

Abstract
A series of non-natural infrared probes is reported that consist of a metal-tricarbonyl modified with a -(CH2)n- linker and cysteine-specific leaving group. They can be site-specifically attached to proteins using mutagenesis and similar protocols for EPR spin labels, which have the same leaving group. We characterize the label’s frequencies and lifetimes using 2D IR spectroscopy in solvents of varying dielectric. The frequency range spans 10 cm−1, and the variation in lifetimes ranges from 6 to 19 ps, indicating that these probes are very sensitive to their environments. Also, we attached probes with -(CH2)-, -(CH2)3-, -(CH2)4- linkers to ubiquitin at positions 6 and 63 and collected spectra in aqueous buffer. The frequencies and lifetimes were correlated for 3C and 4C linkers, as they were in the solvents, but did not correlate for the 1C linker. We concluded that lifetime measures solvation, whereas frequency reflects the electrostatics of the environment, which in the case of the 1C linker is a measure of the protein electrostatic field. We also labeled V71C α-synuclein in buffer and membrane-bound. Unlike most other infrared labels, this label has extremely-strong cross-sections and so can be measured with 2D IR spectroscopy at sub-millimolar concentrations. We expect that these labels will find use in studying the structure and dynamics of membrane-bound, aggregated, and kinetically-evolving proteins for which high signal-to-noise at low protein concentrations is imperative.
Keywords: infrared spectroscopy, 2D IR spectroscopy, vibrational dynamics, Stark effect, protein surface electrostatics, cysteine-selective protein labeling
INTRODUCTION
Vibrational modes are sensitive to their environment. Frequencies scale with the local electrostatic field, while the line widths and lifetimes depend on the electrostatic disorder of the surrounding environment.1–3 As such, vibrational modes are often utilized to report on solvation, electrostatics, and hydrogen bonding.4–8 However, proteins contain so many natural vibrational modes that spectra cannot be quantitatively interpreted without some type of spectroscopic probe to isolate the region of interest. There exist several strategies for obtaining structurally specific information. Isotope labeling a backbone carbonyl with 13C18O or a sidechain with CD isolates a single residue and is structurally non-perturbative.9–13 One can also label pieces or domains of proteins in order to obtain larger-scale structural information or monitor protein-protein contacts.14 Another approach is to utilize non-natural vibrational probes, such as nitriles, nitrophenyl, and thiocyanates.15–20 These probes are minimally perturbative, because they are small and they can be applied to large proteins because their triple-bond character shifts the frequency away from most protein, lipid, and water infrared absorbance.
While these probes are all sensitive to electrostatics and structure, in practice their use is limited by the synthetic methods in which they are incorporated into polypeptides or proteins and by their weak oscillator strengths. Isotope labels and non-natural probes can be straightforwardly integrated into polypeptides synthesized via commonly employed solid-phase techniques, which is typically applicable for polypeptides up to about 50 amino acids in length. For proteins, native chemical ligation can be used to semi-synthesize proteins from pieces in which one piece is a labeled polypeptide.21,22 Non-natural labels can be incorporated site-specifically into recombinantly-produced proteins utilizing genetic manipulations.15,18,19,23 Azido groups can be incorporated by via mutagenesis.24 One drawback, however, is that most of these probes have weak transition dipoles,5,25 which limits their usage to highly stable proteins that can be concentrated to several mM.5,17,19,23,26,27 Even worse, the weak oscillator strength leads to even poorer signal for higher order spectroscopies, such as 2D IR spectroscopy, where signal scales with the fourth power of the transition dipole.28
To improve the concentration requirement while retaining the ease with which protein mutations enable labeling, we designed a labeling scheme that utilizes the strong, isolated absorption of metal carbonyls and straightforward, selective attachment through cysteine residues (Scheme 1). 1a–c were synthesized by derivatizing CpRe(CO)3 with an alkylated methanethiosulfonate, –(CH2)nSSO2CH3, where n is 1, 3, or 4, hereafter referred to as ReL1, ReL3, ReL4, or generally as ReLx. To attach the label to the protein, the methanethiosulfonate, leaving group, common to EPR spectroscopists for nitroxide spin labeling, reacts exclusively with cysteine side chain of a protein.29–32 The labeling reaction robustly withstands the oxygen and water-rich environment of typical protein buffers. As a result, our label can be incorporated into proteins using standard site-specific mutagenesis protocols to generate a single cysteine residue.
Scheme 1.
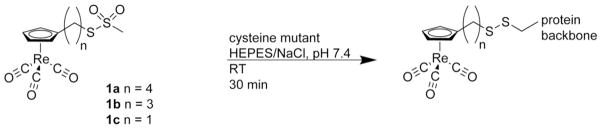
The carbonyl vibrational modes of CpRe(CO)3 absorb in a region of the infrared spectrum that is well resolved from native protein vibrational transitions and water absorbance with an extinction coefficient of ~4,100 M−1cm−1.30 For comparison, the nitrile group, which is the most commonly used non-natural vibrational probe, has an extinction coefficient of 22–240 M−1cm−1.33 Due to the scaling difference for FTIR versus 2D IR signal, obtaining comparable signal-to-noise for CpLx versus a nitrile requires ~40 times lower concentrations for FTIR and ~1600 times lower concentration for 2D IR spectra. In the data reported here, the protein concentration is <1 mM, which is much lower than the concentrations typically used for nitriles.5,17 The signal-to-noise of our measurements demonstrate that protein concentrations <100 μM can be studied with this label. Of course, the metal carbonyl labels are larger and structurally more perturbative, but as we show below, will be good probes for protein sites in analogy to spin labels used for EPR spectroscopy.
