

Abstract
Extracellular signal-regulated kinase 1/2 (ERK1/2) that belongs to a subfamily of mitogen-activated protein kinases (MAPKs) plays diverse roles in the central nervous system. Activation of ERK1/2 has been observed in various types of neuronal excitation, including seizure activity in vivo and in vitro, as well as in NMDA-receptor (NMDA-R)-dependent long-term potentiation in the hippocampus. On the other hand, recent studies in cultured neurons have shown that NMDA-R stimulation could result in either ERK1/2 activation or non-activation, depending on the pharmacological manipulations. To assess NMDA-R-dependent regulation of ERK1/2 activity in vivo, here we examined the effect of NMDA-R-induced seizure activity on ERK1/2 activation by using rat cortical slice preparations. NMDA-R-dependent seizure activity introduced by Mg2+-free condition did not cause ERK1/2 activation. On the other hand, when picrotoxin was added to concurrently suppress GABAA-receptor-mediated inhibition, profound ERK1/2 activation occurred, which was accompanied by strong phospho-ERK1/2-staining in the superficial and deep cortical layer neurons. In this case, prolonged membrane depolarization and enhanced burst action potential firings, both of which were much greater than those in Mg2+-free condition alone, were observed. Differential ERK1/2 activation was supported by the concurrent selective increase in phosphorylation of a substrate protein, phospho-site 4/5 of synapsin I. These results indicate that NMDA-R activation through a release from Mg2+-blockade, which accompanies enhancement of both excitatory and inhibitory synaptic transmission, was not enough, but concurrent suppression of GABAergic inhibition, which leads to a selective increase in excitatory synaptic transmission, was necessary for robust ERK1/2 activation to occur within the cortical network.
Keywords: ERK1/2 MAPK, NMDA-receptor, seizure activity, GABAA-receptor
1. Introduction
Extracellular signal-regulated kinase (ERK) is a subfamily of mitogen-activated protein kinases (MAPKs) that have been implicated in diverse cellular processes (Chang and Karin, 2001; Sweatt, 2001). Aside from its major roles in cellular proliferation and differentiation, ERK1/2 in the central nervous system plays a variety of roles in neuronal survival or death, synaptic plasticity, and learning and memory through phosphorylation of various substrates, such as transcription factors, cytoskeletal proteins, regulatory enzymes and kinases in postmitotic neurons (Hardingham and Bading, 2010; Subramaniam and Unsicker, 2006; Sweatt, 2004; Thomas and Huganir, 2004). A number of classical studies using neuronal cultures showed that ERK1/2 is activated in response to excitatory glutamatergic stimulation and following Ca2+-influx into neurons (Bading and Greenberg, 1991; Fiore et al., 1993; Kurino et al., 1995; Murphy et al., 1994). In addition, tetanic stimulation at the Schaffer collateral-CA1 synapse induces N-methyl-d-aspartate receptor (NMDA-R)-dependent long-term potentiation that requires ERK1/2 activation, and ERK1/2 activation seems to be necessary for hippocampus-dependent memory as well (Atkins et al., 1998; English and Sweatt, 1997; Sweatt, 2004; Thomas and Huganir, 2004).
Recent detailed studies in cultured neurons revealed that NMDA-R activation can cause either stimulatory or inhibitory effects on ERK1/2 activation depending on the level of NMDA-R activation (Chandler et al., 2001) or on the location of activated NMDA-Rs on neuronal cell surface, i.e., synaptic or extrasynaptic (Ivanov et al., 2006; Léveillé et al., 2008). On the other hand, robust activation of ERK1/2 has been observed in various seizure models, implicating a close relationship between neuronal excitation and ERK1/2 activation (Baraban et al., 1993; de Lemos et al., 2010; Gass et al., 1993; Kim et al., 1994; Merlo et al., 2004; Murray et al., 1998; Yamagata et al., 2002). However, how NMDA-R activation regulates ERK1/2 activation at a neuronal network level remains to be established.
