

Abstract
Anaplastic Lymphoma Kinase (ALK) was initially discovered as an oncogene in human lymphoma and other cancers, including neuroblastoma. However, little is known about the physiological function of ALK. We identified the alk ortholog in zebrafish (Danio rerio) and found that it is highly expressed in the developing central nervous system (CNS). Heat-shock inducible transgenic zebrafish lines were generated to over-express alk during early neurogenesis. Its ectopic expression resulted in activation of the MEK/ERK pathway, increased cell proliferation, and aberrant neurogenesis leading to mis-positioning of differentiated neurons. Thus, overexpressed alk is capable of promoting cell proliferation in the nervous system, similar to the situation in ALK-related cancers. Next, we used Morpholino mediated gene knock-down and a pharmacological inhibitor to interfere with expression and function of endogenous Alk. Alk inhibition did not affect neuron progenitor formation but severely compromised neuronal differentiation and neuron survival in the CNS. These data indicate that tightly controlled alk expression is critical for the balance between neural progenitor proliferation, differentiation and survival during embryonic neurogenesis.
Introduction
Anaplastic Lymphoma Kinase (ALK) was initially discovered as an oncogene in human anaplastic large cell lymphomas (ALCL), a subset of T-cell lymphomas [1], [2], [3]. Genetic dissection revealed an hybrid gene (NPM-ALK, also known as p80) at the t(2;5)(p23;q35) chromosomal translocation breakpoint, comprising a fusion of a nucleolar protein gene nucleophosmin (NPM) and a part of a gene coding for ALK, a novel tyrosine kinase. Moreover, another fusion of alk with the gene encoding echinoderm microtubule-associated protein-like4, encoding EML4-ALK hybrid protein, was identified in a subset of non-small-cell lung cancers (NSCLCs) [4]. To date, several more ALK hybrid proteins have been identified in various cancer types, such as TRK-fused gene (TFG)-ALK, tropomyosin 3 (TPM3)-ALK, tropomyosin 4 (TPM4)-ALK, clathrin heavy chain-like 1 (CLTCL1)-ALK, and moesin (MSN)-ALK [5]. In most, if not all, of these hybrid proteins the distal portion of ALK containing the tyrosine kinase domain is fused with proximal parts of the diverse proteins having oligomerization domains. Consequently, the hybrid proteins undergo spontaneous dimerization, leading to autophosphorylation and ultimately constitutive enzymatic activation of the ALK tyrosine kinase domain.
Overexpression of ALK is often observed in human neural tumors, primarily neuroblastoma [6]. Neuroblastomas are embryonic tumors of the peripheral sympathetic nervous system derived from neural crest tissues. It is one of the most frequent pediatric solid tumors, accounting for about 15% of childhood cancer mortality [7]. A clear correlation of ALK deregulation with neuroblastoma development has been established [8], [9], [10], [11], and at least ten ALK mutations, all found in the tyrosine kinase domain, were identified in the neuroblastoma samples. Among these, R1275Q, F1174L and F1245L/V/C occur most frequently [8], [11]. Increased copy numbers and gene amplifications of the ALK locus is also often detected in neuroblastoma patients [8], [10]. These germ line mutations in ALK explain most of the inheritable neuroblastomas, but activating mutations can also be somatically acquired. These mutations lead to constitutive autophosphorylation of mutated ALK, which displays increased kinase activity and excess phosphorylation on downstream targets such as Akt, STAT3 and ERK1/2. Mutated ALK from neuroblastoma samples is capable of transforming interleukin-3-dependent haematopoietic Ba/F3 cells into cytokine-independent growth [9], as well as NIH3T3 fibroblasts into colony and tumors in nude mice [8]. Knock-down of ALK by siRNA [8], [11], or ALK inhibition by small molecules PF-2341066/Crizotinib [10] and TAE684 [9] leads to suppressed cell growth, with decreased proliferation and increased apoptosis in neuroblastoma cells harboring mutated or amplified ALK. Together, these studies in neuroblastomas suggest that in a normal cellular context, appropriate ALK expression and activity levels need to be maintained for cell proliferation and survival.
While a pathogenic role for ALK has been demonstrated for many human cancers, little is known about the physiological function of Alk during development [12], [13], [14], [15]. Expression of ALK has been reported in murine neural tissues [13], [14], [16] but its function, especially in the CNS, remains elusive. A recent report in chicken showed that Alk controls proliferation of sympathetic neurons in the peripheral nervous system (PNS) (Reiff et al., 2011). In zebrafish (Danio rerio), a closely related co-ortholog of Alk, leukocyte tyrosine kinase (ltk) encodes a 1530 amino acids (aa) long protein [17]. ltk is expressed in neural crest cells (NCCs) and iridophores, a pigment cell type widespread in anamniote vertebrates. ltk is mutated in zebrafish shady mutants, which are characterized by the complete lack of iridophores and the loss of a subset of NCCs by apoptosis. Further analysis revealed that zebrafish ltk controls fate specification in a distinct subset of NCCs derived from the multipotent premigratory NCCs [17].
In the present study, we identified the full length zebrafish alk sequence and show that alk is expressed in the developing zebrafish CNS, that its over-expression results in increased proliferation of neural progenitors and that it is required for neuronal differentiation and survival. This is the first study that investigates Alk function is zebrafish and shows a critical role for Alk in the embryonic CNS.
Materials and Methods
Cloning of full-length zebrafish alk
The 5′UTR and N-terminal zebrafish alk sequences were obtained by 5′RACE (Rapid Amplification of cDNA Ends) using a kit from Invitrogen. The full length alk coding sequence was then PCR amplified with Phusion polymerase (Finnzymes), primers ALKatgXhoI (5′- CCGCTCGAGCCACCATGTGTGATAACGCAGCAGAG -3′) and ALKdownXbaI (5′- TGCTCTAGATTACAGCACAGTGGCGTTGT -3′), and cloned into pCS2+ (for capped mRNA preparation) or alk:HSE:cfp (for generation of transgnic fish, see below) using XhoI and XbaI sites present in these vectors.
Generation and maintenance of transgenic fish
All animal experiments were performed in accordance with approved IACUC protocols of the National University of Singapore (protocol numbers 075/07; 082/10; BR19/10). Zebrafish inbred AB wild-type strains were crossed to obtain embryos that were raised in 30% Danieau's solution at 28°C and staged as described previously [18]. For generation of transgenic fish, a alk:HSE:cfp construct was generated that contains the full-length zebrafish alk and cfp under control of a bidirectional heat-shock promoter in addition to flanking I-SceI sites [19], [20]. HSE:cfp was used as control. Plasmid DNA and I-SceI meganuclease (New England Biolabs) were co-injected into AB wild-type embryos at the one-cell stage as described [21]. On the next day, injected embryos were heat shocked for 1 hr at 39C. Embryos with strong CFP expression were raised to adulthood and crossed to WT AB fish. For screening and maintaining these lines, embryos were heat-shocked after 24 hpf to prevent early embryonic defects caused by alk overexpression. F2 or F3 embryos of two independent alk overexpressing lines (alk:HSE:cfp 1 and alk:HSE:cfp 2) and the HSE:cfp control line were used in the experiments described below.
Morpholino and RNA microinjection
The following Morpholino oligonucleotides (MO; from GeneTools, Philomath USA) were used: alk-ATG MO, 5′-TCCTCTGCTGCGTTATCACACATTC-3′; alk-ATG-mismatch control MO, 5′-TCgTCTcCTGCcTTATgACAgATTC-3′ (base substitutions indicated by small letters); splice-site MOs: alk-E3I3 MO, 5′-CATTTATGCAGAGCACCTGGTGATG-3′, and alk-I3E4 MO, 5′-CAAGGCCCCTGCCAGACAGAATGAT-3′; standard control MO, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; p53 MO, 5′-GCGCCATTGCTTTGCAAGAATTG-3′. All MOs were injected at 2 mg/ml, except for MO efficiency tests (5 mg/ml) and for the p53 MO (8 mg/ml).
Capped mRNAs were synthesized in vitro using the mMESSAGE mMACHINE SP6 Kit (Ambion). NPM-ALK mRNA was injected at 50 ng/µl. alkATG-egfp mRNA was injected at 50 ng/µl. To test the translation blocking efficiency of the alk ATG-MO, a target construct alkATG-egfp was generated that contains the alk 5′UTR and the ATG-MO target site, in addition to a 6 bp XhoI linker, in-frame with the egfp coding sequence.
RNA in situ hybridization, immunostaining and Western blotting
Whole mount RNA in situ hybridization and immunostaining were performed as described earlier [22]. An antisense riboprobe comprising the alk sequence from nt 6–885 was used to detect alk expression, and a sense probe covering the same sequence was used as control. The following primary antibodies were used: anti-phospho-Histone 3 (anti-pH 3; Millipore); anti-HuC/HuD (anti-HuC/D; Molecular Probes); anti-ERK1/2 (Santa Cruz); and anti-phospho-ERK1/2 (anti-pERK; Cell Signaling). For detection, either the VECTASTAIN Elite ABC Kit (Vector Labs) for diamino benzidine (DAB; Sigma) staining was used, or Alexa Fluor-488/568 coupled secondary antibodies (Invitrogen) for fluorescence detection. TUNEL assays were performed using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore) or with an anti-DIG primary antibody (Roche) for fluorescence detection. For 5-bromo-2-deoxyuridine (BrdU) labelling of cells in S-phase, embryos were incubated in 10 mM BrdU (Sigma) for 15 min on ice before fixation, followed by detection with an anti-BrdU primary antibody (DSHB, University of Iowa) as described previously [23]. For Western analysis, zebrafish embryos were deyolked and proteins extracted as previously described [24]. Western blotting was performed with anti-ERK and anti-pERK antibodies using standard protocols.
Alk inhibitor treatment
Dechorionated embryos were incubated with the pharmacological Alk inhibitor CEP-26939 [Teva Pharmaceuticals, also known as cmpd 13; 25] at 40 µM with 1% DMSO in Danieau's solution. 1% DMSO in fish medium was used as control. Embryos were treated from 24 hpf to 60 hpf, with changes of medium every 12 hours, and fixed at 60 hpf for further processing.