Previous methods were developed for labeling proteins with metal carbonyls for infrared spectroscopy, radio-pharmacology, and monitoring electron-transfer.34–36 Labels possessing residue-specific functional groups, such as N-hydroxysuccinimide esters, imidoesters, and Fischer-type metallocarbene complexes that attach to primary amines like those on lysine residues.34 However, those compounds are not generally useful as site-specific probes because most soluble proteins contain several lysine residues as well as an amine at the N-terminus. Thus, eliminating all but one lysine to serve as the label site is likely to alter the protein function. Another possible functional group is the maleimide group, which, at neutral pH and room temperature, is cysteine selective.31,37 In fact, CpRe(CO)3 has already been attached to proteins via a maleimide linker.37 However, the maleimide group is bulky with a five-member ring in the linker. Our linker, a methanethiosulfonate derivate, is an analog of a commonly used molecule for site-specific spin labeling in EPR spectroscopy and therefore well characterized.29,32 It is cysteine-specific at neutral pH and room temperature29,32 and is preferable to maleimide, because it results in a flexible, straight-chain linker. Using –(CH2)nSSO2CH3 produces near quantitative reactivity toward cysteine residues under mild conditions. Another method targets histidine-binding to attach the labels.35,36,38–40 Both the histidine targeted here and the cysteine in the label we present are relatively infrequent in proteins, but if present, histidines often play important structural roles and are sometimes charged in proteins, so the elimination of naturally occurring histidines is often detrimental. Moreover, the size of the histidine label and the flexibility of the linker are comparable to our compound with a single –(CH2)–, e.g. ReL1, and histidine labels bind via coordinate bonds whereas our cysteine labels bond covalently. The cysteine label also offers broader applicability to aggregating proteins, since the conditions are milder and metal ion reagents, which are necessary in the histidine labeling scheme and typically accelerate amyloid aggregation, are not involved in the cysteine approach. While these facts must be considered when choosing an appropriate label, metal-carbonyls attached to histidine provided interesting insights into the solvation of hen egg white lysozyme and were used to monitor charge transfer through a protein and to label β-amyloid peptide 1–28.40–42
In this paper, we demonstrate our approach by showing how CpLx is sensitive to protein environments. We first show frequency and lifetime sensitivity of CpLx in solvents of varying polarity. We then utilize the frequency and lifetime sensitivity to examine the hydration and electrostatic surface of ubiquitin. We measure these quantities on ubiquitin for all three lengths of linker, which enables us to vary the extent to which the probe is tethered to the protein surface. Finally, the universal usage of CpLx is demonstrated by studying solution-phase and vesicle-bound CpL4-labeled α-synuclein, a synaptic protein that is natively disordered in solution but adopts an α-helical conformation when associated with a phospholipid bilayer. By examining the labels vibrational dynamics in various protein environments, we begin to illustrate some aspects of the label’s information content. Because most metal-carbonyl molecules are insoluble in water, the full extent to which their frequency, line shapes, or lifetimes are sensitive to environment is unknown. However, inhomogeneous line shapes have been measured in the 2D IR spectra of Ni2(PPh3)2(CO)2 in THF–hexane (1:3),43 and the lifetime of a protein-bound metal carbonyl has recently been shown to be quite sensitive to solvent polarity.40 In a similar manner, we report here that the rhenium carbonyl stretch modes are indeed good reporters of protein environment.
EXPERIMENTAL SECTION
Label Synthesis
Detailed information is provided in the SI. In brief, Re2(CO)10 is oxidized with cyclopentadiene to give CpRe(CO)3 (2).30 A hydrogen atom on the cyclopentadienyl ligand is substituted by activation with n-BuLi in THF at −78°C. For ReL1-label, hydroxymethylation is facilitated via paraformaldehyde. After purification, the hydroxyl group is replaced with an iodide by way of a TMS intermediate to give [η5-(C5H4)CH2I]Re(CO)3 (7). For the ReL3- and ReL4-labels, the route to the iodide is direct from the lithium-activated Cp-ring with diiodopropane or diiodobutane to [η5-(C5H4)(CH2)3I]Re(CO)3 (5) or [η5-(C5H4)(CH2)4I]Re(CO)3 (4), respectively.44 For all iodide products, [η5-(C5H4)(CH2)nI]Re(CO)3 where n=1,3 or 4, the iodide is replaced using NaSSO2CH3 in DMF to give [η5-(C5H4)(CH2)nSSO2CH3]Re(CO)3 where n=1,3 or 4 (1a–c). The product was purified and DMF removed by diluting in water and extracting into ethyl acetate, which was subsequently washed with water and dried to give CpLx.
Protein Synthesis and Purification
The ubiquitin mutants K6C and K63C were provided by the Streiter Research Group in the UW-Madison Chemistry Department. The recombinant V71C α-synuclein mutant was expressed and purified as follows. A codon-optimized gene for wild-type human α-synuclein was purchased from Genscript (Piscataway, NJ) and subcloned into a modified pET32a vector (Novagen) containing a Factor Xa site for His6-tag removal, using Bam HI and Eco RI restriction sites. The V71C mutant was generated by linearization of the plasmid via PCR with 5′-phosphorylated mutagenic primers followed by blunt-ended ligation using T4 ligase under standard reaction conditions. DNA sequencing was used to confirm the presence of the V71C mutant. Protein expression and purification were performed as described previously,45 using a modified cleavage buffer for the Factor Xa reaction (20 mM Tris, 50 mM NaCl, 0.1 M CaCl2, pH 7.5). Prior to reverse phase chromatography purification the protein was incubated in 0.1 mM DL-dithiotheitol at 95°C for 5 minutes to reduce any disulfide bonds. MALDI mass [M+H]+ meas. 14,463.0 m/z (calc. 14,465.1 m/z).
Protein Labeling
Five-fold excess of each label was reacted with 500 μg aliquot of each protein in 1 mL of 10 mM HEPES, 100 mM NaCl buffer (pH 7.4) buffer. The solution was stirred for 1 hour. Labeled protein was purified by size-exclusion chromatography on an HPLC by flowing the reaction solution over a Tricorn 10/600 column packed with Sephadex G-75 gel filtration medium with 10 mM NH4CO3 buffer (pH 7). Elutants containing the labeled protein were lyophilized and characterized by MALDI (see SI).
Sample Preparation for Infrared Measurements
For the non-lipid-containing samples, 65 μg (4.3 nmol) of protein was resuspended in 20 mM deuterated-phosphate buffer (pH 7.4, 5 μL) at 500 μM concentrations. 3:1 POPC/POPS lipid vesicles were prepared by evaporating under high vacuum, 27 μL POPC and 12 μL POPS (in chloroform, 25 mg/mL). The resulting film was resuspended in 20 mM deuterated phosphate buffer (pH 7.4, 5 μL) and sonicated for 30 minutes. The lipid solution was added to the 65 μg (4.3 nmol) of protein and stirred gently. All samples were measured between two 56 micron-spaced CaF2 windows.
