclinonc.2009.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wheatley-Price P, Shepherd FA. Targeting angiogenesis in the treatment of lung cancer. J Thorac Oncol. 2008;3:1173–84. doi: 10.1097/JTO.0b013e318187220f. [DOI] [PubMed] [Google Scholar]
- 66.Dietrich J, Wang D, Batchelor TT. Cediranib: Profile of a novel anti-angiogenic agent in patients with glioblastoma. Expert Opin Investig Drugs. 2009;18:1549–57. doi: 10.1517/13543780903183528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marvania B, Lee PC, Chaniyara R, et al. Design, synthesis and antitumor evaluation of phenyl n-mustard-quinazoline conjugates. Bioorg Med Chem. 2011;19:1987–98. doi: 10.1016/j.bmc.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 68.Kapuriya N, Kakadiya R, Dong H, et al. Design, synthesis, and biological evaluation of novel water-soluble n-mustards as potential anticancer agents. Bioorg Med Chem. 2011;19:471–85. doi: 10.1016/j.bmc.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Chaniyara R, Kapuriya N, Dong H, et al. Novel bifunctional alkylating agents, 5,10-dihydropyrrolo[1,2-b]isoquinoline derivatives, synthesis and biological activity. Bioorg Med Chem. 2011;19:275–86. doi: 10.1016/j.bmc.2010.11.030. [DOI] [PubMed] [Google Scholar]
- 70*.Partin JV, Anglin IE, Kyprianou N. Quinazoline-based alpha 1-adrenoceptor antagonists induce prostate cancer cell apoptosis via tgf-beta signalling and i kappa b alpha induction. Br J Cancer. 2003;88:1615–21. doi: 10.1038/sj.bjc.6600961. Correlation of Doxazosin and TGFβ signaling in inducing apoptosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cells via a death receptor-mediated pathway. Cancer Res. 2006;66:464–72. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shaw YJ, Yang YT, Garrison JB, et al. Pharmacological exploitation of the alpha1-adrenoreceptor antagonist doxazosin to develop a novel class of antitumor agents that block intracellular protein kinase b/akt activation. J Med Chem. 2004;47:4453–62. doi: 10.1021/jm049752k. [DOI] [PubMed] [Google Scholar]
- 73.Garrison JB, Shaw YJ, Chen CS, Kyprianou N. Novel quinazoline-based compounds impair prostate tumorigenesis by targeting tumor vascularity. Cancer Res. 2007;67 :11344–52. doi: 10.1158/0008-5472.CAN-07-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74**.Sakamoto S, Schwarze S, Kyprianou N. Anoikis disruption of focal adhesion-akt signaling impairs renal cell carcinoma. Eur Urol. 2011;59:734–44. doi: 10.1016/j.eururo.2010.12.038. It was shown that Doxazosin derivatives can induce anoikis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sirisoma N, Kasibhatla S, Pervin A, et al. Discovery of 2-chloro-n-(4-methoxyphenyl)-n-methylquinazolin-4-amine (ep12825, mpi-0441138) as a potent inducer of apoptosis with high in vivo activity. J Med Chem. 2008;51:4771–79. doi: 10.1021/jm8003653. [DOI] [PubMed] [Google Scholar]
- 76.Bauer J, Morley J, Spanton S, et al. Identification, preparation, and characterization of several polymorphs and solvates of terazosin hydrochloride. J Pharm Sci. 2006;95 :917–28. doi: 10.1002/jps.20425. [DOI] [PubMed] [Google Scholar]
- 77.Rosini M, Antonello A, Cavalli A, et al. Prazosin-related compounds. Effect of transforming the piperazinylquinazoline moiety into an aminomethyltetrahydroacridine system on the affinity for alpha1-adrenoreceptors. J Med Chem. 2003;46:4895–903. doi: 10.1021/jm030952q. [DOI] [PubMed] [Google Scholar]
- 78.Bolognesi ML, Marucci G, Angeli P, et al. Analogues of prazosin that bear a benextramine-related polyamine backbone exhibit different antagonism toward alpha1-adrenoreceptor subtypes. J Med Chem. 2001;44:362–71. doi: 10.1021/jm000995w. [DOI] [PubMed] [Google Scholar]
- 79.Manetti F, Corelli F, Strappaghetti G, Botta M. Arylpiperazines with affinity toward alpha(1)-adrenergic receptors. Curr Med Chem. 2002;9:1303–21. doi: 10.2174/0929867023369961. [DOI] [PubMed] [Google Scholar]
