

Abstract
The title compound, C8H13NO2Se, crystallizes as a non-merohedral twin with an approximate 9:1 component ratio with two symmetry-independent molecules in the asymmetric unit. Our density-functional theory (DFT) computations indicate that the carboxy C atom is expected to be slightly pyramidal due to an n→ π* interaction, wherein the lone pair (n) of the Se atom overlap with the antibonding orbital (π*) of the carbonyl group. Such pyramidalization is observed in one molecule of the title compound but not the other.
Related literature
For background to hybrid density functional theory (DFT) and natural bond orbital (NBO) analysis, see: Glendening et al. (2001 ▶); Weinhold (1998 ▶); Weinhold & Landis (2005 ▶). For literature related to the synthesis, see: Bhattacharyya & Woollins (2001 ▶) and for NBO studies of the title compound, see: Choudhary & Raines (2011a
▶); DeRider et al. (2002 ▶); Choudhary et al. (2009 ▶, 2010a
▶,b
▶); Jakobsche et al. (2010 ▶); Bartlett et al. (2010 ▶); Choudhary & Raines (2011b
▶). For geometrical checks with ConQuest and Mercury, see: Bruno et al. (2002 ▶). For Gaussian 03 software, see: Frisch (2004 ▶). For puckering parameters, see: Cremer & Pople (1975 ▶).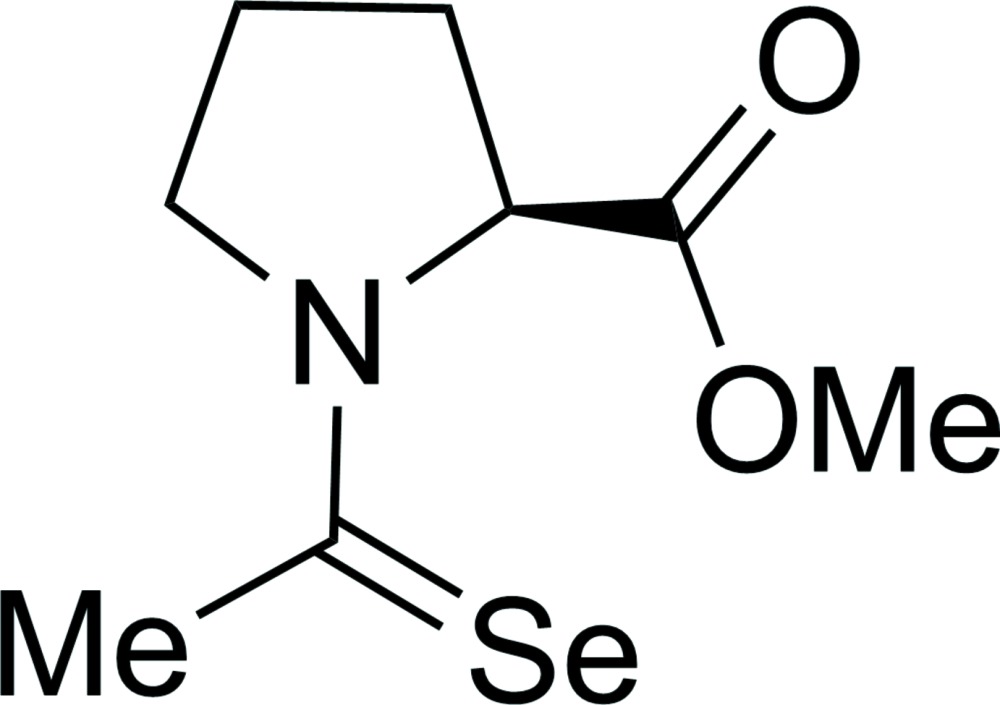
Experimental
Crystal data
C8H13NO2Se
M r = 234.15
Triclinic,
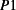
a = 7.050 (3) Å
b = 7.442 (3) Å
c = 10.334 (4) Å
α = 85.166 (6)°
β = 86.220 (6)°
γ = 64.682 (4)°
V = 488.1 (3) Å3
Z = 2
Mo Kα radiation
μ = 3.81 mm−1
T = 105 K
0.47 × 0.37 × 0.35 mm
Data collection
Bruker SMART APEX2 area detector diffractometer
Absorption correction: multi-scan (TWINABS; Bruker, 2007 ▶) T min = 0.268, T max = 0.349
3012 measured reflections
3012 independent reflections
2938 reflections with I > 2σ(I)
Refinement
R[F 2 > 2σ(F 2)] = 0.051
wR(F 2) = 0.135
S = 1.12
3012 reflections
224 parameters
3 restraints
H-atom parameters constrained
Δρmax = 1.46 e Å−3
Δρmin = −0.63 e Å−3
Absolute structure: Classical Flack method preferred over Parsons because s.u. lower.
