

Abstract
Many non-infectious neurodegenerative diseases are associated with the accumulation of fibrillar protein. These diseases all exhibit phenotypic diversity and propagation of pathology that is reminiscent of prionopathies. Furthermore, emerging studies of amyloid-β, α–synuclein, and tau proteins suggest that they share key biophysical and biochemical characteristics with prions. Propagation of protein misfolding in these diseases may therefore occur via mechanisms similar to those underlying prion pathogenesis. If verified in vivo, this will suggest new therapeutic strategies to block propagation of protein misfolding throughout the brain.
Introduction
Prionopathies are unique among neurodegenerative diseases because they are infectious, that is, spontaneous transmission from one individual to another has occurred outside of an experimental setting. Prion diseases derive from protein misfolding, which in rare cases can be due to exposure to exogenous prion species1 – that is, “infection”– but are usually due to events that occur spontaneously within the individual. The prion protein in its nonpathogenic form (PrPC) is expressed in many human cell types2. When PrPC comes into contact with a pathogenic prion protein conformer (PrPSc), PrPC is induced to misfold in a process termed “templated conformation change.” Through this interaction, the conformation of a misfolded PrPSc molecule is communicated to a native PrPC protein3. This interaction may involve other proteins within the cell, and it is unknown whether one or more PrPSc molecules form the pathogenic “seed.”
Recent studies are now highlighting prion-like mechanisms of propagation of protein misfolding in a variety of common, non-infectious (that is, transmission between individuals has never been shown outside experimental conditions), neurodegenerative diseases such as Alzheimer disease (AD), frontotemporal dementia (FTD), Parkinson disease (PD), and polyglutamine diseases. Like prionopathies, all of these diseases are associated with the accumulation of fibrillar aggregates of proteins such as tau, amyloid-β (Aβ), α-synuclein, and polyglutamine proteins. With the exception of polyglutamine diseases, which derive from an unusual mutation that produces a protein with an abnormally long glutamine tract, sporadic cases of these diseases involve the wild-type form of each protein, whereas rarer, autosomal dominant forms of the disease are linked to mis-sense or splicing mutations. Similarly, although prion diseases are defined by their infectivity, most prion disease cases actually arise sporadically from wild-type protein, or through inherited mutations in the prion protein4. This article will highlight two important commonalities between prion and non-prion neurodegenerative diseases—phenotypic diversity and spreading pathology—and will review basic research that is beginning to elucidate the biochemical and cellular basis of these similarities.
Phenotypic Diversity
Most common neurodegenerative diseases manifest myriad disease phenotypes. In AD, the speed of cognitive decline, age of onset, and the location and extent of Aβ plaque load vary considerably 5–7. Aβ aggregates are also present in muscle fibers in inclusion body myositis, a common age-related inflammatory muscle disease8, and in the vascular wall in cerebral amyloid angiopathy9. PD, dementia with Lewy bodies, and multiple system atrophy are all associated with α-synuclein deposition10, yet are strikingly distinct clinical syndromes. PD is associated with α-synuclein mis-sense mutations11, 12, as well as gene amplification13–15. Most remarkably, tau aggregation is a pathological hallmark of more than twenty different neurodegenerative diseases, including AD10 and frontotemporal dementia with Parkinsonism (FTDP-17), a familial form of disease caused by mutations in the tau gene10. Sporadic tauopathies vary considerably in brain region involvement, disease duration, age of onset, and fibril morphology10.
Prion diseases also have very diverse phenotypes, involving both the central and peripheral nervous system, exhibit distinct rates of progression16, 17, and can derive from mutations within the prion protein gene18. The wild-type prion protein (PrP) is the causative agent for Kuru19 and sporadic Creutzfeldt-Jakob Disease (CJD), among others. Prions also cause fatal familial insomnia20–22. Thus, variation in presentation and course of disease defines both prion and non-prion based neurodegeneration. Distinct conformations of pathogenic proteins could play a key role in determining the phenotypic diversity of non-infectious neurodegenerative diseases.