FTIR and 2D IR Spectroscopy
Femtosecond mid-IR pulses (~1965 cm−1 center frequency, 185 cm−1 bandwidth) were generated as follows. The 800 nm output of a 1 kHz Ti:sapphire KMLabs Wyvern regenerative amplifier (1.3 mJ, 45 fs transform-limited pulses) is used to pump a home-built optical parametric amplifier (OPA). Here, 800 nm pulses are downconverted into signal and idler pulses (180 μJ) in a type II BBO crystal (θ = 28.0°, 2 mm thick) in two stages with collinear alignment. Mid-IR pulses with FWHM bandwidths of approximately 185 cm−1 at 5 μm and pulse energies of >5 μJ are then generated by difference frequency mixing of the signal and idler pulses in a type II AgGaS2 crystal (θ = 45.4°, 1 mm thick). The Mid-IR light is then split into pump (97%) and probe (3%) beams, and the pump beam is sent through a pulse shaper to create pulse pairs as described previously.46 The pulse pair was scanned with time delays up to 4 ps in steps of 40 fs. The waiting time was scanned by a calibrated motorized translational stage that incremented in steps of 200–1000 fs up to 50 ps. The probe pulse was detected with a 64 channel MCT array after passing through a spectrometer. The frequency resolution along the pump and probe axis are 4 cm−1 and 3 cm−1, respectively. The pump and probe polarizations were set parallel.
RESULTS AND DISCUSSION
Synthesis of CpLx
Synthetic routes to CpLx are shown in Scheme 2 and occur by way of iodo-intermediates. The iodo-intermediate, 5, was synthesized as previously described.44 Similarly, 4 was produced after activation with n-BuLi at −78°C in THF and subsequent addition of diiodobutane in small excess. The reaction finished at room temperature and was purified as previously described for 5. The 1H and 13C NMR peak shifts and multiplicities agreed with the literature report for 5.44 1H and 13C NMR spectra for 4, not previously reported, agreed with the expected result based on NMR theory. For 7, the CpL1 iodo-intermediate, 2 was alkylated as previously described using paraformaldehyde to produce 6.47 After purifying over an ether flash column, 1H and 13C NMR peaks agreed with those reported in the literature.47 The hydroxyl group in 6 was replaced with iodide by way of a TMS intermediate after addition of NaI and TMSCl in ACN, which resulted in 7, as similarly performed by Olah et al.48 See SI for NMR and EI mass spectra.
Scheme 2.
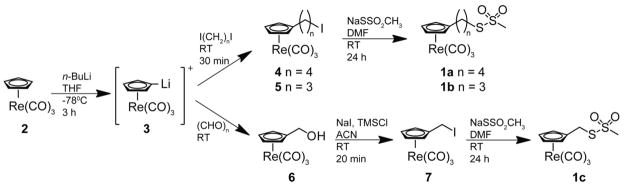
The iodide in 4, 5, and 7 is replaced by adding five molar excess of sodium methanethiosulfonate and stirring in DMF overnight. The product was purified by aqueous extraction. Evidence for CpLx appears in 1H and 13C NMR spectra as the methyl peak appears at ~3.25 ppm and ~51.1 ppm, respectively. Assignment of the methyl peak is supported by HSQC experiments (see SI). The proper mass for compounds CpLx corresponded to peaks observed in EI spectra.
Reaction of CpLx with Mutant Proteins
Prior to labeling reactions with each mutant, DL-dithiothreitol (DTT) reduced the thiol groups to maximize labeling efficiency. Label 1a was dissolved in acetone, and ~5 μL of this stock was added to ~1 mL of the protein in HEPES buffer (pH 7.4). Vials were mixed gently for 30 minutes at room temperature, after which the unreacted label was separated from the protein with size-exclusion chromatography. Observed masses of each labeled protein corresponded to the predicted result. Assuming that the neutrally charged label does not affect ionization, labeling efficiency is ascertained as quantitative from MALDI spectra.
Solvent-Dependent Lifetimes and Frequencies
The 2D IR spectra of ReL1 in DMSO and attached to K63C ubiquitin is shown in Fig. 1 for a waiting time of 0 fs (see SI for spectra of all labels in each solvent and at each mutant position). The diagonal and cross peaks in the spectrum are elongated along the diagonal, thereby creating nodal slopes (dashed lines) that are tilted. The slope of the node between the overtone and fundamental diagonal peak is a measure of the inhomogeneity of the vibrational mode,49 whereas the slope of the cross peak node also depends on the correlation in their vibrational frequency fluctuations.50 In solvents, the nodal slopes are more vertical than when the labels are attached to the protein in buffer, indicating that the modes are more homogeneous (Table 1). Thus, the carbonyl stretch modes are sensitive to solvent polarity and dynamics, similar to the amide I carbonyl stretches of protein backbones that are often used to study protein structure.28 As has been done for the amide I modes, it should be possible to develop a correlation between frequency and solvent electrostatic field,51–53 which would enable a quantitative measure of solvent polarity. In this report we do not dwell on these features, but instead focus on frequencies and lifetimes instead.
Figure 1.

2D IR spectra of the ReL1 in DMSO (a) and covalently bound to the K63C ubiquitin mutant (b).
Table 1.
The frequency, fundamental lifetime, overtone lifetime, and node slope for the symmetric stretch of CpLx in either solvent or after reaction with the specified mutant.
| ω/cm−1 (±0.5) | T01/ps | T12/ps | Node Slope/ωpump÷ωprobe | ||
|---|---|---|---|---|---|
| DMSO | 1C | 2014.0 | 5.55 ± 0.05 | 6.14 ± 0.04 | 2.50 ± 0.03 |
| 3C | 2015.6 | 10.1 ± 0.2 | 9.97 ± 0.13 | 3.40 ± 0.23 | |
| 4C | 2014.2 | 7.91 ± 0.10 | 8.35 ± 0.08 | 4.37 ± 0.09 | |
| CH2Cl2/CCl4 | 1C | 2024.1 | 18.1 ± 0.4 | 16.1 ± 0.4 | 1.98 ± 0.07 |
| 3C | 2024.5 | 19.0 ± 0.6 | 18.9 ± 0.5 | 1.95 ± 0.10 | |
| 4C | 2022.4 | 14.2 ± 0.3 | 14.2 ± 0.2 | 5.13 ± 0.11 | |
| Ubiquitin K6C | 1C | 2022.2 | 9.13 ± 0.17 | 8.47 ± 0.19 | 1.63 ± 0.29 |
| 3C | 2019.4 | 12.8 ± 0.2 | 12.4 ± 0.1 | 1.77 ± 0.04 | |
| 4C | 2015.4 | 5.75 ± 0.12 | 4.67 ± 0.11 | 1.56 ± 0.42 | |
| Ubiquitin K63C | 1C | 2020.8 | 6.97 ± 0.13 | 5.80 ± 0.10 | 1.86 ± 0.21 |
| 3C | 2017.0 | 6.56 ± 0.19 | 5.67 ± 0.10 | 1.73 ± 0.19 | |
| 4C | 2020.4 | 19.7 ± 1.1 | 14.0 ± 0.5 | 1.86 ± 0.10 | |
| α-synuclein V71C buffer (4C Label) | 2020.7 | 11.1 ± 0.7 | 6.74 ± 1.65 | 3.75 ± 0.21 | |
| α-synuclein V71C vesicles (4C Label) | 2019.2 | 14.2 ± 3.1 | 9.09 ± 2.11 | 3.78 ± 0.11 | |
Shown in Fig. 2a is the peak associated with the symmetric stretch mode as illustrated by a slice through the diagonal (ωpump=ωprobe) of the 2D IR spectra for ReL1 in DMSO and 1:1 CH2Cl2/CCl4. There is a 12 cm−1 frequency difference between these two solvents, which is as large as the FWHM of the peaks themselves. The 1:1 CH2Cl2/CCl4 mixture is used here, because the labels are not soluble in CCl4 alone. The other compounds show the same solvent frequency dependence (Table 1. Spectra reported in Fig. S1). Within the same solvent, the frequency varies ~2 cm−1, which we attribute to differences in the degree to which the leaving group interacts with the carbonyl groups. From these results, we conclude that the symmetric stretch is expected to have a frequency of about 2015 cm−1 in polar environments and 2024 cm−1 in non-polar environments.