Here we employed a cortical slice model of seizure activity and showed that NMDA-R activation through a release from Mg2+-blockade did not cause activation of ERK1/2. However, when combined with a blockade of inhibitory γ-aminobutyric acid type A receptor (GABAA-R), NMDA-R activation resulted in robust activation of ERK1/2, which was accompanied by a concurrent increase in substrate phosphorylation. In the latter condition, pyramidal and non-pyramidal neurons revealed longer depolarization with more spike firings than in the former condition, suggesting amplified excitatory glutamatergic synaptic transmission through suppression of the inhibitory cortical network.
2. Results
2.1. ERK1/2 activity during NMDA-R-induced seizure activity in cortical slices
NMDA-R-dependent synchronized seizure activity can be induced in cortical slices by omission of extracellular Mg2+ (Flint and Connors, 1996; Silva et al., 1991; Thomson and West, 1986). In this condition, strong depolarization with many spikes occurred in pyramidal neurons, which was accompanied by a corresponding change in field potentials (Kawaguchi, 2001). According to these studies, we examined the effects of Mg2+-free condition on ERK1/2 activity in cortical slices (Fig. 1). After incubation in Mg2+-free ACSF for 40–60 min, cortical slices showed no change in ERK1/2 activity, compared to control slices incubated in normal ACSF containing 1.2 mM Mg2+ (Fig. 1A, left two columns; 95.6±6.5% of control value, n=5). Immunoblot analysis also revealed that the level of phospho-ERK1/2, the active form, was unchanged, as was the total ERK1/2 level (Fig. 1B, left two panels). The results demonstrate that no activation of ERK1/2 occurred in cortical slices incubated in Mg2+-free condition.
Fig. 1. Marked activation of ERK1/2 in cortical slices in Mg2+-free condition with GABAA-R blockade, but not in Mg2+-free condition alone.
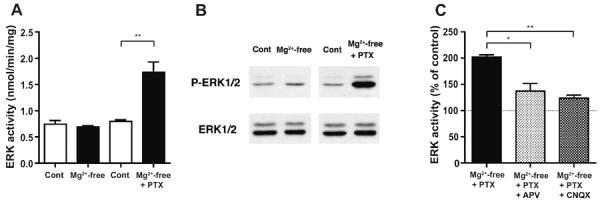
A, ERK1/2 activity measured by kinase activity assay in cortical slices incubated in Mg2+-free ACSF alone and with concurrent GABAA-R blockade by picrotoxin (PTX, 100 μM) (black columns), as compared to control slices incubated in parallel in normal ACSF containing 1.2 mM Mg2+ (Cont, white columns). Kinase activity was unaltered in Mg2+-free condition alone (left two columns), while it was markedly increased with concurrent blockade of GABAA-R by picrotoxin, compared to control condition (right two columns; ** p<0.01, unpaired t-test, n=5). B, Representative immunoblots showing the phospho-ERK1/2 (P-ERK1/2) and total ERK1/2 levels in cortical slices. The phospho-ERK1/2 level was unchanged in Mg2+-free condition, while it showed a marked increase when picrotoxin was included in Mg2+-free medium. The total ERK1/2 level remained unchanged in either condition. C, The effect of NMDA-R and non-NMDA-R inhibition on the increase in ERK1/2 activity in Mg2+-free condition with concurrent blockade of GABAA-R by picrotoxin. d-APV (50 μM), an NMDA-R antagonist or CNQX (10 μM), a non-NMDA-R antagonist was included in the incubation solution. Data are expressed as a percentage of control values from slices incubated in parallel in normal ACSF containing 1.2 mM Mg2+. * p<0.05, ** p<0.01, ANOVA followed by Bonferroni's test; n=3, 6, 4 for the left, middle, right columns, respectively.
Next, we examined the effects of concurrent blockade of GABAA-R on ERK1/2 activity. In a condition where picrotoxin (100 μM) was included in Mg2+-free ACSF, a marked increase in ERK1/2 activity was observed, compared to control slices incubated in normal ACSF (Fig. 1A, right two columns; 217.9±22.0% of control value, n=5). Similarly, the phospho-ERK1/2 level was increased distinctively, while the total ERK1/2 level remained unchanged (Fig. 1B, right two panels). The results demonstrate that robust ERK1/2 activation occurred only when slices were exposed to GABAA-R blockade in addition to Mg2+-free condition.