Underlying mechanisms: conformations and strains?
In the case of the prion diseases, it is thought that distinct conformers, or “strains,” of prion fibril underlie each disease phenotype. Depending upon a variety of factors, including amino acid sequence, post-translational modifications, and aggregation conditions, PrPSc assembles into multiple, individually self-propagating conformations that generate these distinct disease phenotypes in humans and mice22–24. It is not yet possible to predict the specific phenotype that will result from a given PrPSc conformation in mammals. However, the yeast prion Sup35 has helped inform our understanding of mammalian prions. Sup35 alternates between a soluble (active) and aggregated (inactive) state that controls its cellular activity. The aggregation state of Sup35 is transmitted in a heritable, epigenetic fashion from parent to daughter yeast cell. The rate of growth and fibril fragility determines the efficiency with which protein aggregates are passed from mother to daughter cells25, and these biochemical features have been directly linked to fibril structure26. This level of structural detail is not yet available for PrP, but recent work in mouse models indicates that unique prion strains can also be defined by conformational diversity that correlates with the sensitivity of the associated fibrils to in vitro denaturation24.
To what extent can similar conformational diversity explain the diverse phenotypes of the non-infectious neurodegenerative diseases? In fact, conformational diversity of a variety of amyloid proteins is now widely recognized. For example, tau fibrils can exist in distinct structures that are stable over serial seeding reactions27. Wild-type, ΔK280, and a P301L/V337M double mutant fibrils are conformationally distinct when prepared in vitro. When mutant tau seeds are used to induce fibrillization of wild-type monomer, the resulting fibrils closely resemble the conformation of the mutant seed, and are distinct from the wild-type fibril conformation27. Distinct, self-propagating fibril structures have also been documented for Aβ28 and α-synuclein29, putting these proteins in the same biochemical class as prions. Even the growth conditions of in vitro Aβ fibrillization reactions have been shown to specify the conformation of the resultant fibrils28. Aβ fibrils assume one of two distinct conformations, depending on whether the reactions are gently agitated. When incubated with fresh Aβ monomer, each fibril type faithfully propagates the original conformation over successive seeding reactions. The two Aβ fibril conformations have distinct toxicities when added to primary neurons28. Although intriguing, this artificial readout of fibril toxicity is of unknown significance in relation to diversity of human disease, and thus the effect of distinct Aβ conformers on AD phenotypes in vivo is at this stage purely speculative. α-synuclein proteins also exhibit fibrillar conformational diversity, as mis-sense mutations that are responsible for dominantly inherited synucleinopathy produce fibrils that are conformationally distinct from wild-type fibrils29, 30. In vitro studies indicate that mutant fibrils can “transmit” their conformation to wild-type protein, driving it into a new conformation that resembles the original mutant seed29. Again, however, there is yet no evidence that distinct synuclein structures underlie the various synucleinopathy phenotypes.
Taken together, these in vitro studies indicate that tau, Aβ, and α-synuclein are all capable of the same type of templated conformation change that was first described for prions. These data suggest that such proteins might also form distinct conformers in vivo that could cause variation in regional pathology and disease progression. Protein interactions with distinct amyloid conformers could affect pathogenesis in several ways: conformation-specific aggregate interactions with cellular targets could produce unique toxicities; additionally, protein interactions are also likely to affect fibril conformation, and thus the propensity for amplification of protein misfolding (and toxicity) within the cell. The relative ease with which it is possible to generate distinct protein fibril conformers in vitro suggests that there might be even more pathological syndromes than those of which we are currently aware. Until distinct mammalian pathologies are clearly linked to discrete amyloid protein conformations, however, it remains unknown whether prion-like conformational diversity of pathological proteins can in fact account for phenotypic variation in the common neurodegenerative diseases.