Figure 2.

Solvent dependence of the labels. (a) Slices through the 2D IR diagonal (ωpump=ωprobe) and (b) measured lifetimes of each label in either DMSO or 1:1 CHCl2/CCl4.
The lifetime of the ReLx symmetric stretch correlates with its frequency. In the 1:1 CH2Cl2/CCl4 mixture, the ReL1 lifetime is 16.1 ps, which is similar to other metal carbonyl compounds in non-polar solvents.54 In DMSO, the ReL1 lifetime is 6.14 ps. To our knowledge, the lifetime of very few metal carbonyl compounds are known in polar solvents. All the ReLx compounds exhibited lifetimes <10 ps in DMSO and > 14 ps in 1:1 CH2Cl2/CCl4 (Table 1). The lifetime of the v=0→1 and v=1→2 transition can be measured from the intensities of the diagonal absorption and the off-diagonal overtone, respectively. In what follows, we discuss the overtone lifetimes because there is no overlap with sample background scatter, when present, and thus is generally more accurate especially in heterogeneous samples like those involving membranes. The apparent lifetimes and frequency of the antisymmetric stretch absorption band also depends on solvent, but since there are two antisymmetric stretches that are not resolvable, we do not pursue it more here. Thus, we learn from these measurements that there is a very large dynamic range in both the lifetime and the frequency of the symmetric stretch mode. The strong correlation between lifetime and frequency is evident in Fig. 3, which plots both in the same graph.
Figure 3.

Lifetime and frequency for the labels in (a) solvent and (b) covalently linked to two mutants of ubiquitin.
Lifetimes and Frequencies of Labeled Ubiquitin
Having identified an empirical relationship between frequency and lifetimes, we attached them to two different locations on ubiquitin. We attached each of the three labels to each of the two sites on ubiquitin, resulting in a total of six samples. Representative 2D IR spectra are shown in Fig. S1. From these measurements, we extracted the symmetric stretch frequency and the lifetime, which are plotted in Fig. 3. The six frequencies span from 2015.4 to 2022.2 cm−1. On average, they lie closer to the frequency observed for ReLx in DMSO than in the CH2Cl2/CCl4 mixture (Fig. 3). The lifetimes span from 4.67 to 14.0 ps, which is also more similar to the lifetimes of ReLx in DMSO than CH2Cl2/CCl4. These averages are consistent with the protein being solvated in water. We also note that the correlation between frequency and lifetime is retained for linkers ReL3 and ReL4, but not for ReL1. For ReL3 and ReL4, whether the label is attached to K6C or to K63C, a high frequency is accompanied by a long lifetime, as was observed for the model compounds (Fig. 3). However, for ReL1, a high frequency is accompanied by a short lifetime.
We interpret this data with regards to the hydration of the probe and the electrostatic field of the protein. Vibrational frequencies correlate with the surrounding field strength due to the Stark effect.55,56 The Stark effect is utilized in small vibrational probes, like nitriles, to measure the internal and external electric fields of proteins.55,57 The same principle holds true for more complicated vibrations like the amide modes of protein backbones. In contrast, vibrational lifetimes are often dictated by time-dependent fluctuations in the frequency more so than the frequencies themselves. Thus, in general, vibrational frequencies reflect the average electrostatic field of their surroundings while the lifetime is a measure of the rate and magnitude of environment induced frequency fluctuations.58 Of course, solvent resonances can also influence the lifetimes, such as been observed for histidine coordinated metal compounds.40
In DMSO and CH2Cl2/CCl4, the above description of frequencies and lifetimes holds for CpLx. DMSO has, on average, stronger electrostatic fields and larger frequency fluctuations then does the non-polar solvent mixture CH2Cl2/CCl4 (the magnitude of the frequency fluctuations is observed in the broader FWHM of the symmetric stretch in DMSO than CH2Cl2/CCl4 in Fig. 2). Thus, the two are correlated in simple solvents that provide a homogeneous environment around the label. From the protein data, it appears that ReL4 at K6C and ReL3 at K63C sample a homogeneous environment that is similar to water, since both the lifetime and frequency have values close to the label in liquid water. Likewise, the frequency and lifetime of ReL3 at K6C and ReL4 at K63C are correlated, which we interpret to mean that these two labels are also sampling a homogeneous environment, although the environment is less polar than water since both the frequency and lifetime assume intermediate values as in the water and CH2Cl2/CCl4 model studies. In contrast to the ReL3 and ReL4 labels, the ReL1 labels do not follow the same trend. At both positions on ubiquitin, the lifetime is short and the frequency is high, which suggests that the ReL1 label is experiencing rather weak electrostatic fields but very dynamic field fluctuations, which is unlike what is observed in liquids. From these observations, we conclude that the labels held by ReL1 linkers are so short that their frequencies are dominated by the electrostatics of the protein surface, but that the solvent to which they are exposed is still sufficient to cause fast vibrational lifetimes. In contrast, the ReL3 and ReL4 labels are long enough that they can more broadly sample the homogeneous solvent environment.