Microscopy, cell counting and statistical analysis
Confocal images were taken with a Zeiss LSM 510 Meta laser scanning microscope. All other images were taken with a Nikon SMZ1000 or a Nikon ECLIPSE 90i microscope. pH 3 positive cells were counted in confocal projections of 20 continuous optical sections representing a 50 µm thick area within rhombomeres r4 to r6. For cell counting, BODIPY-Texas-Red (Molecular Probes, Invitrogen) was used to counterstain cell membranes of HuC/D positive neurons. Embryos were stained in 20 µM BODIPY-TR for 1 h prior to imaging [26], [27]. Statistical analysis was done with GraphPad Prism 5. Unpaired two-tailed Student's t-test was used in comparisons between treated and control groups, with p<0.05 indicating statistical significance. Bar graphs were plotted with GraphPad Prism 5, showing means with standard error means (SEM) as error bars.
Results
Identification of full-length zebrafish alk
A partial zebrafish anaplastic lymphoma kinase (alk) sequence was available in the NCBI database (GenBank: XM_686872.2). It is located on chromosome 17 (Ensembl: ENSDARG00000095833) and comprises a putative 4164 base pairs (bp) transcript encoding 1387 amino acids (aa). We used 5′RACE to identify a 216 bp 5′ UTR that contains several STOP codons in all three reading frames, and the predicted translation start site of zebrafish alk. The full-length zebrafish alk coding region contains 4167 bp encoding a 1388 aa protein. The zebrafish Alk tyrosine kinase (TK) domain (aa 795–1062) has 89.6% aa identity to that of human ALK (aa 1116–1383; GenBank: NM_004304.4). Key tyrosine residues are present in zebrafish Alk (Y957, Y961 and Y962) that correspond to the human ALK kinase domain activation loop, a YXXXYY motif. Notably, this motif presents as YRASYY in all vertebrate Alk proteins, and slightly modified as YRSDYY in the fruit fly [28]. Moreover, three other residues in the TK domain: F1174, F1245 and R1275 that are most frequently affected by gain-of-function mutations in human neuroblastoma [8], [11] are also conserved in zebrafish Alk (F853, F924 and R954, respectively). Together, these data suggest a critical role for the highly conserved Alk residues in zebrafish.
Zebrafish alk expression during embryonic neurogenesis
We examined Alk mRNA transcript expression by RT-PCR at developmental stages from 6 hours post fertilization (hpf) to 72 hpf. Alk transcripts are first detected at 12 hpf, and expression levels continuously increased with the most profound increase at 48 hpf ( Fig. 1A ). The appearance of alk expression shortly after gastrulation corresponds to the initiation of segmentation and neurulation (10 hpf; [18]. Neurogenesis in the zebrafish hindbrain peaks between 24 hpf and 48 hpf, when the neuron to progenitor ratio changes most significantly [29]. The increase of alk mRNA levels from 12 hpf to 48 hpf therefore coincides with neurogenesis in zebrafish. In adult tissues, alk is highly expressed in brain, with much lower expression in heart, caudal fin, as well as testis, and none detectable in liver ( Fig. 1B ). Abundant expression of alk in adult brain suggests that it may function in adult neurogenesis and neural regeneration, which is prominent in zebrafish [30].
Figure 1. Expression of zebrafish alk.

(A) RT-PCR of cDNA at different embryonic stages. gapdh is used as loading control. (B) RT-PCR of cDNA from different adult tissues. beta-actin is used as loading control. (C) Lateral view of whole embryos at 24 hpf with dorsal up and anterior to the left. Insert shows dorsal view of head region. (D–G) Transverse sections of embryos at 24 hpf, at forebrain (D), midbrain (E), rhombomere 2 (r2; F) and r5 (G) levels, corresponding to positions indicated by lines in (C). (H) Lateral view of whole embryo at 48 hpf with dorsal up and anterior to the left. Insert shows dorsal view of head region. (I–K) Transverse sections of embryos at 48 hpf at forebrain (I), midbrain (J), and r5 (K), corresponding to positions indicated by lines in (D). Scale bars: C,H, 200 µm; D–E,I–K, 100 µm.
To analyze the spatial control of alk expression, whole mount in situ hybridization was performed with two antisense alk riboprobes representing either the alk N- (nt 6–885) or C-terminus (nt 3393–4071) in embryos at 24, 48 and 72 hpf. Both probes showed identical patterns ( Fig. 1C–K ; Fig. S1; and data not shown). Sense probes were used as negative control and showed no staining (Fig. S1). As early as 24 hpf, alk transcripts were broadly expressed in the developing central nervous system (CNS) including the spinal cord, with elevated levels in the brain ( Fig. 1C ). At 48 hpf, strong expression was found in the mid- and hindbrain ( Fig. 1H ), which remained high at 72 hpf (Fig. S1H,I). Transverse sections of embryos at 24 ( Fig. 1D–G ) and 48 hpf ( Fig. 1I–K ) at different brain levels showed that alk is expressed in the entire brain, with higher expression in ventral than dorsal regions of the early forebrain ( Fig. 1D ) and midbrain ( Fig. 1E ). In the hindbrain, expression levels were higher in regions where differentiated neurons locate rather than in regions with newborn progenitors ( Fig. 1F,K ), especially at 48 hpf [29].
Increased neural proliferation and aberrant neurogenesis in embryos ectopically expressing alk
We generated transgenic alk:HSE:cfp zebrafish lines that simultaneously co-express CFP and wild-type Alk under control of a bi-directional heat-shock promoter (Fig. S2)[19]. A control line with only the cfp sequence was also established (Fig. S2A). Embryos showing no CFP signal after heat shock (transgenic negative siblings (Sib); arrows in Fig. S2) were used as negative controls for comparison. In situ hybridization at different time points after heat shock at 24 hpf showed elevated levels of alk mRNA, detectable as soon as 1 hour post heat shock (hph; Fig. S3A), degraded over time (Fig. S3B), and eventually returned to the baseline alk concentration at 22 hph (Fig. S3C). A second heat shock performed at 48 hpf reactivated the transgene (Fig. S3D). CFP negative embryos showed no additional alk RNA (arrows in Fig. S3), confirming that they were transgenic negative siblings. Notably, after a 1.5 hour heat shock at 39.5°C at 10.5 hpf, all transgenic alk expressing embryos in both lines showed marked morphological malformations of the brain (Fig. S2D,E), similar to embryos injected with human NPM-ALK mRNA (data not shown). In contrast, HSE:cfp control embryos showed no defects with the same treatment (Fig. S2C), suggesting that the observed defects are caused by overexpressed Alk.
Defects in embryos heat shocked at 10.5 hpf were investigated at 24 hpf. For quantification of cell numbers in the experiments outlined below, we analyzed the hindbrain between rhombomeres r4 and r6, using the otic vesicles as landmarks in transverse sections ( Fig. 2 ). Importantly, similar findings were also observed in other regions of the CNS where endogenous alk is expressed at 24 hpf ( Fig. 1C ; data not shown). In alk-overexpressing embryos (Tg+), the 4th ventricle, a normally T-shaped opening in the hindbrain (asterisks in Fig. 2A,B,D ), was completely absent. Instead, several small cavities were observed (arrows in Fig. 2E,G , with high magnification in the inset E′). This suggested early neural tube defects, since the 4th ventricle, like other brain ventricles, is formed by proliferation and differentiation in the early neuroepithelium [18]. Accordingly, proliferation is abundant at this stage in the zebrafish hindbrain [29], and cell divisions occur almost exclusively at the most apical ventricular surface [29], [31]. In control siblings, proliferating cells positive for phospho-histone 3 (pH 3) were observed at the apical surface as expected ( Fig. 2A,B,D ). In contrast, in all alk Tg+ embryos of the alk:HSE:cfp 1 line, pH 3 positive cells were localized in a completely randomized manner ( Fig. 2E–G ). Similarly, differentiated postmitotic neurons are normally found in two clusters at each side of the basal and lateral regions of the neural tube (arrowheads in Fig. 2A,D ; [29]. In alk Tg+ embryos, HuC/D positive cells showed ectopic appearance at places, where they never occur in controls ( Fig. 2E–G ), while the patterns of pH3+ and HuC/D+ cells was not affected in HSE:cfp Tg+ embryos ( Fig. 2B ). To rule out possible positional effects of the inserted transgene, embryos of the second line alk:HSE:cfp 2 were examined and showed the same results (Fig. S4B).
Figure 2. Overexpression of alk promotes cell proliferation and affects neurogenesis.

(A,B) Confocal sections of HSE:cfp embryos. Sib (A) and Tg+ (B) embryos show no difference in number and distribution of pH 3 and HuC/D positive cells. (C) Y-axis indicates numbers of pH 3 positive cells counted in a 50 µm confocal stack of the hindbrain. Mean ± SEM, n = 10 embryos in each group. Sib and Tg+ HSE:cfp embryos were not significantly different (p = 0.84). Sib and Tg+ embryos of both alk:HSE:cfp lines were significantly different (***p<0.001). Unpaired two tailed t-test. (D–G) Confocal sections of alk:HSE:cfp 1 embryos. Sib (D) and Tg+ (E–G, from three different embryos) had different neural tube shapes. Dividing cells (pH 3, green) and neurons (HuC/D, red) in Tg+ embryos (E–G) were mispositioned (arrowheads), with aberrant patterns. (E′) High magnification of the boxed area in (E). Asterisk labels 4th ventricle. Arrows label small cavities found in the neural tube. (H,I) Confocal sections of alk:HSE:cfp 1 embryos, with BrdU labelled cells in S-phase. In Sib (H), BrdU positive cells occupy a region between dividing cells and neurons that exited the cell cycle, in a pattern complementary to pH 3 and HuC/D in (A,B,D). In Tg+ (I), BrdU positive cells were randomly positioned. Smaller dimension of samples in (H,I) might be due to HCl treatment in the experiment procedure. (J,K) Manual sections of embryos after in situ hybridization showed expanded ccnd1 expressions in Tg+ (K) in comparison to Sib embryos (J). Sib, transgenic negative siblings. Tg+, transgenic positive embryos. All images are sections perpendicular to neural tube. Scale bars: 50 µm.
Counting pH 3 positive cells revealed that Tg+ embryos of both alk:HSE:cfp 1 and alk:HSE:cfp 2 lines had an average of 44% and 53% more cells in M-phase compared to their Sib control groups, respectively (36.50 ± 2.16 vs. 25.30 ± 0.84; 39.10 ± 1.87 vs. 25.50 ± 0.69) ( Fig. 2C ). Consistently, HSE:cfp control embryos showed no significant difference from Sib embryos (25.40 ± 0.54 vs. 25.60 ± 0.81). This indicates that overexpression of alk but not CFP significantly elevates cell proliferation in the hindbrain. DNA synthesis in cells at S-phase was analyzed by 5-bromo-2-deoxyuridine (BrdU) labelling. In controls, BrdU cells occupy intermediate domains between the ventricular surface division zones and areas rich in postmitotic neurons, in a pattern complementary to pH 3 and HuC/D positive cells ( Fig. 2H ). In alk:HSE:cfp 1 Tg+ embryos, S-phase cells were not restricted to these positions but scattered throughout the hindbrain ( Fig. 2I ). The total area with cells in S-phase was drastically expanded, which made cell counting impossible. Interestingly, an increase of cells in S-phase has also been reported in primary sympathetic ganglia neuron cultures from embryonic chicken transfected with human ALK gene [15].