Spreading Pathology
Neurodegenerative diseases begin with dysfunction in a discrete region, whereas at later stages they typically involve much larger areas of brain. Pathology often occurs within particular neural networks, and progresses in a predictable manner. For example, the transentorhinal region first shows signs of deterioration and tau pathology in AD. Glutamatergic cells in this region project into the entorhinal cortex, which is the next to degenerate. Lesions of the hippocampus, amygdala, and the neocortex follow31. Recent studies of normal and demented patients using functional imaging have corroborated these pathological studies, and have shown that degeneration in distinct neurodegenerative diseases such as AD, corticobasal ganglionic degeneration, and FTD follow patterns of intrinsic neuronal connectivity present in normal individuals32. PD is well known to begin with motor symptoms that are largely caused by the degeneration of dopaminergic neurons within the substantia nigra; however, a substantial fraction of patients go on to develop dementia implying that additional brain regions are involved 33. Likewise, in amyotrophic lateral sclerosis, the progression of symptoms locally within the spinal cord and the combined degeneration of upper and lower motor neurons is well known34. Taken together, these observations suggest a pathogenic link between one affected cell and its neighbor. Despite these patterns of disease progression, there is not yet clear experimental evidence that this progression of neurodegenerative diseases other than prionopathies results from the ‘spread’ of disease from one area to another.. Prionopathies begin with a tiny inoculum, such as a contaminated surgical device or transplanted tissue, or the spontaneous accumulation of PrPSc in a single cell or group of cells. PrPSc propagation depends on a network of PrPC-expressing cells35. Ultimately, however, pathology involves a large area of the nervous system36. This may occur via spread through neuronal networks. In hamsters, orally derived PrPSc appears to spread along the vagus nerve to the medulla, pons, midbrain, cerebellum, and thalamus via neuroanatomical pathways37. And two studies have observed that PrPSc injected into the eye travels along defined neuroanatomical connections to involve larger brain regions38, 39.
Underlying mechanisms: cell to cell transmission?
Propagation of PrP misfolding between cells follows a model in which PrPSc travels from an infected cell to a naïve cell, whereupon it encounters and converts PrPC to PrPSc 39. These features of prion disease suggest that PrPSc may gain access to a connected neuron by traversing the synapse, or that PrPSc released into the extracellular space may be taken up by nearby cells. Cell culture studies support this hypothesis. Cultured primary mouse neurons spontaneously take up fibrillar PrP, which localizes to late endosomes and/or lysosomes40. PrP aggregates have been shown to transfer between cultured cells both through exosomes41 tunneling nanotubes42, and putative cytoplasm connections between mammalian cells43. Whether these events underlie the spread of prion pathology in vivo will require more mechanistic studies involving targeted disruption of these processes.
It is unknown whether non-prion protein aggregates move between cells in people. Pathological studies of PD patients who underwent fetal transplant surgery are provocative, but not conclusive. In these reports, engrafted mesencephalic dopaminergic neurons developed ubiquitin and α-synuclein-positive Lewy bodies, many of which were indistinguishable from lesions in the diseased host44–46. Recent studies in mice have essentially replicated the work in patients, and involve transplantation of synuclein-negative cells into a human synuclein transgenic mouse, where the transplanted cells developed Lewy bodies47. This investigation clearly indicates that synuclein protein is capable of trans-cellular movement in vivo, and has obvious implications for the potential of aggregated protein to spread pathology from cell to cell in humans. Whether aggregates can transfer directly between cells in vivo is unknown, but cell culture studies suggest this is possible.
Aggregates comprised of Aβ, α-synuclein, tau, and polyglutamine proteins are readily internalized by cultured cells40, 47–51. In the case of polyglutamine proteins, the uptake of an aggregate causes the wild-type (unexpanded) form of the protein expressed in the cell to misfold51. Similarly, internalized tau aggregates appear to interact directly with normally folded tau and trigger its fibrillization50. Intracellular tau aggregates are also capable of transferring between co-cultured cells50. Thus, tau and polyglutamine proteins, like prions, can “transmit” a misfolded state from the outside to the inside of a cell. This idea is supported by the observation that the yeast prion, Sup35, can accomplish trans-cellular propagation of aggregates when expressed in mammalian cells52. While these experiments are intriguing, a clearer interpretation awaits definition of basic mechanisms of uptake and cell-cell transfer, and demonstration that this influences propagation of pathology in vivo.