Shown in Fig. 4 is a map of the ubiquitin surface hydrophobicity in each of the two regions to which our labels are attached. Drawn on this map is one possible conformation that each label might adopt, with a circle drawn to illustrate the extent to which the label could sample the protein surface if the linker group is fully extended. K6C lies in a pocket while K63C is located on the edge of the protein. These conformations are not energy minimized and ignore steric overlaps. Thus, the region that the label samples is most likely smaller, but they permit us to evaluate the outer limit of the possible conformations. For example, the metal-carbonyl headgroup cannot extend further than 7.1 Å from the protein surface when attached at K6C and no further than 8.8 Å at position K63C. Thus, it is likely that labels with the ReL1 linker will feel the protein electrostatic field, as deduced from the experiments above. The ReL3 and ReL4 linkers allow the headgroup to sample a much wider range of the protein surface. Presumably, the label will preferentially cluster near hydrophobic regions of the protein (shown in red), since the label is hydrophobic. The carbonyl groups on metal-carbonyl compounds do not form hydrogen bonds. Thus, the carbonyl groups could face towards the protein to be protected from the solvent. However, there may be stabilizing forces between the cyclopentadienyl ring and the phenyl rings of side chains, like phenylalanine, that prefer the carbonyl groups to face towards the solvent. Thus, samples in which frequency and lifetime do not correlate may be an indication of preferential surface orientation.
Figure 4.

Hydrophobicity of the ubiquitin surface. (a) Side view of the two labeled positions of ubiquitin, K6C and K63C with ReL1 label attached. Schematics of distances accessible to ReL1, ReL3, and ReL4 labels attached to (b) K6C and (c) K63C with circles drawn about the cysteine β-carbon. Hydrophobic and hydrophilic residues are shown red and blue, respectively, according to the Kyte-Doolittle hydrophobicity scale, as described in the SI.
2D IR Spectroscopy of α-Synuclein
We collected high-resolution 2D IR spectra for CpL4-labeled V71C α-synuclein in buffer solution in the presence and absence of lipid vesicles (Fig. 5). In solution, α-synuclein is intrinsically disordered and becomes α-helical in the presence of lipid vesicles,32 which is confirmed by a narrowing and red-shift of the amide I absorption band (not shown). Spectra of the label on α-synuclein possess similar features to that of ubiquitin. The symmetric stretch frequencies of the labels attached to α-synuclein are within 2 cm−1 regardless of the presence of the lipids. Similar to the ubiquitin spectra, elongated line shapes are observed in the diagonal peaks indicating inhomogeneity. As commonly observed with broadened line shapes, the node slopes were higher than in the homogeneous limit (Table 1). The 2D line shapes indicate that in solution CpL4 is experiencing a strong electrostatic environment that is slowly evolving on the few picoseconds timescale.
Figure 5.

2D IR spectra of α-synuclein labeled with CpL4 in (a) buffer and (b) with lipids.
Conclusions about dynamics from the line shapes agree with results from the lifetime measurements. Plotted decay of 2D IR intensity as the waiting time is scanned show that lifetime of CpL4-labeled V71C α-synuclein on lipid vesicles is slightly longer than in the absence of lipids (Fig. 7, Fit results are listed in Table 1). As discussed for the solvent/ubiquitin data, vibrational relaxation is faster for the CpL4 exposed to buffer at 6.74 ps compared to 9.09 ps in lipids. The close proximity of CpL4’s frequency and lifetime indicates that the V71C position encounters a similar environment when α-synuclein is bound to the lipid vesicles. The vesicle-bound protein vibrational relaxation is 3 ps slower indicating that the label is experiencing lower electrostatic microenvironments or solvation when bound to the lipids. According to the wheel diagram in Fig. 6, V71C resides at the membrane interface, and previous experiments put the depth at 3.9 Å,32 which lies at the bilayer/buffer interfacial region. Thus, the data is consistent with V71C retaining a slightly lower level of hydration in the buffer and lipids. However, we do not know the propensity for CpLx to solvate in water versus the membrane. The carbonyl groups in CpRe(CO)3 may extend away from V71C according to the length of the flexible linker and the hydrophobicity of the probe and thus may prefer to snorkel into the membrane headgroups or perhaps into the lipid tails. The measured lifetimes and frequencies appear consistent with the label located in the headgroup region, in which it would only be partially solvated.
Figure 7.

Lifetime of the (a) fundamental and (b) overtone features of the symmetric stretch in the 2D IR spectra of CpL4-labeled α-synuclein in buffer and with vesicles. Fits are single exponentials with lifetimes given in Table 1.
Figure 6.
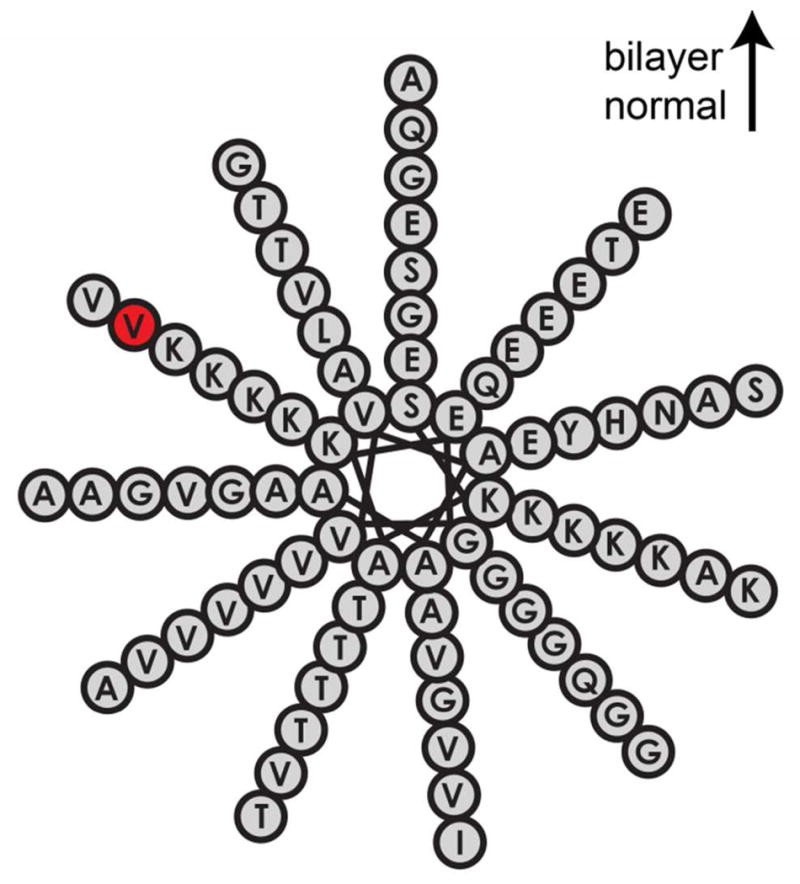
The helical wheel diagram for α-synuclein shows the relative orientation of the protein to the lipid bilayer with the labeled residue (V71, red).