Flack parameter: 0.01 (3)
Data collection: APEX2 (Bruker, 2012 ▶); cell refinement: SAINT-Plus (Bruker, 2007 ▶); data reduction: SAINT-Plus; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: OLEX2 (Dolomanov et al., 2009 ▶) and NBOView (Wendt & Weinhold, 2001 ▶); software used to prepare material for publication: OLEX2, GX and FCF_filter (Guzei, 2012 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536813011112/kj2221sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813011112/kj2221Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536813011112/kj2221Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
This project has been funded in part by NIH grant R01 AR044276.
supplementary crystallographic information
Comment
We have previously reported on extensive studies of geometrical and conformational attributes of several amide bond isosteres (Choudhary & Raines, 2011a). In contrast, selenoamides have not received much attention. Herein we report the crystal structure of the title compound N-selenoacetyl–(2S)-proline methyl ester (Scheme 1, (I)) and the results of a hybrid density functional theory (DFT) and Natural Bond Orbital (NBO) analysis (Glendening et al., 2001, Weinhold, 1998, Weinhold & Landis, 2005) of its geometrical features.
Compound (I), Figure 1, crystallizes as a non-merohedral twin with the minor component contribution of 10 (2)%. The two components are related by 179° degree rotation about the [110] vector. The asymmetric unit in the relatively rare space group P1 contains two symmetry-independent molecules with the same handedness. The absolute structures of both components have been unequivocally established by anomalous dispersion effects: the Flack x parameters for the two components have been refined independently to 0.04 (2) and 0.02 (2). The two molecules have essentially identical geometries and their non-H atoms can be superimposed with a RMS of 0.042 Å. All geometrical parameters in the molecules are typical within experimental error (Bruno et al., 2002). The conformations of the five-membered rings in (I) are characterized by the puckering coordinates (Cremer & Pople, 1975) q2 and φ2 which measured 0.376 (14) Å and 89.8 (19)° for the Se1 molecule and 0.370 (15) Å and 85 (2)° for the Se1a molecule. Whereas the extent of puckering of the rings is the same, the ring in the Se1 molecule is in twisted conformation 3T4 whereas the ring in the other molecule is in the 3T4 conformation with a small 3E envelope character. The envelope character is probably statistically significant.
The key feature of (I) is pyramidalization of atom C7 described with parameters Δ and Θ defined in Figure 2. These parameters are 0.016 (12) Å and 0.06 (5)° for the Se1 molecule and 0.040 (13) Å and 1.5 (5)° for the Se1a molecule. In the Se1 molecule the pyramidalization is not observed whereas in the second molecule the slight pyramidalization is statistically significant. For comparison, in the sulfur analog of (I) the relevant pyramidalization parameters Δ and Θ are 0.0293 (13) Å and 1.10 (5)°, also small and statistically significant.
We conducted DFT and NBO analyses of four low energy conformations of (I) (DeRider et al., 2002, Choudhary et al., 2009, Choudhary et al., 2010b, Choudhary et al., 2010a, Jakobsche et al., 2010) at the B3LYP/6–311+G(2 d,p) level of theory using Gaussian 03 (Frisch et al., 2004) and comment here on the most stable conformer. We have previously reported an interaction in proteins, termed the n→π* interaction, wherein the lone pairs (n) of an oxygen (Oi-1) of a carbonyl group overlap with the antibonding orbital (π*) of Ci=Oi of an adjacent carbonyl group. The similar overlap in (I) between the lone pairs (n) of the selenium and the antibonding orbital (π*) of the carbonyl group is shown in Figure 3. (Bartlett et al., 2010, Choudhary & Raines, 2011b). This interaction resembles the Bürgi–Dunitz trajectory for nucleophilic additions to the carbonyl group and induces pyramidalization of the acceptor carbonyl group (Choudhary et al., 2009). The second-order perturbation theory as implemented in NBO 5.0 suggests n→π* interaction mediated stabilization of the trans conformation by 0.84 kcal/mol. The findings of our crystallographic studies partially support our theoretical findings: molecule Se1A shows pyramidalization whereas molecule Se1 does not. We attribute these results to the twinned nature of the crystals that lead to relatively high e.s.d.'s on geometrical parameters, but it was not possible to isolate a better crystal.