It was previously observed that intracerebral injection of human or mouse AD brain material can initiate Aβ pathology in transgenic mice53. It has also been observed recently that microinjection of brain extracts from mice transgenic for human tau protein will induce misfolding of tau in recipient mice. It was suggested that the induced tau misfolding propagated beyond the site of injection, although this is uncertain since it is hard to rule out diffusion of injected material. However, tau protein must be present in the injected material for this effect to manifest, which hints at a “prion-like” mechanism54. Although sporadic neurodegenerative diseases certainly don’t derive from injected brain extracts, these studies indicate that misfolding can somehow be communicated from the extracellular to the intracellular space, as was previously observed with tau in tissue culture50. So far, it has not yet been demonstrated in vivo that a misfolded protein within one cell can directly trigger misfolding in a connected cell, which would more directly test the idea that AD, tauopathy, or synucleinopathies involve prion-like mechanisms. And there is absolutely no evidence that these disorders have ever been responsible for transmission of disease between individuals as bona fide prionopathies.
Distinctions between diseases
Crucial distinctions remain between the prionopathies and common neurodegenerative diseases. Most importantly, there is no evidence, despite decades of study, of true, spontaneous “infectivity” for any sporadic disease such as AD, FTD, or PD. The biophysical properties that allow a protein to be eaten, passage through the digestive system, and then be absorbed to replicate in the host before infectious particles make their way to the brain clearly sets prion proteins apart from any other known amyloid protein associated with neurodegenerative disease. However, Serum amyloidosis A (SAA) has been studied as another potentially infectious amyloid disease55 derived from misfolding of the serum amyloid A protein56. Although this amyloidosis is not associated with neurodegeneration, it has many features of transmission similar to prionopathies, including an oral route57.
In addition, most prionopathies exhibit relatively rapid progression within the CNS, with sCJD averaging 4–6 months from symptom onset to death58, whereas common neurodegenerative diseases generally progress over many years. Finally, whereas PrP is a transmembrane protein, which could in theory more easily allow trans-cellular propagation, tau and synuclein normally function within the cell, and thus it is more difficult to understand how they could accomplish trans-cellular movement.
A common model of propagation?
Even taking into account these distinctions, increasing experimental evidence is now suggesting that basic cellular mechanisms of trans-cellular prion propagation may be applicable to a wide range of protein pathologies. In this model (Fig. 1), fibrillar protein “seeds” from adjacent or synaptically connected cells may be taken up to induce aggregation of otherwise normally structured protein. The propensity for this to occur could be influenced by splice isoforms and post-translational modifications of the proteins involved. This model could explain both for the phenotypic diversity observed in sporadic neurodegenerative disease, in which a single protein underlies a variety of conditions, and for the inexorable spread of pathology, in which aggregates are capable of moving between cells to propagate misfolding. It also can explain the involvement of neuronal networks in neurodegeneration. If these ideas are fully validated in animal models, they will suggest an important new conceptual framework with which to consider pathogenesis of an enormous range of neurodegenerative diseases.
Figure 1. Potential mechanisms for trans-cellular propagation of protein misfolding.