One such experimental example of environmental conditions are vesicle samples, which give rise to large amounts of background scatter. Yet, in this paper, we present high signal-to-noise spectra, illustrating the utility of these samples to be used on inhomogeneous systems. Although not reported here, 2D IR spectroscopy can be implemented in a time-resolved fashion to probe dynamics on picoseconds to milliseconds to seconds timescale.4,5,12,20,27,28,38 We believe that these compounds provide a new method for protein structure, dynamics, and environment on systems such as membrane bound and aggregating proteins.
Conformational Sampling and Vibrational Heterogeneity of the Probes
A central question regarding these probes are the conformational sampling allowed by the linkers and the solvation preference. The probes themselves are hydrophobic, which is why they do not dissolve in water unless attached to a soluble molecule like a protein. As a result, the metal carbonyl probably associates with the most hydrophobic region assessable by the length of its linker. Possible regions include a hydrophobic pocked on the surface of the protein or the hydrophobic interior of a membrane bilayer. A similar situation is encountered in EPR probes, which are also hydrophobic, and their structural distributions and propensities have been characterized over the past decade.59,60 Even our shortest label, ReL1 that has a single –(CH2)–, still has conformational freedom, similar to the conformational freedom of the histidine labeling method. Of course, one could crystallize the protein with the linker to definitively ascertain its conformation,40 but we envision using these labels in situations that cannot be crystallized, like kinetically aggregating proteins. In those cases, the ability to change the linker length will help characterize the type of environment in which the probe resides. For example, if a label is snorkeling into the membrane, then shortening the linker length would alter the lifetimes and frequencies if the label is drawn into the solvent. Thus, future studies will characterize the solvation propensity of these probes under controlled situations, as has been done for EPR labels.61
Another interesting observable, which has not been discussed here, are the 2D linewidths of the metal carbonyl modes. 2D linewidths provide information about solvation and structural heterogeneities. For example, they have been used to obtain the secondary structure and depth of a membrane bound antibiotic and spectral diffusion times have identified water in amyloid fibers. Similarly, it is clear from 2D IR work on the hen egg white lysozyme that metal carbonyls line shapes are also sensitive to hydration dynamics and structural conformational distributions. Thus, waiting time experiments that probe the vibrational dynamics should also help reveal the binding site of the probes and identify their environment. One distinct difference between EPR and FTIR or 2D IR spectroscopies, is that infrared spectra can be calculated from molecular dynamics simulations of only a few microseconds.53 Thus, by generating a realistic distribution of conformations, understanding the microscopic origins of the 2D IR spectra reported here can be accomplished by comparison of simulations.12,62–65
CONCLUSION
We have observed an empirical relationship between lifetime and frequency as related to solvation and electrostatics for this novel set of infrared probes that we synthesized for simple covalent attachment to recombinantly-produced proteins. As illustrated in these experiments, these probes reflect the microscopic structure and environment of the protein surface. Additional insights into the conformational propensities of these labels will come from additional site mutations as well as molecular dynamics simulations. A useful feature of infrared spectroscopy is that correlations between electrostatic fields and 2D IR spectra can be obtained, which can also be used to calculate lifetimes. The theory will have to account for the normal mode behavior of these compounds, since all three carbonyl stretches contribute to the observables. Nonetheless, it will be possible to quantitatively link the experimental observables to the atomic level features of the protein and its solvent. Steps along these lines are already underway for other metal carbonyl groups.66 The utility of the labels presented here, is that they can be applied to site-specifically to a wide range of proteins at very low concentrations. Application to V71C mutated α-synuclein demonstrates that high signal-to-noise spectra can be obtained even at low sample concentrations and in membrane environments that cause background scatter. We anticipate that they will be especially useful in studies of membrane bound proteins and kinetically evolving proteins such as occurs during amyloid fiber formation.
In future studies, we will characterize the hydrophobicity of this label and its effect on protein structure by varying the mutation site on the protein and the length of the linker. The ability to choose a variety of flexible linkers will foster these studies, since long linkers will allow the probe to reside in environments favorable to its hydrophobicity while short linkers will force it to lie close to the protein surface. The probes reported here are larger than other non-natural vibrational probes, but comparable in size to EPR spin probes. Like EPR probes, they will be useful in characterizing protein structure or monitoring dynamics through residues on the outsides of proteins. Long linker lengths may make the labels less perturbative by allowing the headgroup to reside on the protein surface. Due to their high extinction coefficients, samples at <100 μM concentrations can be studied, which is much lower than possible for existing non-natural vibrational probes and thus applicable to a much broader range of proteins. Although not reported here, 2D IR spectroscopy can be implemented in a time-resolved fashion to probe dynamics on picoseconds to milliseconds to seconds timescale.67–70 We believe that these compounds provide a new method for protein structure, dynamics, and environment that will be especially useful for studying membrane bound and aggregating proteins.