Experimental
Compound (I) was synthesized following from its oxygen congener by using (PhPSe2)2 (Woolins' reagent) following a procedure reported previously (Bhattacharyya & Woollins, 2001). A small amount of (I) was dissolved in hexanes with a minimal amount of ethyl acetate. Slow evaporation of the solution afforded X-ray quality crystals of (I) after ~4 days.
Refinement
All H-atoms were placed in idealized locations and refined as riding with appropriate thermal displacement coefficients Uiso(H) = 1.2 or 1.5 times Ueq(bearing atom).
Figures
Fig. 1.
Molecular structure of (I). The thermal ellipsoids are shown at 50% probability level.
Fig. 2.

Pyramidalization parameters Δ and Θ of an n→π* interaction in (I).
Fig. 3.
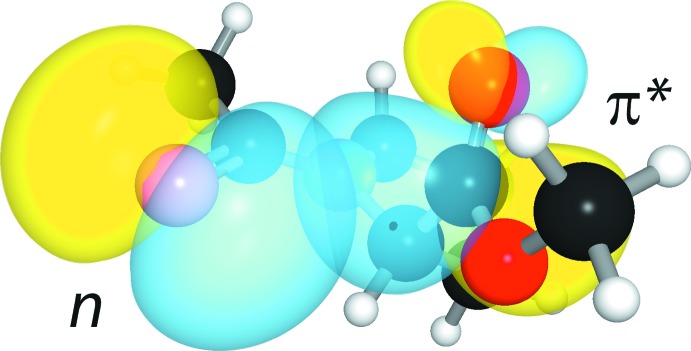
An NBO depiction of the n→π* orbital overlap in (I) generated with NBOView (Wendt & Weinhold, 2001)
Crystal data
| C8H13NO2Se | Z = 2 |
| Mr = 234.15 | F(000) = 236 |
| Triclinic, P1 | Dx = 1.593 Mg m−3 |
| a = 7.050 (3) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 7.442 (3) Å | Cell parameters from 746 reflections |
| c = 10.334 (4) Å | θ = 3.0–29.0° |
| α = 85.166 (6)° | µ = 3.81 mm−1 |
| β = 86.220 (6)° | T = 105 K |
| γ = 64.682 (4)° | Block, colourless |
| V = 488.1 (3) Å3 | 0.47 × 0.37 × 0.35 mm |
Data collection
| Bruker SMART APEX2 area detector diffractometer | 3012 measured reflections |
| Radiation source: microfocus sealed X-ray tube, Incoatec Iµs | 3012 independent reflections |
| Mirror optics monochromator | 2938 reflections with I > 2σ(I) |
| Detector resolution: 7.9 pixels mm-1 | θmax = 25.1°, θmin = 2.0° |
| 0.5° ω and 0.5° φ scans | h = −8→8 |
| Absorption correction: multi-scan (TWINABS; Bruker, 2007) | k = −8→8 |
| Tmin = 0.268, Tmax = 0.349 | l = −12→12 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.051 | w = 1/[σ2(Fo2) + (0.0947P)2 + 1.2373P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.135 | (Δ/σ)max < 0.001 |
| S = 1.12 | Δρmax = 1.46 e Å−3 |
| 3012 reflections | Δρmin = −0.63 e Å−3 |
| 224 parameters | Absolute structure: Classical Flack method preferred over Parsons because s.u. lower. |
| 3 restraints | Flack parameter: 0.01 (3) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refined as a 4-component twin. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Se1 | 1.02636 (11) | 0.03198 (10) | 0.80590 (9) | 0.0268 (3) | |