To examine whether such an increase in ERK1/2 activity was indeed dependent on NMDA-R activation, we performed additional experiments using an NMDA-R antagonist, d-2-amino-5-phosphonovaleric acid (d-APV). When d-APV (50 μM) was included in Mg2+-free ACSF in addition to picrotoxin, the level of ERK1/2 activation was significantly reduced compared to that without d-APV (Fig. 1C; Compare the left and middle columns). A similar result was observed when a non-NMDA-R antagonist, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) was instead included (Fig. 1C; Compare the left and right columns). These results indicate that ERK1/2 activation observed in Mg2+-free condition with picrotoxin was dependent on NMDA-Rs, as well as on non-NMDA-Rs
We also examined the distribution of phospho-ERK1/2-positive neurons by immunohistochemistry (Fig. 2). In slices incubated in Mg2+-free ACSF with picrotoxin, the number of neurons positive with phospho-ERK1/2 was increased and the extent of staining was markedly enhanced in the superficial and deep cortical layers, compared to control slices incubated in normal ACSF (Fig. 2B), while ERK1/2-staining was unchanged in either condition (Fig. 2A). Among phospho-ERK1/2-positive neurons, pyramidal neurons were prominent, which are characterized by a pyramidal-shaped soma and an extending apical dendrite arising from the pial side of the soma (Fig. 2C).
Fig. 2. Increased phospho-ERK1/2-staining in cortical slices in Mg2+-free condition with GABAA-R blockade.
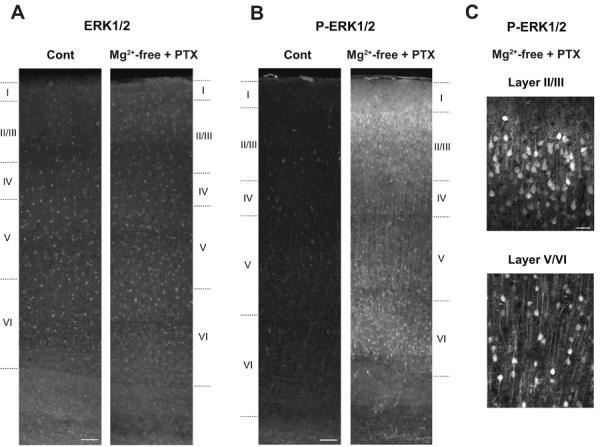
Representative staining with anti-ERK1/2 (A) and with anti-phospho-ERK1/2 (P-ERK1/2) (B) in control slices incubated in normal ACSF containing 1.2 mM Mg2+ (Cont) and in stimulated slices incubated in Mg2+-free ACSF with concurrent GABAA-R blockade by picrotoxin (PTX, 100 μM). When comparing the sections from control and stimulated slices, images were taken consecutively across the cortical layers using the same exposure time between control and stimulated slices, and were reconstructed afterwards. The number of phospho-ERK1/2-positive neurons and the extent of staining were markedly enhanced in the superficial and deep cortical layers of stimulated slices, compared to control slices (B), while ERK1/2-staining was unchanged in either condition (A). Similar results were obtained in three independent experiments (n=3). Cortical layers are indicated based on Nissl staining of adjacent slices. C, Enlarged confocal images of phospho-ERK1/2-staining in stimulated slices in the superficial (Layer II/III) and deep (Layer V/VI, the border between layers V and VI) cortical layers. Among phospho-ERK1/2-positive neurons, pyramidal neurons are prominent, which are characterized by a pyramidal-shaped soma and an extending apical dendrite arising from the pial side of the soma. Scale bars, 100 μm in A and B, 20 μm in C.