Supplementary Material
Acknowledgments
We thank Chuck Casey, Alex Clemens, Steve Burke, Charlie Fry, and Tehshik Yoon. For the ubiquitin mutants, we are grateful to Eric Streiter and Ellen Valkevich for the gift of the two cytochrome c mutants. The National Science Foundation (NSF) supported the majority of this research through a Collaborative Research in Chemistry (CRC) Grant 0832584, with additional support by the National Institutes of Health (NIH) through the Molecular Biophysics Training Grant T32 GM08293 and the NIDDK via DK79895. Within the UW-Madison Chemistry Department, we used the NMR facility, supported by grants NIH 1 S10 RR13866-01, NSF CHE-9629688, and NSF CHE-9208463, and the mass spectrometry facility, supported by NSF CHE-9304546. This study also made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA. The protein graphics were generated with UCSF Chimera software, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
ABBREVIATIONS
- ReLx
, [η5-(C5H4)(CH2)nSSO2CH3]Re(CO)3 where n, 1, 3, or 4
- ReL1
[η5-(C5H4)(CH2)1SSO2CH3]Re(CO)3
- ReL3
[η5-(C5H4)(CH2)3SSO2CH3]Re(CO)3
- ReL4
[η5-(C5H4)(CH2)4SSO2CH3]Re(CO)3
Footnotes
Supporting Information. Experimental details, synthetic characterization including 1H, 13C, and HSQC NMR spectra, FTIR spectra and MALDI spectra, 2D IR spectra, and 2D IR waiting time intensities. This information is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Berne BJ, Pecora R. Dynamic Light Scattering: with Applications to Chemistry, Biology, and Physics. Wiley-Interscience; 1975. [Google Scholar]
- 2.Kubo R. Adv Chem Phys. John Wiley & Sons, Inc; A Stochastic Theory of Line Shape. [Google Scholar]
- 3.Hamm P, Zanni M. Concepts and Methods of 2d Infrared Spectroscopy. Cambridge University Press; 2011. [Google Scholar]
- 4.DeCamp MF, DeFlores L, McCracken JM, Tokmakoff A, Kwac K, Cho M. The Journal of Physical Chemistry B. 2005;109:11016–11026. doi: 10.1021/jp050257p. [DOI] [PubMed] [Google Scholar]
- 5.Waegele MM, Culik RM, Gai F. J Phys Chem Lett. 2011;2:2598–2609. doi: 10.1021/jz201161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee H, Lee G, Jeon J, Cho M. J Phys Chem A. 2011;116:347–357. doi: 10.1021/jp209709e. [DOI] [PubMed] [Google Scholar]
- 7.King JT, Ross MR, Kubarych KJ. J Phys Chem B. 2012;116:3754–3759. doi: 10.1021/jp2125747. [DOI] [PubMed] [Google Scholar]
- 8.Greve C, Preketes NK, Fidder H, Costard R, Koeppe B, Heisler IA, Mukamel S, Temps F, Nibbering ETJ, Elsaesser T. J Phys Chem A. 2013;117:594–606. doi: 10.1021/jp310177e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naraharisetty SRG, Kurochkin DV, Rubtsov IV. Chem Phys Lett. 2007;437:262–266. [Google Scholar]
- 10.Rubtsov IV. Acc Chem Res. 2009;42:1385–1394. doi: 10.1021/ar900008p. [DOI] [PubMed] [Google Scholar]
- 11.Schade M, Moretto A, Crisma M, Toniolo C, Hamm P. J Phys Chem B. 2009;113:13393–13397. doi: 10.1021/jp906363a. [DOI] [PubMed] [Google Scholar]
- 12.Woys AM, Lin YS, Reddy AS, Xiong W, de PJJ, Skinner JL, Zanni MT. J Am Chem Soc. 2010;132:2832–2838. doi: 10.1021/ja9101776. [DOI] [PubMed] [Google Scholar]
- 13.Nydegger MW, Rock W, Cheatum CM. PCCP. 2011;13:6098–6104. doi: 10.1039/c0cp01087a. [DOI] [PubMed] [Google Scholar]
- 14.Moran SD, Woys AM, Buchanan LE, Bixby E, Decatur SM, Zanni MT. Proc Natl Acad Sci USA. 2012;109:3329–3334. S3329/3321–S3329/3324. doi: 10.1073/pnas.1117704109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fafarman AT, Webb LJ, Chuang JI, Boxer SG. J Am Chem Soc. 2006;128:13356–13357. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marek P, Mukherjee S, Zanni MT, Raleigh DP. J Mol Biol. 2010;400:878–888. doi: 10.1016/j.jmb.2010.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urbanek DC, Vorobyev DY, Serrano AL, Gai F, Hochstrasser RM. J Phys Chem Lett. 2010;1:3311–3315. doi: 10.1021/jz101367d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith EE, Linderman BY, Luskin AC, Brewer SH. J Phys Chem B. 2011;115:2380–2385. doi: 10.1021/jp109288j. [DOI] [PubMed] [Google Scholar]
- 19.Alfieri KN, Vienneau AR, Londergan CH. Biochemistry. 2011;50:11097–11108. doi: 10.1021/bi200903p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutta S, Li YL, Rock W, Houtman JCD, Kohen A, Cheatum CM. The Journal of Physical Chemistry B. 2011;116:542–548. doi: 10.1021/jp208677u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dawson P, Muir T, Clark-Lewis I, Kent S. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 22.Flavell RR, Muir TW. Acc Chem Res. 2008;42:107–116. doi: 10.1021/ar800129c. [DOI] [PubMed] [Google Scholar]
- 23.Thielges MC, Axup JY, Wong D, Lee HS, Chung JK, Schultz PG, Fayer MD. J Phys Chem B. 2011;115:11294–11304. doi: 10.1021/jp206986v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagarajan S, Taskent-Sezgin H, Parul D, Carrico I, Raleigh DP, Dyer RB. J Am Chem Soc. 2011;133:20335–20340. doi: 10.1021/ja2071362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Choi JH, Cho M. PCCP. 2010;12:12658–12669. doi: 10.1039/c0cp00214c. [DOI] [PubMed] [Google Scholar]
- 26.Choi JH, Raleigh D, Cho M. J Phys Chem Lett. 2011;2:2158–2162. doi: 10.1021/jz200980g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagchi S, Boxer SG, Fayer MD. J Phys Chem B. 2012;116:4034–4042. doi: 10.1021/jp2122856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim YS, Hochstrasser RM. J Phys Chem B. 2009;113:8231–8251. doi: 10.1021/jp8113978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berliner LJ, Grunwald J, Hankovszky HO, Hideg K. Anal Biochem. 1982;119:450–455. doi: 10.1016/0003-2697(82)90612-1. [DOI] [PubMed] [Google Scholar]
- 30.Agbossou F, Ramsden JA, Huang YH, Arif AM, Gladysz JA. Organometallics. 1992;11:693–701. [Google Scholar]