| O1 | 0.6582 (13) | 0.5319 (12) | 0.9097 (8) | 0.0258 (17) | |
| O2 | 0.6687 (15) | 0.5834 (13) | 0.6919 (8) | 0.0267 (19) | |
| N1 | 0.6036 (14) | 0.1857 (14) | 0.8793 (9) | 0.0209 (18) | |
| C1 | 0.807 (2) | −0.108 (2) | 1.0113 (14) | 0.024 (3) | |
| H1A | 0.7617 | −0.0430 | 1.0937 | 0.036* | |
| H1B | 0.9523 | −0.2093 | 1.0174 | 0.036* | |
| H1C | 0.7156 | −0.1708 | 0.9932 | 0.036* | |
| C2 | 0.7948 (17) | 0.0453 (16) | 0.9034 (12) | 0.023 (2) | |
| C3 | 0.4089 (18) | 0.2096 (19) | 0.9573 (12) | 0.024 (3) | |
| H3A | 0.4309 | 0.2039 | 1.0515 | 0.029* | |
| H3B | 0.3636 | 0.1052 | 0.9401 | 0.029* | |
| C4 | 0.2498 (18) | 0.4141 (18) | 0.9102 (13) | 0.027 (3) | |
| H4A | 0.1058 | 0.4219 | 0.9172 | 0.032* | |
| H4B | 0.2550 | 0.5190 | 0.9608 | 0.032* | |
| C5 | 0.3161 (18) | 0.436 (2) | 0.7695 (13) | 0.028 (3) | |
| H5A | 0.2615 | 0.5776 | 0.7380 | 0.034* | |
| H5B | 0.2662 | 0.3647 | 0.7132 | 0.034* | |
| C6 | 0.5599 (17) | 0.3388 (16) | 0.7724 (10) | 0.020 (2) | |
| H6 | 0.6241 | 0.2781 | 0.6885 | 0.024* | |
| C7 | 0.6369 (17) | 0.4933 (16) | 0.8033 (11) | 0.022 (2) | |
| C8 | 0.734 (3) | 0.740 (2) | 0.7072 (15) | 0.031 (3) | |
| H8A | 0.6159 | 0.8558 | 0.7411 | 0.046* | |
| H8B | 0.7828 | 0.7783 | 0.6228 | 0.046* | |
| H8C | 0.8493 | 0.6918 | 0.7681 | 0.046* | |
| Se1A | 1.07753 (15) | 0.71492 (14) | 0.39148 (11) | 0.0314 (4) | |
| O1A | 0.5592 (13) | 1.1157 (14) | 0.2797 (8) | 0.0279 (18) | |
| O2A | 0.5329 (13) | 1.1213 (14) | 0.4976 (9) | 0.0264 (19) | |
| N1A | 0.9670 (14) | 1.1015 (14) | 0.2881 (9) | 0.0212 (19) | |
| C1A | 1.259 (2) | 0.861 (2) | 0.1724 (16) | 0.026 (3) | |
| H1AA | 1.1935 | 0.9144 | 0.0886 | 0.038* | |
| H1AB | 1.3372 | 0.7162 | 0.1715 | 0.038* | |
| H1AC | 1.3562 | 0.9199 | 0.1874 | 0.038* | |
| C2A | 1.0943 (16) | 0.9115 (16) | 0.2781 (11) | 0.020 (2) | |
| C3A | 0.9718 (18) | 1.2692 (17) | 0.2000 (12) | 0.025 (2) | |
| H3AA | 0.9716 | 1.2428 | 0.1077 | 0.030* | |
| H3AB | 1.0970 | 1.2921 | 0.2141 | 0.030* | |
| C4A | 0.771 (2) | 1.446 (2) | 0.2387 (14) | 0.031 (3) | |
| H4AA | 0.7858 | 1.5722 | 0.2220 | 0.037* | |
| H4AB | 0.6505 | 1.4540 | 0.1904 | 0.037* | |
| C5A | 0.7423 (19) | 1.4030 (19) | 0.3828 (13) | 0.031 (3) | |
| H5AA | 0.5938 | 1.4753 | 0.4120 | 0.037* | |
| H5AB | 0.8312 | 1.4416 | 0.4341 | 0.037* | |