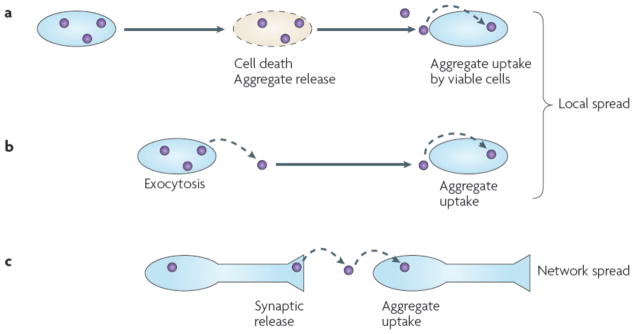
(A) Intracellular protein aggregation leads to cell death. This results in release of protein aggregates into the extracellular space, which are subsequently taken up and corrupt protein folding in vulnerable cells. (B) As part of the physiology of a living cell, protein aggregates may be released, potentially via exosomes or exocytosis. This results in protein aggregates in the extracellular space that may be taken up by adjacent cells. This mechanism can account for local propagation of misfolding, along with (A). (C) Aggregates might cross synapses. Release could be due to local degeneration of a synapse, normal synaptic physiology, or as part of an exocytic process as in (B). This mechanism can explain network degeneration in neurodegenerative diseases.
Implications for Treatment
If non-infectious neurodegenerative diseases and prionopathies have similar mechanisms of progression, this will have important therapeutic implications. Current therapies for neurodegenerative diseases generally target non-specific mechanisms to prevent cell death and promote neuron survival, or they focus on disease-specific events that govern stability and clearance of target proteins inside and outside of cells. If protein misfolding in one cell can trigger similar events in a neighbor, then new therapeutic strategies based on halting non-cell autonomous effects will be required. For example, stem cell therapies may have limited utility unless it is possible to render the transplanted cells resistant to the effects of misfolded protein from the host. On the other hand, new approaches based on antibody therapies may have much wider application than previously realized, where the main focus as been on extracellular Aβ. Indeed, vaccination of mice in experimental models of tauopathy and synucleinopathy (which involve intracellular proteins) has been reported to ameliorate pathology59, 60. Finally, as mechanisms of aggregate uptake and cell-to-cell transmission are determined, it may be possible to design novel pharmacological interventions that block disease progression (Fig. 2).
Figure 2. New therapeutic approaches.
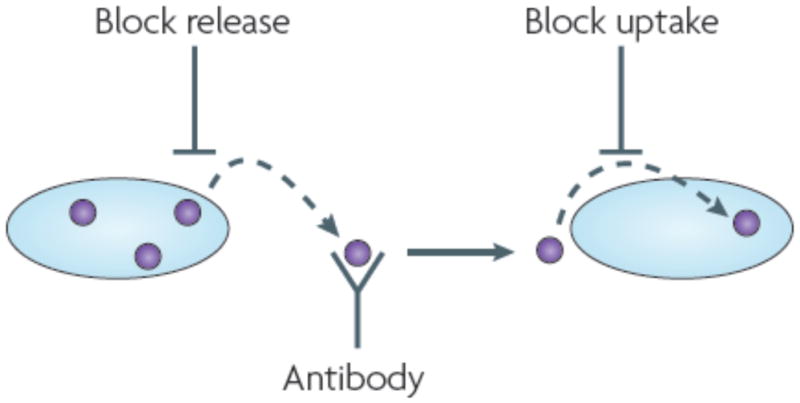
If trans-cellular propagation of protein misfolding occurs, then new strategies could supplement existing approaches to promote cell survival and block intracellular accumulation of misfolded species. As the cellular mechanisms of aggregate release and uptake are delineated, it may be possible to inhibit these events pharmacologically or genetically. Antibody-based therapies might also be expanded to target protein aggregates that are generated inside a cell and released into the extracellular space.