- 31.Rudolf B, Palusiak M, Zakrzewski J, Salmain M, Jaouen G. Bioconjugate Chem. 2005;16:1218–1224. doi: 10.1021/bc050073d. [DOI] [PubMed] [Google Scholar]
- 32.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Proc Natl Acad Sci USA. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jo H, Culik RM, Korendovych IV, DeGrado WF, Gai F. Biochemistry. 2010;49:10354–10356. doi: 10.1021/bi101711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salmain M, Jaouen G. CR Chim. 2003;6:249–258. [Google Scholar]
- 35.Blanco-Rodríguez AM, Busby M, Grǎdinaru C, Crane BR, Di Bilio AJ, Matousek P, Towrie M, Leigh BS, Richards JH, Vlček A, Gray HB. J Am Chem Soc. 2006;128:4365–4370. doi: 10.1021/ja057451+. [DOI] [PubMed] [Google Scholar]
- 36.Warren JJ, Ener ME, Vlček A, Jr, Winkler JR, Gray HB. Coord Chem Rev. 2012;256:2478–2487. doi: 10.1016/j.ccr.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haquette P, Salmain M, Svedlung K, Martel A, Rudolf B, Zakrzewski J, Cordier S, Roisnel T, Fosse C, Jaouen G. ChemBioChem. 2007;8:224–231. doi: 10.1002/cbic.200600387. [DOI] [PubMed] [Google Scholar]
- 38.Binkley SL, Leeper TC, Rowlett RS, Herrick RS, Ziegler CJ. Metallomics. 2011;3:909–916. doi: 10.1039/c1mt00065a. [DOI] [PubMed] [Google Scholar]
- 39.Santos-Silva T, Mukhopadhyay A, Seixas JD, Bernardes GJL, Romão CC, Romão MJ. J Am Chem Soc. 2011;133:1192–1195. doi: 10.1021/ja108820s. [DOI] [PubMed] [Google Scholar]
- 40.King JT, Arthur EJ, Brooks CL, Kubarych KJ. J Phys Chem B. 2012;116:5604–5611. doi: 10.1021/jp300835k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Bilio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vlček A, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760–1762. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 42.Valensin D, Anzini P, Gaggelli E, Gaggelli N, Tamasi G, Cini R, Gabbiani C, Michelucci E, Messori L, Kozlowski H, Valensin G. Inorg Chem. 2010;49:4720–4722. doi: 10.1021/ic902593e. [DOI] [PubMed] [Google Scholar]
- 43.Shim SH, Zanni MT. PCCP. 2009;11:748–761. doi: 10.1039/b813817f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mavunkal IJ, Moss JR, Bacsa J. J Organomet Chem. 2000;593–594:361–368. [Google Scholar]
- 45.Westerlund K, Moran SD, Privett HK, Hay S, Jarvet J, Gibney BR, Tommos C. Protein Eng Des Sel. 2008;21:645–652. doi: 10.1093/protein/gzn043. [DOI] [PubMed] [Google Scholar]
- 46.Middleton CT, Woys AM, Mukherjee SS, Zanni MT. Methods. 2010;52:12–22. doi: 10.1016/j.ymeth.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herberhold M, Biersack M. J Organomet Chem. 1995;503:277–287. [Google Scholar]
- 48.Olah GA, Narang SC, Gupta BGB, Malhotra R. J Org Chem. 1979;44:1247–1251. [Google Scholar]
- 49.Kwak K, Rosenfeld DE, Fayer MD. J Chem Phys. 2008;128:204505/204501–204505/204510. doi: 10.1063/1.2927906. [DOI] [PubMed] [Google Scholar]
- 50.Hayashi T, Mukamel S. J Chem Phys. 2006;125:194510/194511–194510/194511. doi: 10.1063/1.2348865. [DOI] [PubMed] [Google Scholar]
- 51.Hayashi T, Mukamel S. J Mol Liq. 2008;141:149–154. doi: 10.1016/j.molliq.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin YS, Shorb JM, Mukherjee P, Zanni MT, Skinner JL. J Phys Chem B. 2009;113:592–602. doi: 10.1021/jp807528q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, Middleton CT, Zanni MT, Skinner JL. J Phys Chem B. 2011;115:3713–3724. doi: 10.1021/jp200745r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heilweil EJ, Cavanagh RR, Stephenson JC. Chem Phys Lett. 1987;134:181–188. [Google Scholar]
- 55.Suydam IT, Snow CD, Pande VS, Boxer SG. Science. 2006;313:200–204. doi: 10.1126/science.1127159. [DOI] [PubMed] [Google Scholar]
- 56.Choi JH, Oh KI, Cho M. J Chem Phys. 2008;129:174512/174511–174512/174511. doi: 10.1063/1.3001915. [DOI] [PubMed] [Google Scholar]
- 57.Hu W, Webb LJ. J Phys Chem Lett. 2011;2:1925–1930. [Google Scholar]
- 58.Eaves JD, Tokmakoff A, Geissler PL. J Phys Chem A. 2005;109:9424–9436. doi: 10.1021/jp051364m. [DOI] [PubMed] [Google Scholar]
- 59.Warshaviak DT, Serbulea L, Houk KN, Hubbell WL. The Journal of Physical Chemistry B. 2010;115:397–405. doi: 10.1021/jp108871m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berliner LJ Stable Radicals, editor. Spin Labeling: A Modern Perspective. John Wiley & Sons, Ltd; [Google Scholar]
- 61.Volkov A, Dockter C, Bund T, Paulsen H, Jeschke G. Biophys J. 2009;96:1124–1141. doi: 10.1016/j.bpj.2008.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hahn S, Kim SS, Lee C, Cho M. J Chem Phys. 2005;123:084905/084901–084905/084910. doi: 10.1063/1.1997151. [DOI] [PubMed] [Google Scholar]
- 63.Zhuang W, Abramavicius D, Voronine DV, Mukamel S. Proc Natl Acad Sci USA. 2007;104:14233–14236. doi: 10.1073/pnas.0700392104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, Middleton CT, Singh S, Reddy AS, Woys AM, Strasfeld DB, Marek P, Raleigh DP, de PJJ, Zanni MT, Skinner JL. J Am Chem Soc. 2011;133:16062–16071. doi: 10.1021/ja204035k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dijkstra AG, Jansen TLC, Knoester J. J Phys Chem B. 2011;115:5392–5401. doi: 10.1021/jp109431a. [DOI] [PubMed] [Google Scholar]
- 66.Baiz CR, Kubarych KJ, Geva E, Sibert EL. The Journal of Physical Chemistry A. 2011;115:5354–5363. doi: 10.1021/jp201641h. [DOI] [PubMed] [Google Scholar]
- 67.Hamm P, Helbing J, Bredenbeck J. Annu Rev Phys Chem. 2008;59:291–317. doi: 10.1146/annurev.physchem.59.032607.093757. [DOI] [PubMed] [Google Scholar]
- 68.Baiz CR, Kubarych KJ. J Am Chem Soc. 2010;132:12784–12785. doi: 10.1021/ja1043853. [DOI] [PubMed] [Google Scholar]
- 69.Dunkelberger EB, Buchanan LE, Marek P, Cao P, Raleigh DP, Zanni MT. J Am Chem Soc. 2012;134:12658–12667. doi: 10.1021/ja3039486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jones KC, Peng CS, Tokmakoff A. Proc Natl Acad Sci U S A, Early Ed. 2013. pp. 1–6.pp. 6 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.