| C6A | 0.8117 (17) | 1.1760 (18) | 0.3963 (11) | 0.022 (2) | |
| H6A | 0.8771 | 1.1185 | 0.4819 | 0.027* | |
| C7A | 0.6233 (17) | 1.1279 (17) | 0.3787 (11) | 0.023 (2) | |
| C8A | 0.347 (2) | 1.085 (3) | 0.4944 (16) | 0.040 (4) | |
| H8AA | 0.2399 | 1.1934 | 0.4433 | 0.060* | |
| H8AB | 0.2912 | 1.0772 | 0.5832 | 0.060* | |
| H8AC | 0.3832 | 0.9585 | 0.4546 | 0.060* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Se1 | 0.0198 (6) | 0.0286 (7) | 0.0363 (8) | −0.0150 (6) | 0.0045 (5) | −0.0029 (5) |
| O1 | 0.028 (4) | 0.027 (4) | 0.026 (5) | −0.016 (4) | 0.000 (3) | −0.004 (3) |
| O2 | 0.030 (5) | 0.030 (5) | 0.029 (5) | −0.021 (4) | −0.007 (4) | 0.000 (3) |
| N1 | 0.020 (5) | 0.021 (5) | 0.026 (5) | −0.014 (4) | 0.002 (3) | −0.001 (3) |
| C1 | 0.019 (6) | 0.023 (7) | 0.027 (7) | −0.007 (6) | −0.004 (5) | −0.001 (5) |
| C2 | 0.016 (5) | 0.019 (5) | 0.037 (7) | −0.008 (5) | 0.000 (4) | −0.006 (4) |
| C3 | 0.020 (6) | 0.022 (6) | 0.035 (6) | −0.012 (5) | 0.001 (5) | −0.004 (5) |
| C4 | 0.015 (5) | 0.021 (6) | 0.047 (7) | −0.011 (5) | 0.001 (5) | −0.004 (5) |
| C5 | 0.021 (6) | 0.020 (6) | 0.045 (7) | −0.009 (5) | −0.010 (5) | 0.002 (5) |
| C6 | 0.021 (5) | 0.022 (5) | 0.024 (5) | −0.015 (5) | −0.006 (4) | −0.002 (4) |
| C7 | 0.018 (5) | 0.023 (6) | 0.029 (6) | −0.011 (5) | −0.003 (4) | 0.001 (4) |
| C8 | 0.039 (8) | 0.025 (7) | 0.039 (8) | −0.025 (6) | 0.000 (6) | 0.001 (5) |
| Se1A | 0.0285 (7) | 0.0247 (7) | 0.0453 (9) | −0.0164 (6) | −0.0005 (6) | 0.0041 (6) |
| O1A | 0.019 (4) | 0.039 (5) | 0.030 (5) | −0.016 (4) | −0.001 (3) | −0.005 (4) |
| O2A | 0.019 (4) | 0.035 (5) | 0.033 (5) | −0.019 (4) | −0.005 (3) | 0.002 (3) |
| N1A | 0.018 (5) | 0.029 (5) | 0.023 (5) | −0.018 (4) | −0.002 (3) | 0.002 (4) |
| C1A | 0.018 (6) | 0.027 (7) | 0.039 (8) | −0.018 (6) | 0.003 (5) | 0.000 (6) |
| C2A | 0.015 (5) | 0.022 (6) | 0.028 (6) | −0.011 (5) | −0.012 (4) | 0.000 (4) |
| C3A | 0.022 (6) | 0.019 (5) | 0.039 (7) | −0.015 (5) | 0.002 (5) | 0.007 (5) |
| C4A | 0.026 (7) | 0.016 (7) | 0.051 (8) | −0.011 (6) | −0.002 (6) | 0.003 (6) |
| C5A | 0.022 (6) | 0.030 (7) | 0.043 (8) | −0.013 (6) | 0.004 (5) | −0.009 (6) |
| C6A | 0.017 (5) | 0.029 (7) | 0.024 (6) | −0.012 (5) | −0.006 (4) | 0.000 (5) |
| C7A | 0.020 (6) | 0.027 (6) | 0.026 (6) | −0.014 (5) | 0.004 (4) | −0.006 (4) |