To test whether the observed increased proliferation is associated with upregulation of the cell cycle machinery, we evaluated the expression of cyclin D (ccnd1), which is known to promote proliferation by regulating cyclin dependent kinases CDK4 and CDK6 [32]. cyclin D is overexpressed in many cancers [32] including ALK positive neuroblastomas [5]. In non-transgenic control embryos, ccnd1 expression was observed in two stripes of the dorsal hindbrain ( Fig. 2J ). In contrast, alk:HSE:cfp 1 Tg+ embryos exhibited an expansion of ccnd1 expression into other regions of the hindbrain ( Fig. 2K ).
Overexpression of alk activates MAPK signaling in vivo
Activation of several signaling pathways has been reported in human ALK-related cancers including MEK/ERK pathway involved in cell proliferation [5]. In Drosophila, Alk functions through ERK activation during visceral mesoderm development [33], [34], [35], [36]. To test whether the MEK/ERK pathway is affected by the alk overexpression in zebrafish, we examined phosphorylation of ERK1/2 (pERK) and expression of total ERK levels using anti-pERK and anti-ERK antibodies, respectively ( Fig. 3 ; Fig. S4).
Figure 3. MAPK signaling is activated upon alk overexpression.

(A,A′) phospho-ERK (pERK) immunostaining of a 24 hpf wild-type embryo without heat shock. Endogenous MAPK activation is evident in the caudal most hindbrain and spinal cord, but low in other parts of the CNS. (B,B′,C,C′) Both Sib (B,B′) and Tg+ embryos (C,C′) of the HSE:cfp control line were normal and no additional pERK was observed. (D,D′,E,E′) Ectopic pERK (arrows) was evident in Tg+ (E,E′) of the alk:HSE:cfp 1 line compared to Sib (D,D′). (F,F′,G,G′) Total ERK was ubiquitously distributed in both Sib (F,F′) and Tg+ (G,G′) embryos of the alk:HSE:cfp 1 line and showed no difference. (A′–G′) Transverse sections from embryos at r5 level as indicated by black lines in (A–G). Sib, transgenic negative siblings. Tg+, transgenic positive embryos. ERK, total ERK1/2. pERK, phosphorylated-ERK1/2. Scale bars: 50 µm. (H) Western blot analysis showing more pERK protein in NPM-ALK mRNA injected embryos than in WT at 11 hpf. Each lane represents protein content from five embryos of a 20–50 embryo pool. (I) Western blots showing increased pERK levels in Tg+ versus Sib embryos at 22 hpf in both alk:HSE:cfp lines. Each lane contains proteins equivalent to five embryos of a 20–50 embryo pool. Only dissected anterior parts of embryos were used to eliminate endogenous pERK originating from spinal cord and tail bud.
In 24 hpf WT embryos without heat shock, increased pERK concentration could only be found in forebrain, mid-hindbrain boundary, caudal-most hindbrain, spinal cord ( Fig. 3A ), as well as the tail bud (image not shown). There were only few individual pERK positive cells in the hindbrain ( Fig. 3A ). This distribution was found in both Sib and Tg+ embryos of the HSE:cfp line, with no additional pERK signals ( Fig. 3B,C ). In cross sections at the level of r5, usually no pERK positive cells could be seen ( Fig. 3A ′,B′,C′). This demonstrated that neither heat shock nor overexpression of CFP activated ERK phosphorylation. In contrast, in Tg+ embryos of alk:HSE:cfp lines, pERK was strongly expressed across the entire CNS albeit in a mosaic pattern ( Fig. 3E ,E′; Fig. S4D,D′), while the total ERK concentration and distribution were similar to the control Sib line ( Fig 3F ,F′,G,G′). Consistently, Western blot analysis showed increased pERK levels but comparable ERK expression in Tg+ alk:HSE:cfp embryos at 22 hpf when compared to Sib controls ( Fig. 3H ). The fusion protein NPM-ALK is a known highly oncogenic form of ALK capable of activating MEK-ERK pathway (Chiarle et al., 2008). Accordingly, embryos at 11 hpf injected with human NPM-ALK mRNA showed increased pERK compared to WT controls, with similar levels of total ERK protein ( Fig. 3H ). Taken together, overexpression of alk causes up-regulation of pERK while total ERK levels remain unchanged. Ubiquitous MEK/ERK activation in the CNS upon alk overexpression is consistent with the observed deregulation of cell proliferation and aberrant neurogenesis.
Increased apoptosis but normal cell proliferation in the hindbrain after alk knock-down
Morpholino oligonucleotides capable of blocking zebrafish alk translation (ATG-MO) and a pair of splice blocking MOs (Spl-MO) were designed to knock-down alk (Fig. S5A). As shown in Fig. S5B, the alk ATG-MO suppressed GFP expression from an alkATG-egfp target construct, while a corresponding alk ATGmismatch-MO with 5 nt substitutions failed to block GFP expression (Fig. S5C–F). Two splice MOs were designed to prevent splicing of intron 3, which results in a 289 bp insertion leading to frame shift with nine possible premature STOP codons (Fig. S5G). RT-PCR with RNA from alk Spl-MO injected embryos at 24 hpf showed a size increase as expected (Fig. S5H).
alk ATG-MO or Spl-MO were injected into zebrafish embryos at the one- or two-cell stage, and apoptosis was examined at 22 hpf using a transferase mediated dUTP nick end labeling (TUNEL) assay. This showed increased apoptosis in the CNS ( Fig. 4B,C,E ; Fig. S6C,F) when compared to uninjected WT ( Fig. 4A ; Fig. S6A). Also, the size of the neural tube appeared smaller after alk knock-down when compared to WT ( Fig. 4A ). Neither a standard control MO nor the alk ATGmismatch-MO resulted in increased apoptosis (Fig. S6B,E), suggesting that the increased apoptosis was specific to alk deficiency. To further exclude MO off-target effects due to p53 activation [37], a p53 MO [38] was co-injected with alk MOs. This did not rescue the apoptosis defects caused by alk ATG-MO or alk Spl-MO (Fig. S6D,G) indicating that the alk MO knock-down effects are specific. Thus, alk depletion seems to promote apoptosis in the CNS independent of the p53 pathway. Interestingly, increased apoptosis was almost exclusively observed in the CNS, where endogenous alk is expressed broadly during early stages (24 hpf; Fig. 1C ) thus further supporting the specificity of the observed defects.
Figure 4. Knock-down of alk does not affect proliferation but induces apoptosis in the hindbrain.

(A–C) Confocal sections of 22 hpf embryos after immunostaining with pH 3 (green) and TUNEL (red). Compared to WT (A), both alk ATG-MO injected (B) or alk Spl-MO injected (C) embryos show pH 3 positive cells at normal positions, but more TUNEL positive cells are evident. Note that the size of neural tube in (B,C) is smaller than in (A). (D) Y-axis indicates numbers of pH 3 positive cells in a 50 µm thick confocal stack of hindbrain. Numbers are not significantly different in alk ATG-MO or alk Spl-MO from WT. (E) Y-axis indicates numbers of TUNEL positive cells in the same samples. Both alk ATG-MO and alk Spl-MO numbers were different from that in WT with high significance (***p<0.001). Mean ± SEM, n = 5 embryos in each group. Unpaired two tailed t-test. Scale bars: 50 µm.
We also tested whether alk inhibition affected cell proliferation. In embryos injected with either alk ATG-MO or Spl-MO, pH 3 positive cells were positioned normally ( Fig. 4B,C ). Counting of pH 3 positive cells within the r4 - r6 hindbrain region ( Fig. 4D ) revealed that neither alk ATG-MO (23.80 ± 0.86) nor alk Spl-MO (24.60 ± 0.93) resulted in a significant difference when compared to WT (25.00 ± 0.84). In contrast, within the same samples, counting of TUNEL positive cells ( Fig. 4E ) showed that both alk ATG-MO (40.60 ± 2.93) and alk Spl-MO (44.00 ± 4.56) injected embryos had significantly increased number of apoptotic cells as compared to controls (5.40 ± 1.44). These findings suggest that alk deficiency may not affect dividing cells in M-phase. Since the majority of cell divisions at this stage represent proliferative neural progenitors, the increased apoptosis observed in alk morphants most likely does not affect these progenitor cells, but rather other, possibly more mature neuronal cell types.
Knock-down of alk impairs neuronal differentiation and reduces neuron numbers in the hindbrain
As direct targets of Notch signaling, Hes genes (Hes1 and Hes5 in mammals) are abundantly expressed in undifferentiated neural progenitors and essential for maintaining stem cell identity [39]. We found no expression change of hairy-related 6 (her6), the zebrafish Hes1 homolog [40], in alk morphants ( Fig. 5B ,B′,B″; Fig. S7B) suggesting that proliferation of undifferentiated neural progenitors is not affected by alk knock-down. In differentiating neural cells, proneural gene expression is up-regulated as Notch-Hes signaling is switched off. Combined expression of different proneural genes restricts neuronal lineages, forces cell cycle exit, initiates neuronal differentiation and promotes neuron survival [41], [42], [43]. Expression of several zebrafish proneural genes was analyzed to study the effect of alk knock-down on neuronal precursor differentiation. Neurogenin 1 (neurog1, neurod3) expression was unchanged or only slightly reduced in alk morphants ( Fig. 5D ,D′,D″; Fig. S7D). On the other hand, expression of the zebrafish atonal ortholog, neurogenic differentiation 4 (neurod4, zath3) was significantly reduced throughout the CNS, including the hindbrain ( Fig. 5F ,F′,F″; Fig. S7F). The achaete-scute complex like (ascl or ash) genes belong to another proneural gene family that can act independently from atonal-related genes in vertebrates [44]. Expression of ascl1b was also reduced in alk morphants ( Fig. 5H ,H′,H″; Fig. S7H). Moreover, delta A (dla), a Notch ligand highly expressed in differentiating neurons, was also reduced ( Fig. 5J ,J′,J″; Fig. S7J). Finally, glial fibrillary acidic protein (gfap) expression was not changed in alk morphants ( Fig. 5L ,L′,L″; Fig. S7L). Of note, gfap is expressed in radial glia cells that overlap with neural progenitors at this embryonic stage. All of the genes mentioned above were expressed normally in alk ATGmismatch-MO injected embryos (Fig. S7A,C,E,G,I,K). Taken together, we observed a down-regulation of several proneural genes in alk morphants. At the same time, progenitor marker expression was unchanged and cell proliferation was not altered. This indicates that progenitor cells do not require alk activity. Instead, we propose that alk knock-down affects early neuronal precursors that fail to differentiate and instead undergo programmed cell death.