2.2. Substrate phosphorylation during NMDA-R-induced seizure activity in cortical slices
To examine whether such ERK1/2 activation was accompanied by an increase in substrate phosphorylation, we measured the phosphorylation state of an ERK1/2 substrate, synapsin I that has multiple phosphorylation sites (Greengard et al., 1993; Hilfiker et al., 1999; Cesca et al., 2010) (Fig. 3). The level of phospho-site 4/5 of synapsin I that is dependent on ERK1/2 was decreased in slices incubated in Mg2+-free ACSF, compared to control slices incubated in normal ACSF containing 1.2 mM Mg2+ (Fig. 3A, left panels; B, left graph). On the other hand, it showed a large increase in slices incubated in Mg2+-free ACSF containing picrotoxin, compared to control slices incubated in normal ACSF (Fig. 3A, right panels; B, right graph). We also checked the level of phospho-site 3 that is dependent on Ca2+/calmodulin-dependent protein kinase II (CaMKII), but not on ERK1/2, in the same conditions. The phospho-site 3 level showed no change in Mg2+-free condition alone, while it showed a profound decrease in Mg2+-free condition with picrotoxin (Fig. 3A, B). The total synapsin I level remained unchanged in either condition. These results demonstrate that not only ERK1/2 activity, but also substrate phosphorylation, was increased in slices exposed to GABAA-R blockade in addition to Mg2+-free condition.
Fig. 3. Increased phospho-site 4/5 level of synapsin I in cortical slices in Mg2+-free condition with GABAA-R blockade, but not in Mg2+-free condition alone.
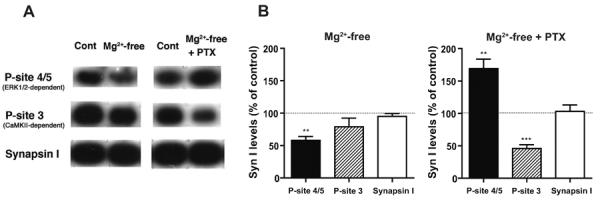
A, Representative immunoblots showing the phospho-site 4/5 (P-site 4/5), phospho-site 3 (P-site 3) and total levels of synapsin I in cortical slices incubated in different conditions. The ERK1/2-dependent-phospho-site 4/5 level was decreased in slices incubated in Mg2+-free condition alone, while it showed a marked increase in slices treated with concurrent GABAA-R blockade by picrotoxin (PTX, 100 μM), compared to control slices incubated in parallel in normal ACSF (Cont). In contrast, the phospho-site 3 level that is dependent on CaMKII, but not on ERK1/2, was unaltered in Mg2+-free condition alone, and largely decreased in Mg2+-free condition with picrotoxin. The total synapsin I level remained unchanged in either condition. B, Quantitative data obtained by immunoblot analyses, expressed as a percentage of control levels. ** p<0.01, *** p<0.001, one group t-test, n=5.
2.3. Electrophysiological changes during NMDA-R-induced seizure activity in cortical slices
We next performed electrophysiological recording from the same slice preparations to examine the actual changes in neuronal activity during the above-mentioned seizure-inducing conditions. When slices were incubated in Mg2+-free ACSF, layer V pyramidal neurons (input resistance, 130±11.6 MΩ, n=6) (Fig. 4A) continuously exhibited phasic spontaneous depolarization with spike firings in whole cell current-clamp recordings (n=39) (Fig. 4B, Mg2+-free). Such neuronal activities were not observed when slices were incubated in normal ACSF containing 1.2 mM Mg2+ (4 cells from 3 slices) (Fig. 4C). Bath-application of d-APV (50 μM), an NMDA-R antagonist completely abolished spontaneous depolarization with spike firings that was observed in Mg2+-free condition (5 cells from 5 slices) (Fig. 4B, APV). Washing d-APV out of the bathing solution resulted in an abrupt appearance of large depolarization with burst firings followed by phasic regular-spike firings (Fig. 4B, Wash of APV). These results demonstrate the dependence of such spontaneous neuronal activities on NMDA-Rs. Layer II/III pyramidal neurons also exhibited rhythmic spontaneous depolarization with spike firings when incubated in Mg2+-free ACSF (n=5) (Fig. 4D), but not in normal ACSF (5 cells from 4 slices) (Fig. 4E). The results confirmed enhanced neuronal activity in different cortical layers, supporting synchronized seizure activity (Kawaguchi, 2001).