- 80.Betti L, Floridi M, Giannaccini G, et al. Design, synthesis, and alpha(1)-adrenoceptor binding properties of new arylpiperazine derivatives bearing a flavone nucleus as the terminal heterocyclic molecular portion. Bioorg Med Chem. 2004;12:1527–35. doi: 10.1016/j.bmc.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 81.Minarini A, Budriesi R, Chiarini A, et al. Search for alpha 1-adrenoceptor subtypes selective antagonists: Design, synthesis and biological activity of cystazosin, an alpha 1d-adrenoceptor antagonist. Bioorg Med Chem Lett. 1998;8:1353–58. doi: 10.1016/s0960-894x(98)00217-0. [DOI] [PubMed] [Google Scholar]
- 82.Giardina D, Gulini U, Massi M, et al. Structure-activity relationships in prazosin-related compounds. 2. Role of the piperazine ring on alpha-blocking activity. J Med Chem. 1993;36:690–8. doi: 10.1021/jm00058a005. [DOI] [PubMed] [Google Scholar]
- 83.Giardina D, Crucianelli M, Melchiorre C, et al. Receptor binding profile of cyclazosin, a new alpha 1b-adrenoceptor antagonist. Eur J Pharmacol. 1995;287:13–6. doi: 10.1016/0014-2999(95)00471-7. [DOI] [PubMed] [Google Scholar]
- 84.Sagratini G, Angeli P, Buccioni M, et al. Synthesis and alpha(1)-adrenoceptor antagonist activity of derivatives and isosters of the furan portion of (+)-cyclazosin. Bioorg Med Chem. 2007;15:2334–45. doi: 10.1016/j.bmc.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 85.Giardina D, Martarelli D, Sagratini G, et al. Doxazosin-related alpha1-adrenoceptor antagonists with prostate antitumor activity. J Med Chem. 2009;52:4951–4. doi: 10.1021/jm8016046. [DOI] [PubMed] [Google Scholar]
- 86.el-Sabbagh OI, Shabaan MA, Kadry HH, Al-Din ES. New octahydroquinazoline derivatives: Synthesis and hypotensive activity. Eur J Med Chem. 2010;45:5390–6. doi: 10.1016/j.ejmech.2010.08.064. [DOI] [PubMed] [Google Scholar]
- 87*.Bremner JB, Griffith R, Coban B. Ligand design for alpha(1) adrenoceptors. Curr Med Chem. 2001;8:607–20. doi: 10.2174/0929867013373110. Development of pharmacophores for ligand design of α1-ARs. [DOI] [PubMed] [Google Scholar]
- 88.MacDougall IJ, Griffith R. Selective pharmacophore design for alpha1-adrenoceptor subtypes. J Mol Graph Model. 2006;25:146–57. doi: 10.1016/j.jmgm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 89.Nobili S, Landini I, Mazzei T, Mini E. Overcoming tumor multidrug resistance using drugs able to evade p-glycoprotein or to exploit its expression. Med Res Rev. 2011 doi: 10.1002/med.20239. [DOI] [PubMed] [Google Scholar]
- 90.Dey S, Ramachandra M, Pastan I, et al. Evidence for two nonidentical drug-interaction sites in the human p-glycoprotein. Proc Natl Acad Sci U S A. 1997;94 :10594–9. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seidman CE, Hess HJ, Homcy CJ, Graham RM. Synthesis and characterization of a radioiodinated photoaffinity probe for the alpha 1-adrenergic receptor. Hypertension. 1984;6:I 7–11. doi: 10.1161/01.hyp.6.2_pt_2.i7. [DOI] [PubMed] [Google Scholar]
- 92.Andrus MB, Mettath SN, Song C. A modified synthesis of iodoazidoaryl prazosin. J Org Chem. 2002;67:8284–6. doi: 10.1021/jo026217o. [DOI] [PubMed] [Google Scholar]
- 93.Shi Z, Peng XX, Kim IW, et al. Erlotinib (tarceva, osi-774) antagonizes atp-binding cassette subfamily b member 1 and atp-binding cassette subfamily g member 2-mediated drug resistance. Cancer Res. 2007;67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 94.Dohse M, Scharenberg C, Shukla S, et al. Comparison of atp-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos. 2010;38:1371–80. doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.El-Azab AS, Al-Omar MA, Abdel-Aziz AA, et al. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. Eur J Med Chem. 2010;45:4188–98. doi: 10.1016/j.ejmech.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 96.Keledjian K, Garrison JB, Kyprianou N. Doxazosin inhibits human vascular endothelial cell adhesion, migration, and invasion. J Cell Biochem. 2005;94:374–88. doi: 10.1002/jcb.20240. [DOI] [PubMed] [Google Scholar]
- 97.Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Aspects Med. 2010;31:205–14. doi: 10.1016/j.mam.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kyprianou N. Doxazosin and terazosin suppress prostate growth by inducing apoptosis: Clinical significance. J Urol. 2003;169:1520–25. doi: 10.1097/01.ju.0000033280.29453.72. [DOI] [PubMed] [Google Scholar]