Figure 5. Knock-down of alk impairs neuronal differentiation.

(A–L) In situ hybridization of neuronal marker genes with wild-type (A,C,E,G,I,K), and alk Spl-MO injected embryos (B,D,F,H,J,L) at 22 hpf. Images in first row show dorsal views of head region with anterior to the left. (A′–L′,A″ –L″) Transverse cross sections at the level of r2 (A′–L′) and r5 (A″ –L″) in WT embryos or alk Spl-MO injected morphants. (A,A′A″,B,B′B″) her6 expression was unchanged in alk morphants compared to WT. (C,C′,C″,D,D′,D″) neurog1 expression was unchanged or only slightly reduced in morphants. (E,E′,E″,F,F′,F″) neurod4 expression was strongly reduced in morphants in several regions including the hindbrain. (G,G′,G″,H,H′,H″) Similarly, ascl1b expression in alk morphants was also significantly reduced. (I,I′,I″,J,J′,J″) dla expression in alk morphants was also reduced. (K,K′,K″,L,L′,L″) Glia marker gfap expression was unchanged. Scale bars: 50 µm. Lines at r2 and r5 indicate levels of cross sections.
MOs elicit their effects from earliest embryonic stages onward. To achieve alk deficiency at later embryonic stages, a pharmacological ALK inhibitor, CEP-26939 was used. Both CEP-26939 and its closely related analog ALK inhibitor, CEP-28122, have been reported to lead to growth inhibition in a variety of cultivated human ALK-related cancer cell lines, including neuroblastoma, through caspase 3/7 activation and apoptosis [25], [45]. In our experiment, zebrafish embryos were incubated in fish medium containing CEP-26939, or DMSO alone as a negative control, starting at 24 hpf. Embryos were fixed at 60 hpf and examined for her6, neurog1, neurod4 and ascl1b expression, as well as apoptosis ( Fig. 6 ). Similar to the findings in Alk morphants, TUNEL staining revealed increased apoptosis in the CNS of the ALK inhibitor treated embryos ( Fig. 6A,B ), which was also confirmed by Acridine Orange staining in living embryos at 48 hpf (data not shown). Consistent with the MO experiments, no difference in her6 expression was seen between inhibitor- and DMSO-treated groups ( Fig. 6C,D ). Furthermore, only a weak reduction of neurog1 expression was observed with no major change in its overall pattern ( Fig. 6E,F ). On the other hand, expression of neurod4 and ascl1b was strongly reduced and almost completely absent in the mid- and hindbrain in CEP-26939 treated embryos ( Fig. 6G,H,I,J ). Significantly, in all regions where neurog1, neurod4 or ascl1b expression was reduced, the number of apoptotic cells was strongly increased. Thus, this pharmacological ALK inhibitor fully recapitulated the effects observed after MO-mediated ALK depletion providing further evidence that ALK activity is required for the differentiation and survival of early neural precursors in the developing CNS.
Figure 6. Inhibition of Alk activity by CEP-26939 phenocopies alk morphant defects.

(A,C,E,G,I) Embryos at 60 hpf treated with 1% DMSO from 24 hpf were used as negative controls. (B,D,F,H,J) 60 hpf embryos treated with 40 µM CEP-26939 from 24 hpf. (A,B) Inhibitor treated embryos showed more TUNEL-positive apoptotic cells in mid- and hindbrain (B) when compared to DMSO treated embryos (A). (C,D) No significant change was observed for her6 expression in inhibitor treated embryos. (E,F) neurog1 was only slightly reduced in inhibitor treated (F) compared to DMSO treated embryos (E). (G,H,I,J) Both neurod4 and ascl1b expression domains in mid- and hindbrain were almost completely absent in inhibitor treated embryos (H,J). All images show dorsal views with the focal plane on mid- and hindbrain, with anterior to the left. Numbers in each image indicate individuals similar as that shown as representatives and total numbers of embryos investigated. Scale bars: 100 µm.
The observed reduction of proneural gene expression suggested compromised neuronal differentiation and survival. To test to what degree alk deficiency affects the final steps of neurogenesis, i.e. the formation of fully differentiated neurons, expression of the pan-neuronal marker HuC/D [46], [47] was analyzed. In the hindbrain at 31 hpf, areas where postmitotic HuC/D+ neurons are located were significantly reduced in size in alk morphants ( Fig. 7B ,B′,C,C′) when compared to WT ( Fig. 7A ,A′) or ATGmismatch-MO injected embryos ( Fig. 7D ,D′). In contrast, proliferative ventricular and subventricular zones appeared normal, when BODIPY-Texas Red was used to label cells ( Fig. 7E ), an average of 92.25 ± 2.94 HuC/D positive neurons was counted per confocal single plane in controls (representing a 5 µm optical section). Cell numbers in the alk ATGmismatch-MO injected group (85.14 ± 2.71) were not significantly different from WT. In contrast, numbers of HuC/D positive neurons were reduced to 50.50 ± 3.43 in alk ATG-MO injected embryos, and 46.36 ± 2.64 in alk Spl-MO injected embryos. In conclusion, alk ATG-MO or alk Spl-MO injected embryos show a significant reduction in the number of differentiated neurons in the hindbrain when compared to WT. Thus, alk is required for differentiation and survival or early neural precursors in order to maintain the correct number of fully differentiated hindbrain neurons at later stages.
Figure 7. Knock-down of alk reduces the number of differentiated hindbrain neurons.

(A–D) Confocal sections of 31 hpf embryos at r5 level immunostained with the pan-neuronal marker HuC/D (green) and co-stained with bodipy-Texas Red (TR, red). The HuC/D channel (green) is separately shown in (A′–D′). Arrowheads point to emerging neurons in the ear, serving as landmark to indicate that all sections were at the same position. In both alk ATG-MO (B,B′) or alk Spl-MO (C,C′) injected embryos, the areas of postmitotic neurons were clearly smaller than in WT (A,A′) or alk ATGmismatch-MO injected embryos (D,D′). The proliferative ventricular and subventricular zones were normal in size. (E) Y-axis indicates numbers of HuC/D expressing cells on sections. Both alk ATG-MO and alk Spl-MO groups were significantly different from WT (***p<0.001), whereas the change in alk ATGmismatch-MO group was not significant (p = 0.088). Mean ± SEM, N = 12 in WT and ATG-MO, N = 14 in Spl-MO and ATGmismatch-MO. Unpaired two tailed t-test. Scale bars: 50 µm.
Discussion
This is the first study reporting structure and functional characterization of zebrafish alk. It is also the first analysis of the physiological role of Alk in the vertebrate central nervous system (CNS). We show that zebrafish alk is expressed in the developing CNS and is required for differentiation and survival of neural cells. The full length zebrafish alk transcript encodes a deduced 1388 amino acids (aa) protein that matches a predicted partial zebrafish alk sequence in the NCBI database (GenBank: XM_686872.2), except an additional Glutamine (Q1280) in a Q(13) repeat located at the C-terminal end, and a Serine (S309) to Alanine (A309) replacement in a S(4) repeat located in its N-terminus. These differences most likely reflect polymorphisms between different zebrafish wild-type strains. Interestingly, the N-terminus of zebrafish Alk (aa 1-708) shares only 25.3% aa identity with its human counterpart (aa 1-1029) and seems to be significantly shorter, while the TK domain shows 89.6% identity. Similarly, chicken Alk (GenBank: XM_419364.3) also has a conserved TK domain (aa 970–1237; 95.5% identical to human), but a much shorter N-terminus (aa 1–883) with only 53.3% identity. When Alk sequences from 17 vertebrate species and Drosophila melanogaster were compared, the TK domain was the most conserved part among all Alk's. In contrast, the Alk N-terminus showed much more variation, consistent with the idea that the N-terminus of Alk has undergone extensive divergence during vertebrate evolution. In mammals, ALK and LTK can be clearly distinguished by their extracellular domains, with respect to its length (1030 aa vs. 424 aa) and the presence or absence of MAM domains, respectively [48], [49]. Phylogenetic analyses suggested that the MAM domain is a feature of a common Alk/Ltk ancestor, and has been retained in Ltk in zebrafish and chicken but not mammals [17]. It is likely that Ltk and Alk evolved their N-termini differently in mammals and in teleosts. As a consequence, mammalian Ltk lost some of the fish specific Ltk functions, such as for iridophore development [17].
Expression of alk in the central nervous system
In mouse, Alk is prominently expressed in the diencephalon, the neuroepithelium surrounding the fourth ventricle, the ventricular and subventricular zone of the cortex, the midbrain, and the medulla oblongata from day 10.5 to day 16.5 [14]. Similar to mouse, alk is expressed in most of the central nervous system (CNS) in zebrafish. However, in mouse and chicken, Alk expression is also detected in the spinal cord [12], [14]. In contrast, zebrafish alk could not be detected in the spinal cord under the experimental conditions used, especially after 24 hpf ( Fig. 1C,H ). This could possibly reflect species-specific differences in Alk expression among different vertebrates. In mouse, rat, and chicken, Alk is also expressed in the peripheral nervous system (PNS) including the trigeminal ganglia, facial ganglia, vestibulocochlear ganglia, inferior ganglia of the vagus nerve, as well as dorsal root ganglia and sympathetic ganglia [12], [13], [14]. In our study, we could not detect expression of zebrafish alk in the PNS. On the other hand, the similar spatiotemporal expression patterns of Alk in the early CNS in mouse [14], chicken [12], and zebrafish (this study) suggest that the activity and function of the highly related Alk tyrosine kinase domain is conserved during CNS neurogenesis.