| C8A | 0.027 (7) | 0.068 (12) | 0.043 (8) | −0.038 (8) | 0.010 (6) | −0.013 (8) |
Geometric parameters (Å, º)
| Se1—C2 | 1.831 (11) | Se1A—C2A | 1.835 (11) |
| O1—C7 | 1.194 (14) | O1A—C7A | 1.170 (14) |
| O2—C7 | 1.337 (14) | O2A—C7A | 1.354 (15) |
| O2—C8 | 1.450 (16) | O2A—C8A | 1.454 (16) |
| N1—C2 | 1.329 (15) | N1A—C2A | 1.319 (15) |
| N1—C3 | 1.495 (14) | N1A—C3A | 1.493 (14) |
| N1—C6 | 1.465 (14) | N1A—C6A | 1.477 (15) |
| C1—H1A | 0.9800 | C1A—H1AA | 0.9800 |
| C1—H1B | 0.9800 | C1A—H1AB | 0.9800 |
| C1—H1C | 0.9800 | C1A—H1AC | 0.9800 |
| C1—C2 | 1.504 (19) | C1A—C2A | 1.490 (18) |
| C3—H3A | 0.9900 | C3A—H3AA | 0.9900 |
| C3—H3B | 0.9900 | C3A—H3AB | 0.9900 |
| C3—C4 | 1.516 (18) | C3A—C4A | 1.522 (18) |
| C4—H4A | 0.9900 | C4A—H4AA | 0.9900 |
| C4—H4B | 0.9900 | C4A—H4AB | 0.9900 |
| C4—C5 | 1.514 (19) | C4A—C5A | 1.51 (2) |
| C5—H5A | 0.9900 | C5A—H5AA | 0.9900 |
| C5—H5B | 0.9900 | C5A—H5AB | 0.9900 |
| C5—C6 | 1.555 (16) | C5A—C6A | 1.541 (17) |
| C6—H6 | 1.0000 | C6A—H6A | 1.0000 |
| C6—C7 | 1.529 (15) | C6A—C7A | 1.541 (15) |
| C8—H8A | 0.9800 | C8A—H8AA | 0.9800 |
| C8—H8B | 0.9800 | C8A—H8AB | 0.9800 |
| C8—H8C | 0.9800 | C8A—H8AC | 0.9800 |
| C7—O2—C8 | 114.7 (10) | C7A—O2A—C8A | 113.3 (10) |
| C2—N1—C3 | 124.9 (10) | C2A—N1A—C3A | 125.3 (9) |
| C2—N1—C6 | 123.3 (9) | C2A—N1A—C6A | 123.3 (10) |
| C6—N1—C3 | 111.8 (9) | C6A—N1A—C3A | 111.1 (9) |
| H1A—C1—H1B | 109.5 | H1AA—C1A—H1AB | 109.5 |
| H1A—C1—H1C | 109.5 | H1AA—C1A—H1AC | 109.5 |
| H1B—C1—H1C | 109.5 | H1AB—C1A—H1AC | 109.5 |
| C2—C1—H1A | 109.5 | C2A—C1A—H1AA | 109.5 |
| C2—C1—H1B | 109.5 | C2A—C1A—H1AB | 109.5 |
| C2—C1—H1C | 109.5 | C2A—C1A—H1AC | 109.5 |
| N1—C2—Se1 | 122.0 (9) | N1A—C2A—Se1A | 122.3 (9) |
| N1—C2—C1 | 115.8 (10) | N1A—C2A—C1A | 117.2 (11) |
| C1—C2—Se1 | 122.2 (8) | C1A—C2A—Se1A | 120.4 (9) |
| N1—C3—H3A | 111.1 | N1A—C3A—H3AA | 111.1 |
| N1—C3—H3B | 111.1 | N1A—C3A—H3AB | 111.1 |
| N1—C3—C4 | 103.2 (10) | N1A—C3A—C4A | 103.2 (9) |
| H3A—C3—H3B | 109.1 | H3AA—C3A—H3AB | 109.1 |
| C4—C3—H3A | 111.1 | C4A—C3A—H3AA | 111.1 |
| C4—C3—H3B | 111.1 | C4A—C3A—H3AB | 111.1 |
| C3—C4—H4A | 111.0 | C3A—C4A—H4AA | 111.0 |
| C3—C4—H4B | 111.0 | C3A—C4A—H4AB | 111.0 |
| H4A—C4—H4B | 109.0 | H4AA—C4A—H4AB | 109.0 |
| C5—C4—C3 | 104.0 (10) | C5A—C4A—C3A | 103.9 (11) |