Conclusions
It is now clear that a variety of disease-associated proteins, not just PrP, exhibit templated conformational change that might underlie certain aspects of phenotypic diversity of disease. Given the clear predictions of this model, future studies should be able to clearly test this idea. The phenomenon of cell-cell transfer of protein aggregates is now well established in cell culture, and mouse models suggest that such transfer may also take place in vivo. It is still uncertain whether proteins distinct from PrP can trigger a true “propagation” of misfolding in the manner of prions – that is, whether aggregated species released from one cell and taken up by another will lead to further aggregation of natively folded species in the recipient cell, and so on. The cellular mechanisms of aggregate release and uptake remain to be elucidated, and it will need to be determined whether the same mechanisms apply to all aggregation-prone proteins. Similarly, the true spectrum of these phenomena in other neurodegenerative diseases associated with protein misfolding is unknown. For example, will TDP-43 or SOD-1, which are both associated with amyotrophic lateral sclerosis, also exhibit such cell to cell transfer? It is also unknown what role glia and proposed cellular mechanisms might play in vivo. Can transfer of aggregates occur across synapses, and can this account for the propagation of pathology along neural networks? As the current studies are extended and augmented by existing knowledge of prion pathogenesis, a new unifying model that melds cell autonomous and non-cell autonomous mechanisms of protein misfolding in neurodegenerative diseases seems likely.
Table.
Common features of mammalian proteins associated with neurodegenerative diseases.
| Protein | Conformational Diversity? | Trans-Cellular Aggregate Movement in Culture? | Aggregate Propagation in Vivo? |
|---|---|---|---|
| prion | yes21, 22 | yes40–42 | yes4 |
| Aβ | yes28 | extracellular aggregates are taken up by cells40 | yes: inoculation of brain triggers further aggregation53 |
| tau | yes27 | extracellular aggregates are taken up by cells and transfer of intracellular aggregates occurs50 | yes: extracellular inoculation with aggregates triggers uptake of aggregates and induces further intracellular tau misfolding54 |
| α-synuclein | yes29 | protein is released by cells and taken up by co-cultured cells47 | possibly: in humans, transplanted cells develop Lewy bodies44–46; transplanted cells in mice take up protein from host and form inclusions47 |
| polyglutamine | yes61 | aggregates are taken up by cultured cells and trigger misfolding of wild- type protein; aggregates can move between cells51 | no: not demonstrated |
Footnotes
The authors declare no conflict of interest.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Caughey B, Race RE, Chesebro B. Detection of prion protein mRNA in normal and scrapie-infected tissues and cell lines. J Gen Virol. 1988;69 ( Pt 3):711–6. doi: 10.1099/0022-1317-69-3-711. [DOI] [PubMed] [Google Scholar]
- 3.Pan KM, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90:10962–6. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williamson J, Goldman J, Marder KS. Genetic aspects of Alzheimer disease. Neurologist. 2009;15:80–6. doi: 10.1097/NRL.0b013e318187e76b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong RA, Nochlin D, Bird TD. Neuropathological heterogeneity in Alzheimer’s disease: a study of 80 cases using principal components analysis. Neuropathology. 2000;20:31–7. doi: 10.1046/j.1440-1789.2000.00284.x. [DOI] [PubMed] [Google Scholar]
- 7.Chui HC, Teng EL, Henderson VW, Moy AC. Clinical subtypes of dementia of the Alzheimer type. Neurology. 1985;35:1544–50. doi: 10.1212/wnl.35.11.1544. [DOI] [PubMed] [Google Scholar]
- 8.Askanas V, Engel WK. Inclusion-body myositis: a myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology. 2006;66:S39–48. doi: 10.1212/01.wnl.0000192128.13875.1e. [DOI] [PubMed] [Google Scholar]
- 9.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 10.Goedert M, et al. From genetics to pathology: tau and alpha-synuclein assemblies in neurodegenerative diseases. Philos Trans R Soc Lond B Biol Sci. 2001;356:213–27. doi: 10.1098/rstb.2000.0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 12.Zarranz JJ, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 13.Ibanez P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–71. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 14.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 15.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 16.Ironside JW, Ritchie DL, Head MW. Phenotypic variability in human prion diseases. Neuropathol Appl Neurobiol. 2005;31:565–79. doi: 10.1111/j.1365-2990.2005.00697.x. [DOI] [PubMed] [Google Scholar]