In vivo activity of over-expressed, non-mutated Alk
Receptor protein tyrosine kinases (RTKs) can undergo spontaneous dimerization, transphosphorylation of monomers, and activation. This process is promoted and stabilized by the presence of RTK ligands [50], [51]. In our alk overexpression experiments, Alk proteins could have spontaneously formed active dimers, resulting in RTK activation. Such autophosphorylation of ALK in absence of any ligand was also reported in cell culture [52]. The observed activation of MEK/ERK pathway confirmed that the transgenic Alk proteins are expressed and enzymatically active. Interestingly, we observed a high degree of mosaicism of MEK/ERK activation although the Alk transgene was ubiquitously expressed. While the exact reason for this remains unclear, it suggests a dynamic regulation of intracellular signaling when Alk is activated in this in vivo context. Overexpression of alk in these transgenic lines resulted in enhanced cell proliferation with increased numbers of cells in M-phase and up-regulation of ccnd1 expression. Furthermore, aberrant neurogenesis patterns were observed with ectopic localization of differentiated neurons. We therefore propose that ectopic activation of Alk leads to excess MAPK activity resulting in increased proliferation of neuronal progenitors, randomized cell cycle exit and differentiation, and consequently mis-positioning of differentiating neurons. Triggering alk overexpression at a later stage (24 hpf) also enhanced proliferation but strikingly to a much lesser extent and in a spatially more restricted fashion (data not shown) when compared to overexpression at 10.5 hpf ( Fig. 2 ). Recent studies suggested that overexpression of mutated ALK works cooperatively with MYCN overexpression to promote the severity of induced neuroblastoma [53], [54]. Interestingly, zebrafish nmyc1, the ortholog of human MYCN, is expressed ubiquitously at high levels in the CNS before 24 hpf, but becomes restricted thereafter [55]. This could explain why proliferative effects of overexpressed Alk were higher at earlier embryonic stages.
Alk is required for differentiation and survival in the CNS but not proliferation
We show that an alk knock-down does not affect cell proliferation in the hindbrain at 24 hpf. Interestingly, decreased cell proliferation was reported in in vitro cultured neuroblastoma cell lines where ALK has been depleted by siRNA [8], [11]. Also, a recent report using cultured chicken sympathetic neurons derived from PNS sympathetic ganglia showed that the knock-down of endogenous chicken Alk results in reduced proliferation [15]. This suggests possible differences of Alk function in different tissues and cell types. Alternatively, it could also reflect differences between in vivo and in vitro cell culture conditions suggesting more complicated scenarios that underlay Alk mediated cell cycle control, such as impact of the microenvironment or functional redundancy with other kinases that cannot be fully mimicked in cell culture settings.
In alk deficient embryos, generated by MO-mediated ALK depletion or pharmacological inhibition of its enzymatic activity, a universal increase in neural cell apoptosis and reduction in expression of selected proneural genes was observed. Since progenitor marker expression was unchanged and cell proliferation was not affected in hindbrain, this indicates that progenitor cells are not impaired by alk deficiency and that the observed apoptosis does not originate from neural progenitors. On the other hand, reduced proneural gene expression in differentiating neuronal precursors suggests defects in differentiation eventually leading to cell death. There are several possibilities how alk deficiency could affect cell survival. First, differentiating neural cells could undergo apoptosis because Alk is a survival factor and functions as a repressor of cell death. Thus, reduced proneural gene expression could simply reflect a reduction in the numbers of differentiating cells. Alternatively, Alk could be a specification factor, which is required to promote proneural gene expression. In morphants, cells that have exited the progenitor state would therefore fail to differentiate and consequently commence apoptosis. It remains unclear at present whether Alk induced apoptosis is the cause or consequence of impaired neurogenesis. However, our study showed that the extent of apoptosis in embryos deficient for Alk is relatively mild compared to the extensive changes in neurogenic gene expression. This observation favours a scenario where Alk deficiency leads to impaired neurogenesis, which subsequently results in apoptosis.
Unfortunately, Alk knock-out animals other than the embryos presented here are lacking in order to address which of the scenarios explains increased apoptosis after alk knock-down. Interestingly and in agreement with a role as specification factor, the Alk co-ortholog Ltk is involved in cell fate specification of multipotent precursors in the iridophore lineage (Lopes et al., 2008). On the other hand, siRNA-based approaches recently reported increased apoptosis upon Alk knock-down in neuroblastoma cell lines [56] supporting the idea of Alk as a survival factor. It is well established that proneural genes are required for cell survival during neuronal differentiation [57], [58], [59]. Based on this and our own observations, a combination of both scenarios therefore seems likely. We propose that Alk is required for the survival of neural precursors that have exited the progenitor state and are about to enter different neural differentiation fates.
Supporting Information
Sense controls and expression of zebrafish alk at 72 hpf. (A–D) Negative controls at 24 hpf using a sense probe, corresponding to Fig. 1D–G, respectively. (E–G) Negative controls at 48 hpf using a sense probe, corresponding to Fig. 1I–K, respectively. (H) Lateral view of embryo at 72 hpf with dorsal up and anterior to the left. (I) Dorsal high magnification views of head region at 72 hpf. (J) Negative sense probe control of (I). Scale bars: A–G, 100 µm; H–J, 200 µm.
(JPG)
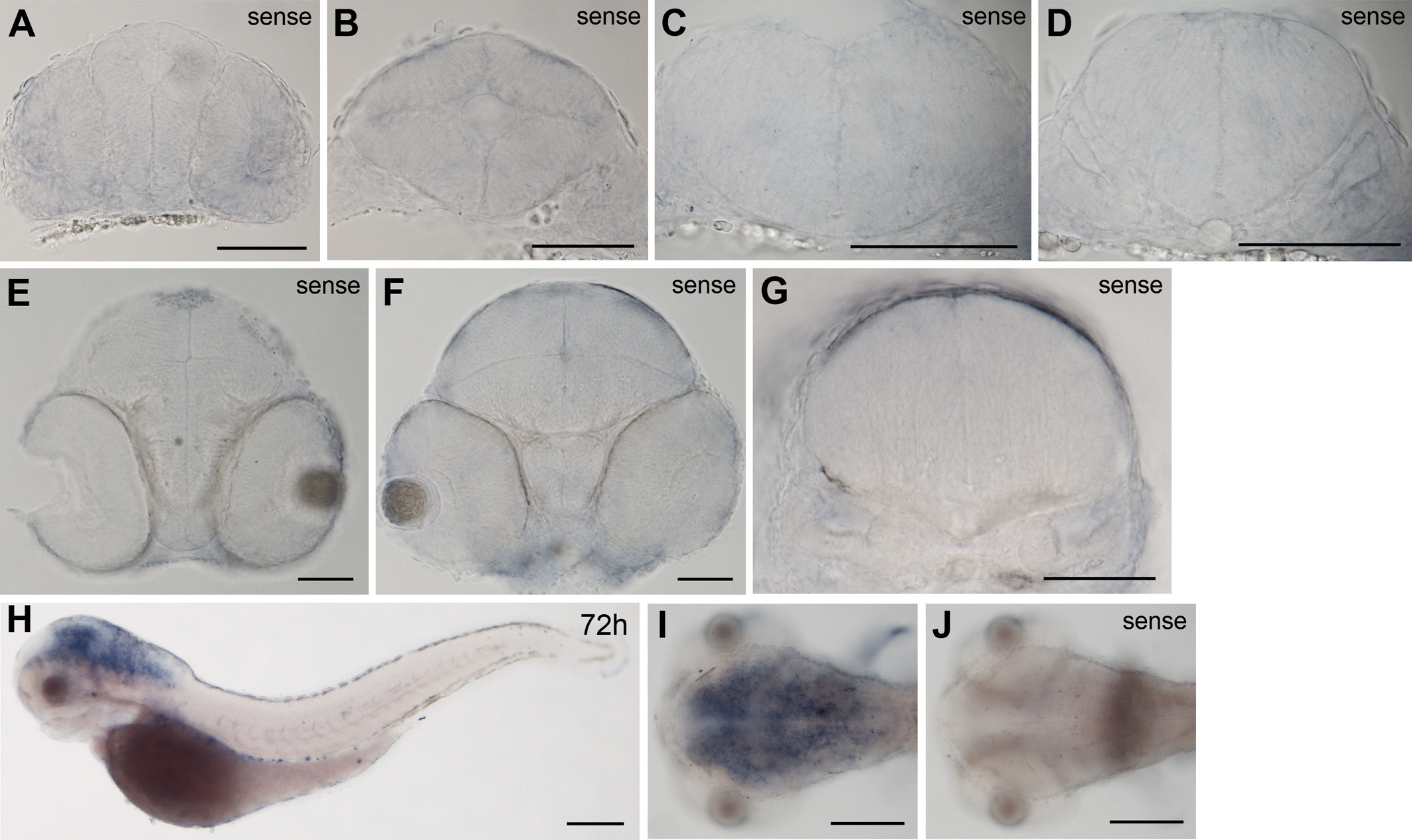{kind=link}
Heat-shock inducible transgenic zebrafish lines. (A,B) Diagrams illustrating transgenic cassettes for control line (A) and alk overexpression lines (B). The construct reported by Bajoghli et al. contains eight heat shock element repeats (HSE) with bidirectional promoter activity. This construct was engineered in such a way that alk and cfp were put on each side (B), or just cfp in the control line (A). (C–E) 24hpf embryos from the control line (HSE:cfp, C), alk overexpression line 1 (alk:HSE:cfp 1, D), and independent alk overexpression line 2 (alk:HSE:cfp 2, E). All embryos received a 1.5 hour long heat shock at 39.5°C and at 10.5 hpf. Bright field images on top and images of the same embryos in CFP channel below. Inserts show representative transgene positive embryos in each line. Arrows point to transgenic negative siblings in each line. They are morphologically normal and not visible in the CFP channel.