| C5—C4—H4A | 111.0 | C5A—C4A—H4AA | 111.0 |
| C5—C4—H4B | 111.0 | C5A—C4A—H4AB | 111.0 |
| C4—C5—H5A | 111.1 | C4A—C5A—H5AA | 111.0 |
| C4—C5—H5B | 111.1 | C4A—C5A—H5AB | 111.0 |
| C4—C5—C6 | 103.3 (10) | C4A—C5A—C6A | 103.8 (9) |
| H5A—C5—H5B | 109.1 | H5AA—C5A—H5AB | 109.0 |
| C6—C5—H5A | 111.1 | C6A—C5A—H5AA | 111.0 |
| C6—C5—H5B | 111.1 | C6A—C5A—H5AB | 111.0 |
| N1—C6—C5 | 102.9 (9) | N1A—C6A—C5A | 103.7 (9) |
| N1—C6—H6 | 111.1 | N1A—C6A—H6A | 110.9 |
| N1—C6—C7 | 110.4 (8) | N1A—C6A—C7A | 110.0 (9) |
| C5—C6—H6 | 111.1 | C5A—C6A—H6A | 110.9 |
| C7—C6—C5 | 110.0 (9) | C5A—C6A—C7A | 110.1 (9) |
| C7—C6—H6 | 111.1 | C7A—C6A—H6A | 110.9 |
| O1—C7—O2 | 125.6 (10) | O1A—C7A—O2A | 125.9 (11) |
| O1—C7—C6 | 125.5 (10) | O1A—C7A—C6A | 126.3 (10) |
| O2—C7—C6 | 108.8 (9) | O2A—C7A—C6A | 107.6 (9) |
| O2—C8—H8A | 109.5 | O2A—C8A—H8AA | 109.5 |
| O2—C8—H8B | 109.5 | O2A—C8A—H8AB | 109.5 |
| O2—C8—H8C | 109.5 | O2A—C8A—H8AC | 109.5 |
| H8A—C8—H8B | 109.5 | H8AA—C8A—H8AB | 109.5 |
| H8A—C8—H8C | 109.5 | H8AA—C8A—H8AC | 109.5 |
| H8B—C8—H8C | 109.5 | H8AB—C8A—H8AC | 109.5 |
| N1—C3—C4—C5 | 31.5 (11) | N1A—C3A—C4A—C5A | 32.7 (12) |
| N1—C6—C7—O1 | −25.3 (15) | N1A—C6A—C7A—O1A | −30.5 (16) |
| N1—C6—C7—O2 | 157.1 (9) | N1A—C6A—C7A—O2A | 155.3 (9) |
| C2—N1—C3—C4 | 167.0 (10) | C2A—N1A—C3A—C4A | 169.8 (10) |
| C2—N1—C6—C5 | 169.2 (10) | C2A—N1A—C6A—C5A | 166.9 (9) |
| C2—N1—C6—C7 | −73.5 (12) | C2A—N1A—C6A—C7A | −75.3 (12) |
| C3—N1—C2—Se1 | −177.9 (8) | C3A—N1A—C2A—Se1A | 179.8 (8) |
| C3—N1—C2—C1 | 5.1 (16) | C3A—N1A—C2A—C1A | 1.6 (15) |
| C3—N1—C6—C5 | −11.6 (11) | C3A—N1A—C6A—C5A | −8.4 (11) |
| C3—N1—C6—C7 | 105.7 (10) | C3A—N1A—C6A—C7A | 109.4 (10) |
| C3—C4—C5—C6 | −39.0 (11) | C3A—C4A—C5A—C6A | −38.3 (12) |
| C4—C5—C6—N1 | 30.9 (11) | C4A—C5A—C6A—N1A | 28.6 (11) |
| C4—C5—C6—C7 | −86.8 (11) | C4A—C5A—C6A—C7A | −89.0 (11) |
| C5—C6—C7—O1 | 87.6 (14) | C5A—C6A—C7A—O1A | 83.2 (15) |
| C5—C6—C7—O2 | −90.1 (11) | C5A—C6A—C7A—O2A | −91.0 (11) |
| C6—N1—C2—Se1 | 1.2 (14) | C6A—N1A—C2A—Se1A | 5.2 (14) |
| C6—N1—C2—C1 | −175.8 (11) | C6A—N1A—C2A—C1A | −173.0 (11) |
| C6—N1—C3—C4 | −12.1 (12) | C6A—N1A—C3A—C4A | −15.0 (12) |
| C8—O2—C7—O1 | 0.0 (18) | C8A—O2A—C7A—O1A | 3.5 (18) |
| C8—O2—C7—C6 | 177.7 (10) | C8A—O2A—C7A—C6A | 177.7 (11) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: KJ2221).