- 17.Wadsworth JD, Collinge J. Update on human prion disease. Biochim Biophys Acta. 2007;1772:598–609. doi: 10.1016/j.bbadis.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Hsiao K, et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338:342–5. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 19.Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197:943–60. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 20.Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278:245–51. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- 21.Bessen RA, et al. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375:698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 22.Telling GC, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–82. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 23.Safar J, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–65. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 24.Legname G, et al. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103:19105–10. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–9. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 26.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–7. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 27.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009;284:3546–51. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petkova AT, et al. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–5. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 29.Yonetani M, et al. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–50. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Bergen M, et al. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A. 2000;97:5129–34. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 32.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobson P, Meara J. The detection of dementia and cognitive impairment in a community population of elderly people with Parkinson’s disease by use of the CAMCOG neuropsychological test. Age Ageing. 1999;28:39–43. doi: 10.1093/ageing/28.1.39. [DOI] [PubMed] [Google Scholar]
- 34.Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx. 2004;1:273–83. doi: 10.1602/neurorx.1.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glatzel M, Aguzzi A. PrP(C) expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol. 2000;81:2813–21. doi: 10.1099/0022-1317-81-11-2813. [DOI] [PubMed] [Google Scholar]
- 36.Brown P, Preece MA, Will RG. “Friendly fire” in medicine: hormones, homografts, and Creutzfeldt-Jakob disease. Lancet. 1992;340:24–7. doi: 10.1016/0140-6736(92)92431-e. [DOI] [PubMed] [Google Scholar]
- 37.Beekes M, McBride PA, Baldauf E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol. 1998;79 ( Pt 3):601–7. doi: 10.1099/0022-1317-79-3-601. [DOI] [PubMed] [Google Scholar]
- 38.Fraser H. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature. 1982;295:149–50. doi: 10.1038/295149a0. [DOI] [PubMed] [Google Scholar]
- 39.Brandner S, et al. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci U S A. 1996;93:13148–51. doi: 10.1073/pnas.93.23.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magalhaes AC, et al. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci. 2005;25:5207–16. doi: 10.1523/JNEUROSCI.0653-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fevrier B, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–8. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gousset K, et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11:328–36. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 43.Gerdes HH, Carvalho RN. Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol. 2008;20:470–5. doi: 10.1016/j.ceb.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Li JY, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 45.Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008;23:2303–6. doi: 10.1002/mds.22369. [DOI] [PubMed] [Google Scholar]
- 46.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–6. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 47.Desplats P, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–5. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–17. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 49.Lee HJ, et al. Assembly-dependent endocytosis and clearance of extracellular alphasynuclein. Int J Biochem Cell Biol. 2008;40:1835–49. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 50.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krammer C, et al. The yeast Sup35NM domain propagates as a prion in mammalian cells. Proc Natl Acad Sci U S A. 2009;106:462–7. doi: 10.1073/pnas.0811571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 54.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Werdelin O, Ranlov P. Amyloidosis in mice produced by transplantation of spleen cells from casein-treated mice. Acta Pathol Microbiol Scand. 1966;68:1–18. doi: 10.1111/apm.1966.68.1.1. [DOI] [PubMed] [Google Scholar]
- 56.Westermark GT, Westermark P. Serum amyloid A and protein AA: molecular mechanisms of a transmissible amyloidosis. FEBS Lett. 2009;583:2685–90. doi: 10.1016/j.febslet.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 57.Lundmark K, et al. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A. 2002;99:6979–84. doi: 10.1073/pnas.092205999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mendez OE, Shang J, Jungreis CA, Kaufer DI. Diffusion-weighted MRI in Creutzfeldt-Jakob disease: a better diagnostic marker than CSF protein 14-3-3? J Neuroimaging. 2003;13:147–51. [PubMed] [Google Scholar]
- 59.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masliah E, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–68. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 61.Nekooki-Machida Y, et al. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:9679–84. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]