(JPG)
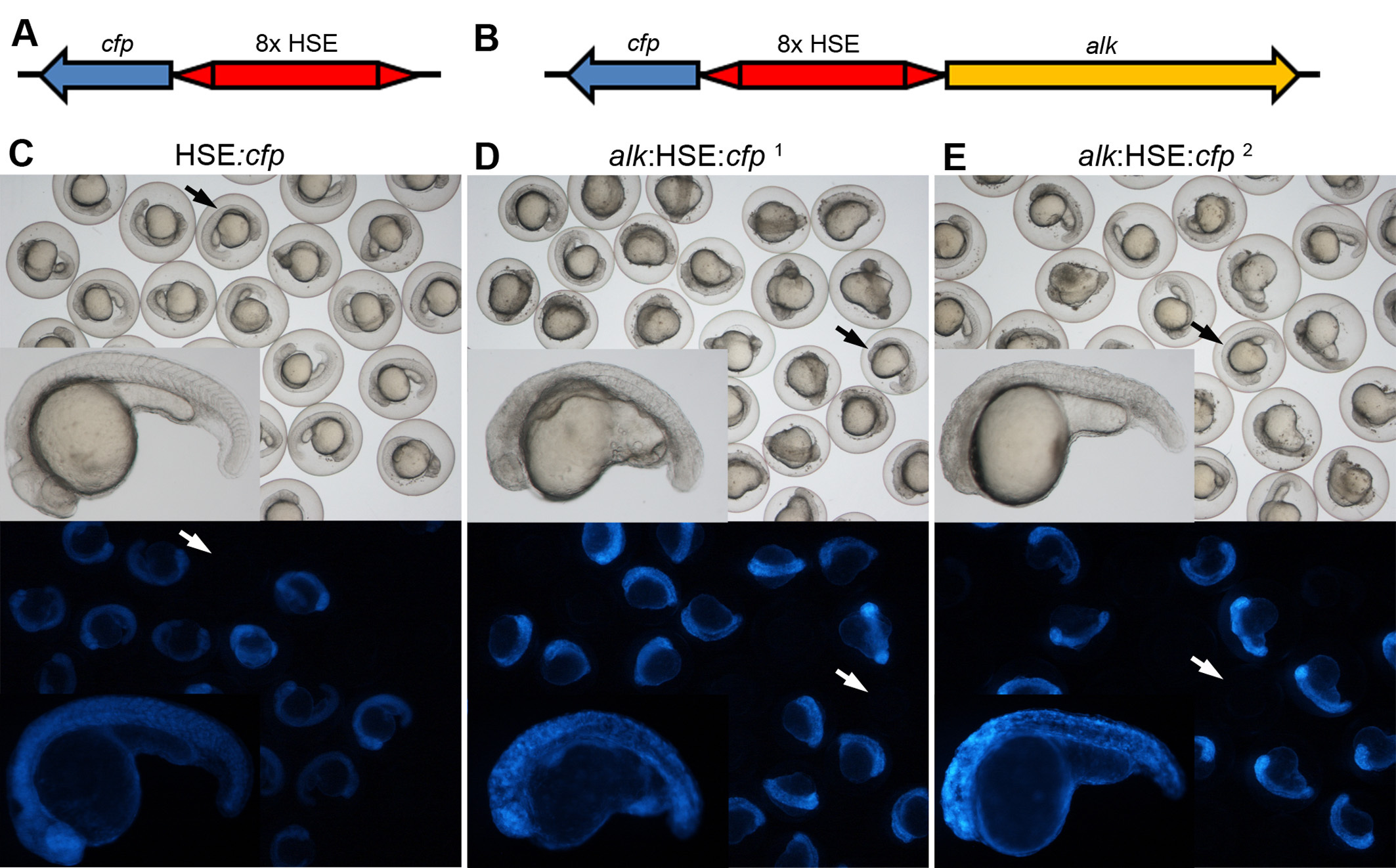{kind=link}
In situ hybridization to detect alk overexpression in alk :HSE: cfp transgenic embryos. A heat shock was performed at 24 hpf. All in situ hybridizations were done using an alk antisense RNA probe, at 1 hour post heat shock (hph) (A), 6 hph (B), 22 hph (C), and shortly after a second HS performed at 48 hpf (D). Inserts show representative transgenic positive embryos. As soon as 1 hour after heat shock, high levels of exogenous alk RNA could be detected in the entire embryo. Reduced staining at 6 hours after HS indicated RNA degradation. Staining almost completely disappeared at 22 hours after HS. A second HS re-activated transcription of overexpressed alk. Arrows points to transgene negative siblings where no exogenous alk was detected. Endogenous alk expression was not detected because of short staining time.
(JPG)
{kind=link}
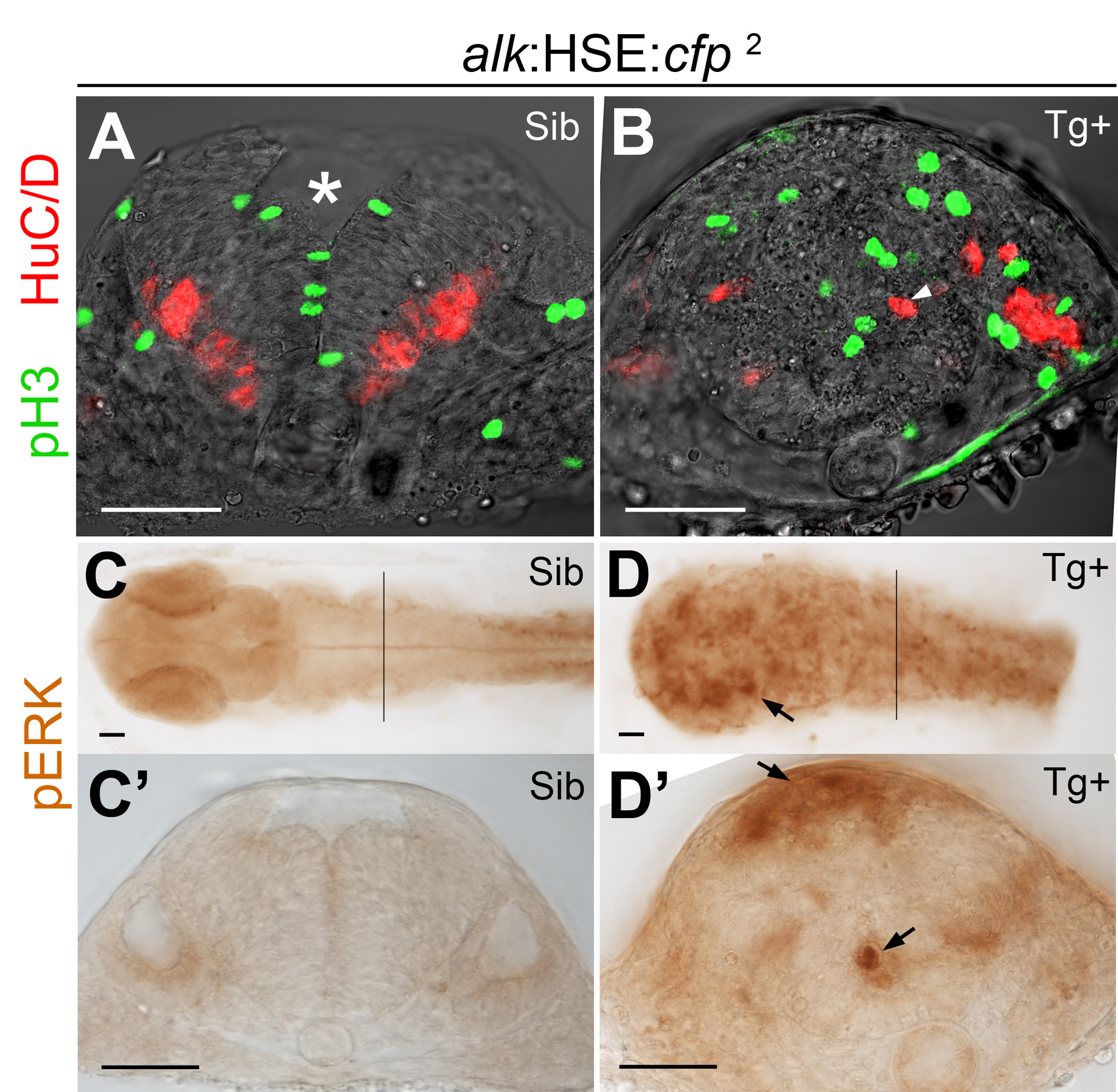
Proliferation and differentiation in the alk :HSE: cfp 2 line. (A,B) Confocal sections of 24hpf embryos of the alk:HSE:cfp 2 line. Sib (A) and Tg+ (B) had different neural tube shapes. Dividing cells (pH 3, green) and neurons (HuC/D, red) in Tg+ embryos (B) were both mispositioned (arrowhead), identical to defects observed in the alk:HSE:cfp 1 line (Fig. 2). Asterisk labels 4th ventricle. (C,C′,D,D′) Ectopic pERK (arrows) in Tg+ (D,D′) of the alk:HSE:cfp 2 line compared to Sib (C,C′), identical to defects observed in alk:HSE:cfp 1 (Fig. 3). pERK, phosphorylated-ERK1/2. Sib, transgene negative siblings. Tg+, transgenic embryos. Scale bars: 50 µm.
(JPG)
{kind=link}
alk MO efficiency test experiments. (A) A schematic diagram showing arrangement of the first four alk exon-intron boundaries and MO binding sites. Numbers show exon/intron sizes in basepairs. (B) A construct containing egfp inserted in frame with the alk ATG site was generated to test the binding efficiency of ATG-MO. (C–F) Injection of this mRNA in combination with MOs resulted in different EGFP translation levels. Embryos at 8 hpf, with bright field images on top and fluorescent images below. Injection of the mRNA resulted in EGFP signal (D). Co-injection with alk ATG-MO blocked its translation (E). alk ATGmismatch-MO did not block its translation (F). Embryos labeled with asterisks in (C) and (E) were taken from the group shown in (D), indicating sufficient fluorescent excitation. (G) A diagram showing aberrant splice products with retention of intron 3, when alk pre-mRNA splicing is blocked by splicing MOs. (H) RT-PCRs were used to test splicing MO efficiency by primers indicated in (A). In Spl-MO injected 24 hpf embryos, the PCR product size was increased by approximately 300 bp (2nd lane) when compared to uninjected wild-type (1st lane), indicating retention of intron 3. -RT controls (3rd and 4th lanes, without reverse transcriptase in cDNA synthesis) excluded contamination by genomic DNA. gapdh was used as loading control.
(JPG)
{kind=link}
Increased apoptosis in alk morphants. TUNEL staining of embryos at 22 hpf. Images show lateral views on top with anterior to the left, and dorsal views of head region below with anterior to the top. WT (A) or Std-MO injected (B) embryos had only few scattered TUNEL positive cells. In alk ATG-MO (C) or Spl-MO injected embryos (F), TUNEL positive cells increased in number, while ATGmismatch-MO injected (E) looked normal. Co-injections with p53-MO (D,G) failed to attenuate the apoptosis defect indicating specificity of the effect. Scale bars: 100 µm.