References
- Bartlett, G. J., Choudhary, A., Raines, R. T. & Woolfson, D. N. (2010). Nat. Chem. Biol. 6, 615–620. [DOI] [PMC free article] [PubMed]
- Bhattacharyya, P. & Woollins, J. D. (2001). Tetrahedron Lett. 42, 5949–5951.
- Bruker (2007). TWINABS and SAINT-Plus Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2012). APEX2 Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002). Acta Cryst. B58, 389–397. [DOI] [PubMed]
- Choudhary, A., Fry, C. G. & Raines, R. T. (2010a). ARKIVOC, pp. 251–262. [PMC free article] [PubMed]
- Choudhary, A., Gandla, D., Krow, G. R. & Raines, R. T. (2009). J. Am. Chem. Soc. 131, 7244–7246. [DOI] [PMC free article] [PubMed]
- Choudhary, A., Pua, K. H. & Raines, R. T. (2010b). Amino Acids, 39, 181–186. [DOI] [PMC free article] [PubMed]
- Choudhary, A. & Raines, R. T. (2011a). ChemBioChem, 12, 1801–1807. [DOI] [PMC free article] [PubMed]
- Choudhary, A. & Raines, R. T. (2011b). Protein Sci. 20, 1077–1087. [DOI] [PMC free article] [PubMed]
- Cremer, D. & Pople, J. A. (1975). J. Am. Chem. Soc. 97, 1358–1367.
- DeRider, M. L., Wilkens, S. J., Waddell, M. J., Bretscher, L. E., Weinhold, F., Raines, R. T. & Markley, J. L. (2002). J. Am. Chem. Soc. 124, 2497–2505. [DOI] [PubMed]
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Frisch, M. J., et al. (2004). GAUSSIAN03 Gaussian Inc., Wallingford, CT, USA.
- Glendening, E. D., Badenhoop, J. K., Reed, A. E., Carpenter, J. E., Bohmann, J. A., Morales, C. M. & Weinhold, F. (2001). NBO Theoretical Chemistry Institute, University of Wisconsin, Madison, USA.
- Guzei, I. A. (2012). GX and FCF_filter Department of Chemistry University of Wisconsin–Madison, Madison, Wisconsin, USA.
- Jakobsche, C. E., Choudhary, A., Raines, R. T. & Miller, S. J. (2010). J. Am. Chem. Soc. 132, 6651–6653. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Weinhold, F. (1998). Encyclopedia of Computational Chemistry, edited by P. v. R. Schleyer, N. L. Allinger, T. Clark, J. Gasteiger, P. A. Kollman, H. F. Shaefer III & P. R. Schreiner, pp. 1792–1811. Chichester, UK: John Wiley & Sons.
- Weinhold, F. & Landis, C. R. (2005). In Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective Cambridge University Press.
- Wendt, M. & Weinhold, F. (2001). NBOView Theoretical Chemistry Institute, University of Wisconsin–Madison: Madison, Wisconsin, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536813011112/kj2221sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536813011112/kj2221Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536813011112/kj2221Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report