(JPG)
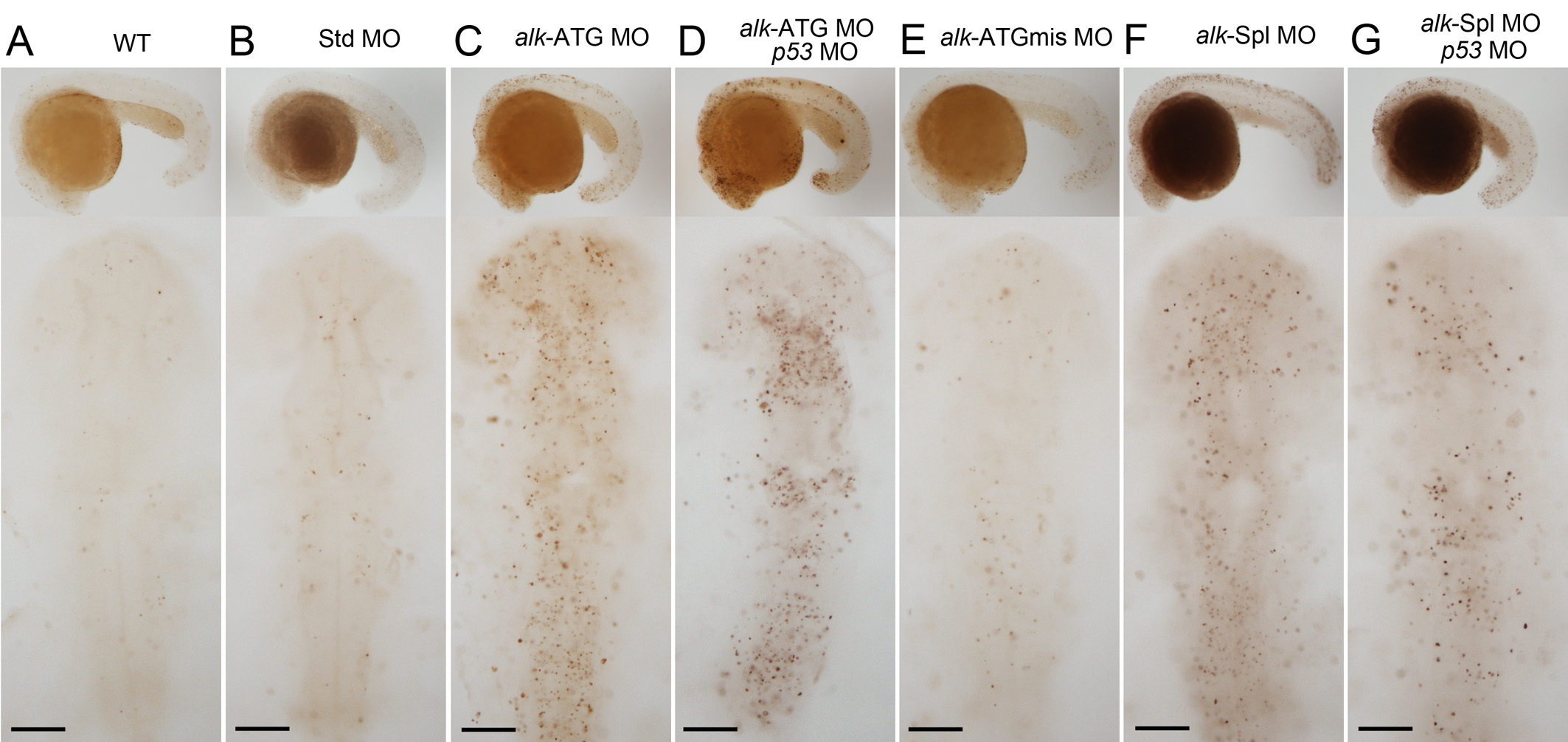{kind=link}
An alk ATG-MO leads to reduced proneural gene expression. (A,C,E,G,I,K) Embryos injected with alk ATGmismatch-MO showed no difference when compared to WT in Fig. 5. (B,D,F,H,J,L) alk ATG-MO injected embryos show the same phenotype as alk Spl-MO injected embryos (Fig. 5). Embryos showed no change in her6, neurog1 and gfap expression, but reduced expression of neurod4, ascl1b and dla identical to alk Spl-MO injected embryos shownin Fig. 5. Images show dorsal views of head regions with anterior to the left. Scale bars: 50 µm.
(JPG)
{kind=link}
Acknowledgments
We thank Drs. Thomas Czerny, Vladimir Korzh and Steven Fong for providing plasmids, and Dr. Liou Yih Cherng for providing ERK antibodies.
Funding Statement
This project was supported by a Singapore Ministry of Education-Academic Research Fund grant (R-154-000-478-112) to CW, an United Kingdom Medical Research Council (MRC) grant (G0400806) to RK, and a grant from the National Cancer Institute (NCI) R01-CA96856 to MW. SY is a National University of Singapore -Department of Biological Sciences graduate scholar. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Benz-Lemoine E, Brizard A, Huret JL, Babin P, Guilhot F, et al. (1988) Malignant histiocytosis: a specific t(2;5)(p23;q35) translocation? Review of the literature. Blood 72: 1045–1047. [PubMed] [Google Scholar]
- 2. Fischer P, Nacheva E, Mason DY, Sherrington PD, Hoyle C, et al. (1988) A Ki-1 (CD30)-positive human cell line (Karpas 299) established from a high-grade non-Hodgkin's lymphoma, showing a 2;5 translocation and rearrangement of the T-cell receptor beta-chain gene. Blood 72: 234–240. [PubMed] [Google Scholar]
- 3. Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi T, et al. (1995) Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood 86: 1954–1960. [PubMed] [Google Scholar]
- 4. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, et al. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448: 561–566. [DOI] [PubMed] [Google Scholar]
- 5. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G (2008) The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 8: 11–23. [DOI] [PubMed] [Google Scholar]
- 6. Dirks WG, Fahnrich S, Lis Y, Becker E, MacLeod RA, et al. (2002) Expression and functional analysis of the anaplastic lymphoma kinase (ALK) gene in tumor cell lines. Int J Cancer 100: 49–56. [DOI] [PubMed] [Google Scholar]
- 7. Maris JM, Hogarty MD, Bagatell R, Cohn SL (2007) Neuroblastoma. Lancet 369: 2106–2120. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y, Takita J, Choi YL, Kato M, Ohira M, et al. (2008) Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455: 971–974. [DOI] [PubMed] [Google Scholar]
- 9. George RE, Sanda T, Hanna M, Frohling S, Luther W (2008) Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455: 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, et al. (2008) Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455: 967–970. [DOI] [PubMed] [Google Scholar]
- 11. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, et al. (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455: 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hurley SP, Clary DO, Copie V, Lefcort F (2006) Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J Comp Neurol 495: 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Degoutin J, Brunet-de Carvalho N, Cifuentes-Diaz C, Vigny M (2009) ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur J Neurosci 29: 275–286. [DOI] [PubMed] [Google Scholar]
- 14. Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, et al. (2006) Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns 6: 448–461. [DOI] [PubMed] [Google Scholar]
- 15. Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, et al. (2011) Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development 138: 4699–4708. [DOI] [PubMed] [Google Scholar]
- 16. Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, et al. (1997) Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 14: 439–449. [DOI] [PubMed] [Google Scholar]
- 17. Lopes SS, Yang X, Muller J, Carney TJ, McAdow AR, et al. (2008) Leukocyte tyrosine kinase functions in pigment cell development. PLoS Genet 4: e1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 19. Bajoghli B, Aghaallaei N, Heimbucher T, Czerny T (2004) An artificial promoter construct for heat-inducible misexpression during fish embryogenesis. Dev Biol 271: 416–430. [DOI] [PubMed] [Google Scholar]
- 20. To TT, Witten PE, Renn J, Bhattacharya D, Huysseune A, et al. (2012) Rankl-induced osteoclastogenesis leads to loss of mineralization in a medaka osteoporosis model. Development 139: 141–150. [DOI] [PubMed] [Google Scholar]
- 21. Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, et al. (2002) I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech Dev 118: 91–98. [DOI] [PubMed] [Google Scholar]
- 22. Liedtke D, Winkler C (2008) Midkine-b regulates cell specification at the neural plate border in zebrafish. Dev Dyn 237: 62–74. [DOI] [PubMed] [Google Scholar]
- 23. Dolbeare F (1995) Bromodeoxyuridine: a diagnostic tool in biology and medicine, Part I: Historical perspectives, histochemical methods and cell kinetics. Histochem J 27: 339–369. [PubMed] [Google Scholar]
- 24. Link V, Shevchenko A, Heisenberg CP (2006) Proteomics of early zebrafish embryos. BMC Dev Biol 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ott GR, Tripathy R, Cheng MG, McHugh R, Anzalone AV, et al. (2010) Discovery of a Potent Inhibitor of Anaplastic Lymphoma Kinase with in Vivo Antitumor Activity. Acs Medicinal Chemistry Letters 1: 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cooper MS, Szeto DP, Sommers-Herivel G, Topczewski J, Solnica-Krezel L, et al. (2005) Visualizing morphogenesis in transgenic zebrafish embryos using BODIPY TR methyl ester dye as a vital counterstain for GFP. Dev Dyn 232: 359–368. [DOI] [PubMed] [Google Scholar]
- 27. Godinho L (2011) Using intravital dyes to ubiquitously label embryonic zebrafish. Cold Spring Harb Protoc 2011: 877–878. [DOI] [PubMed] [Google Scholar]
- 28. Palmer RH, Vernersson E, Grabbe C, Hallberg B (2009) Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 420: 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lyons DA, Guy AT, Clarke JD (2003) Monitoring neural progenitor fate through multiple rounds of division in an intact vertebrate brain. Development 130: 3427–3436. [DOI] [PubMed] [Google Scholar]
- 30. Grandel H, Kaslin J, Ganz J, Wenzel I, Brand M (2006) Neural stem cells and neurogenesis in the adult zebrafish brain: origin, proliferation dynamics, migration and cell fate. Dev Biol 295: 263–277. [DOI] [PubMed] [Google Scholar]
- 31. Dong Z, Yang N, Yeo SY, Chitnis A, Guo S (2012) Intralineage directional notch signaling regulates self-renewal and differentiation of asymmetrically dividing radial glia. Neuron 74: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL (2011) Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11: 558–572. [DOI] [PubMed] [Google Scholar]
- 33. Englund C, Loren CE, Grabbe C, Varshney GK, Deleuil F, et al. (2003) Jeb signals through the Alk receptor tyrosine kinase to drive visceral muscle fusion. Nature 425: 512–516. [DOI] [PubMed] [Google Scholar]
- 34. Lee HH, Norris A, Weiss JB, Frasch M (2003) Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature 425: 507–512. [DOI] [PubMed] [Google Scholar]
- 35. Loren CE, Englund C, Grabbe C, Hallberg B, Hunter T, et al. (2003) A crucial role for the Anaplastic lymphoma kinase receptor tyrosine kinase in gut development in Drosophila melanogaster. EMBO Rep 4: 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loren CE, Scully A, Grabbe C, Edeen PT, Thomas J, et al. (2001) Identification and characterization of DAlk: a novel Drosophila melanogaster RTK which drives ERK activation in vivo. Genes Cells 6: 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robu ME, Larson JD, Nasevicius A, Beiraghi S, Brenner C, et al. (2007) p53 activation by knockdown technologies. PLoS Genet 3: e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langheinrich U, Hennen E, Stott G, Vacun G (2002) Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr Biol 12: 2023–2028. [DOI] [PubMed] [Google Scholar]
- 39. Kageyama R, Ohtsuka T, Kobayashi T (2008) Roles of Hes genes in neural development. Dev Growth Differ 50 Suppl 1S97–103. [DOI] [PubMed] [Google Scholar]
- 40. Pasini A, Henrique D, Wilkinson DG (2001) The zebrafish Hairy/Enhancer-of-split-related gene her6 is segmentally expressed during the early development of hindbrain and somites. Mech Dev 100: 317–321. [DOI] [PubMed] [Google Scholar]
- 41. Diez del Corral R, Storey KG (2001) Markers in vertebrate neurogenesis. Nat Rev Neurosci 2: 835–839. [DOI] [PubMed] [Google Scholar]
- 42. Bertrand N, Castro DS, Guillemot F (2002) Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3: 517–530. [DOI] [PubMed] [Google Scholar]
- 43. Kageyama R, Ohtsuka T, Hatakeyama J, Ohsawa R (2005) Roles of bHLH genes in neural stem cell differentiation. Exp Cell Res 306: 343–348. [DOI] [PubMed] [Google Scholar]
- 44. Lo L, Dormand E, Greenwood A, Anderson DJ (2002) Comparison of the generic neuronal differentiation and neuron subtype specification functions of mammalian achaete-scute and atonal homologs in cultured neural progenitor cells. Development 129: 1553–1567. [DOI] [PubMed] [Google Scholar]
- 45. Cheng M, Quail MR, Gingrich DE, Ott GR, Lu L, et al. (2012) CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol Cancer Ther 11: 670–679. [DOI] [PubMed] [Google Scholar]
- 46. Kim CH, Ueshima E, Muraoka O, Tanaka H, Yeo SY, et al. (1996) Zebrafish elav/HuC homologue as a very early neuronal marker. Neurosci Lett 216: 109–112. [DOI] [PubMed] [Google Scholar]
- 47. Park HC, Hong SK, Kim HS, Kim SH, Yoon EJ, et al. (2000) Structural comparison of zebrafish Elav/Hu and their differential expressions during neurogenesis. Neurosci Lett 279: 81–84. [DOI] [PubMed] [Google Scholar]
- 48. Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, et al. (1997) ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 14: 2175–2188. [DOI] [PubMed] [Google Scholar]
- 49. Roll JD, Reuther GW (2012) ALK-activating homologous mutations in LTK induce cellular transformation. PLoS One 7: e31733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225. [DOI] [PubMed] [Google Scholar]
- 51. Weiss A, Schlessinger J (1998) Switching signals on or off by receptor dimerization. Cell 94: 277–280. [DOI] [PubMed] [Google Scholar]
- 52. Perez-Pinera P, Zhang W, Chang Y, Vega JA, Deuel TF (2007) Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. J Biol Chem 282: 28683–28690. [DOI] [PubMed] [Google Scholar]
- 53. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, et al. (2012) The ALK(F1174L) Mutation Potentiates the Oncogenic Activity of MYCN in Neuroblastoma. Cancer Cell 22: 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhu S, Lee JS, Guo F, Shin J, Perez-Atayde AR, et al. (2012) Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell 21: 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Loeb-Hennard C, Kremmer E, Bally-Cuif L (2005) Prominent transcription of zebrafish N-myc (nmyc1) in tectal and retinal growth zones during embryonic and early larval development. Gene Expr Patterns 5: 341–347. [DOI] [PubMed] [Google Scholar]
- 56. Di Paolo D, Ambrogio C, Pastorino F, Brignole C, Martinengo C, et al. (2011) Selective therapeutic targeting of the anaplastic lymphoma kinase with liposomal siRNA induces apoptosis and inhibits angiogenesis in neuroblastoma. Mol Ther 19: 2201–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tomita K, Moriyoshi K, Nakanishi S, Guillemot F, Kageyama R (2000) Mammalian achaete-scute and atonal homologs regulate neuronal versus glial fate determination in the central nervous system. EMBO J 19: 5460–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pogoda HM, von der Hardt S, Herzog W, Kramer C, Schwarz H, et al. (2006) The proneural gene ascl1a is required for endocrine differentiation and cell survival in the zebrafish adenohypophysis. Development 133: 1079–1089. [DOI] [PubMed] [Google Scholar]
- 59. Yi SH, Jo AY, Park CH, Koh HC, Park RH, et al. (2008) Mash1 and neurogenin 2 enhance survival and differentiation of neural precursor cells after transplantation to rat brains via distinct modes of action. Mol Ther 16: 1873–1882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sense controls and expression of zebrafish alk at 72 hpf. (A–D) Negative controls at 24 hpf using a sense probe, corresponding to Fig. 1D–G, respectively. (E–G) Negative controls at 48 hpf using a sense probe, corresponding to Fig. 1I–K, respectively. (H) Lateral view of embryo at 72 hpf with dorsal up and anterior to the left. (I) Dorsal high magnification views of head region at 72 hpf. (J) Negative sense probe control of (I). Scale bars: A–G, 100 µm; H–J, 200 µm.
(JPG)
Heat-shock inducible transgenic zebrafish lines. (A,B) Diagrams illustrating transgenic cassettes for control line (A) and alk overexpression lines (B). The construct reported by Bajoghli et al. contains eight heat shock element repeats (HSE) with bidirectional promoter activity. This construct was engineered in such a way that alk and cfp were put on each side (B), or just cfp in the control line (A). (C–E) 24hpf embryos from the control line (HSE:cfp, C), alk overexpression line 1 (alk:HSE:cfp 1, D), and independent alk overexpression line 2 (alk:HSE:cfp 2, E). All embryos received a 1.5 hour long heat shock at 39.5°C and at 10.5 hpf. Bright field images on top and images of the same embryos in CFP channel below. Inserts show representative transgene positive embryos in each line. Arrows point to transgenic negative siblings in each line. They are morphologically normal and not visible in the CFP channel.
(JPG)
In situ hybridization to detect alk overexpression in alk :HSE: cfp transgenic embryos. A heat shock was performed at 24 hpf. All in situ hybridizations were done using an alk antisense RNA probe, at 1 hour post heat shock (hph) (A), 6 hph (B), 22 hph (C), and shortly after a second HS performed at 48 hpf (D). Inserts show representative transgenic positive embryos. As soon as 1 hour after heat shock, high levels of exogenous alk RNA could be detected in the entire embryo. Reduced staining at 6 hours after HS indicated RNA degradation. Staining almost completely disappeared at 22 hours after HS. A second HS re-activated transcription of overexpressed alk. Arrows points to transgene negative siblings where no exogenous alk was detected. Endogenous alk expression was not detected because of short staining time.
(JPG)
Proliferation and differentiation in the alk :HSE: cfp 2 line. (A,B) Confocal sections of 24hpf embryos of the alk:HSE:cfp 2 line. Sib (A) and Tg+ (B) had different neural tube shapes. Dividing cells (pH 3, green) and neurons (HuC/D, red) in Tg+ embryos (B) were both mispositioned (arrowhead), identical to defects observed in the alk:HSE:cfp 1 line (Fig. 2). Asterisk labels 4th ventricle. (C,C′,D,D′) Ectopic pERK (arrows) in Tg+ (D,D′) of the alk:HSE:cfp 2 line compared to Sib (C,C′), identical to defects observed in alk:HSE:cfp 1 (Fig. 3). pERK, phosphorylated-ERK1/2. Sib, transgene negative siblings. Tg+, transgenic embryos. Scale bars: 50 µm.
(JPG)
alk MO efficiency test experiments. (A) A schematic diagram showing arrangement of the first four alk exon-intron boundaries and MO binding sites. Numbers show exon/intron sizes in basepairs. (B) A construct containing egfp inserted in frame with the alk ATG site was generated to test the binding efficiency of ATG-MO. (C–F) Injection of this mRNA in combination with MOs resulted in different EGFP translation levels. Embryos at 8 hpf, with bright field images on top and fluorescent images below. Injection of the mRNA resulted in EGFP signal (D). Co-injection with alk ATG-MO blocked its translation (E). alk ATGmismatch-MO did not block its translation (F). Embryos labeled with asterisks in (C) and (E) were taken from the group shown in (D), indicating sufficient fluorescent excitation. (G) A diagram showing aberrant splice products with retention of intron 3, when alk pre-mRNA splicing is blocked by splicing MOs. (H) RT-PCRs were used to test splicing MO efficiency by primers indicated in (A). In Spl-MO injected 24 hpf embryos, the PCR product size was increased by approximately 300 bp (2nd lane) when compared to uninjected wild-type (1st lane), indicating retention of intron 3. -RT controls (3rd and 4th lanes, without reverse transcriptase in cDNA synthesis) excluded contamination by genomic DNA. gapdh was used as loading control.
(JPG)
Increased apoptosis in alk morphants. TUNEL staining of embryos at 22 hpf. Images show lateral views on top with anterior to the left, and dorsal views of head region below with anterior to the top. WT (A) or Std-MO injected (B) embryos had only few scattered TUNEL positive cells. In alk ATG-MO (C) or Spl-MO injected embryos (F), TUNEL positive cells increased in number, while ATGmismatch-MO injected (E) looked normal. Co-injections with p53-MO (D,G) failed to attenuate the apoptosis defect indicating specificity of the effect. Scale bars: 100 µm.
(JPG)
An alk ATG-MO leads to reduced proneural gene expression. (A,C,E,G,I,K) Embryos injected with alk ATGmismatch-MO showed no difference when compared to WT in Fig. 5. (B,D,F,H,J,L) alk ATG-MO injected embryos show the same phenotype as alk Spl-MO injected embryos (Fig. 5). Embryos showed no change in her6, neurog1 and gfap expression, but reduced expression of neurod4, ascl1b and dla identical to alk Spl-MO injected embryos shownin Fig. 5. Images show dorsal views of head regions with anterior to the left. Scale bars: 50 µm.
(JPG)