Fig. 4. NMDA-R-dependent seizure activity caused by Mg2+-free condition in pyramidal neurons in cortical slices.
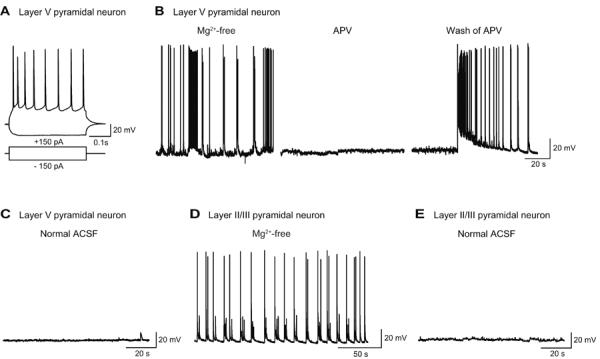
A, Representative whole-cell current-clamp recording from a layer V pyramidal neuron. Current injection of −150 and +150 pA induced hyperpolarization and action potential firings, respectively, at resting membrane potential of −63 mV. B, Recording from another layer V pyramidal neuron. This neuron showed repetitive spontaneous depolarization with accompanying spike firings in Mg2+-free condition (Mg2+-free), which was abolished by bath-application of d-APV (50 μM), an NMDA-R antagonist for five min (APV), but resumed by washing d-APV out of the bathing medium (Wash of APV). C, Recording from a layer V pyramidal neuron in control condition containing 1.2mM Mg2+ (Normal ACSF). The neuron was silent in control condition. D, Recording from a layer II/III pyramidal neuron exhibiting rhythmic spontaneous depolarization with regular-spikings in Mg2+-free condition. E, Recording from a layer II/III pyramidal neuron, which was silent, in control condition.
In addition, non-pyramidal neurons in layer V (input resistance, 353.9±11.7 MΩ, n=8) (Fig. 5A, left) exhibited repetitive events of spontaneous depolarization with intense burst spike firings in Mg2+-free condition (6 cells from 5 slices) (Fig. 5A, Mg2+-free). Such neuronal activities were not observed in normal ACSF containing 1.2 mM Mg2+ (6 cells from 6 slices) (Fig. 5B, Normal ACSF). Non-pyramidal neurons in layer II/III also exhibited spontaneous depolarization with spike discharges in Mg2+-free condition (Fig. 5B, Mg2+-free). These spontaneous spike firings almost completely disappeared during bath-application of d-APV (50 μM) (4 cells from 4 slices) (Fig. 5B, APV). Thus, Mg2+-free condition enhanced neuronal activity, not only in the major excitatory pyramidal neurons, but also in non-pyramidal neurons including inhibitory GABAergic interneurons, in an NMDA-R-dependent manner, leading to an elevation of GABAergic inhibition.
Fig. 5. NMDA-R-dependent seizure activity caused by Mg2+-free condition in non-pyramidal neurons in cortical slices.
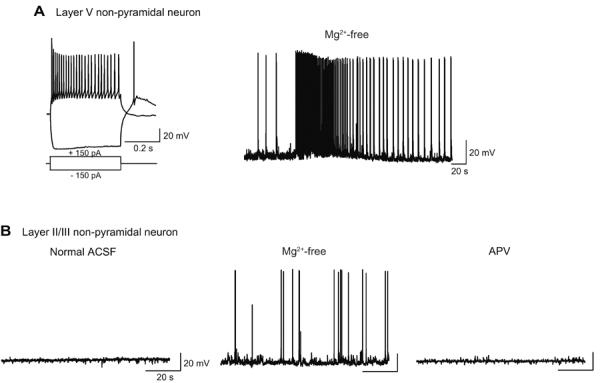
A, Representative recording from a layer V non-pyramidal neuron. Current injection of −150 and +150 pA induced hyperpolarization and action potential firings, respectively, at resting membrane potential of −66 mV (left). Non-pyramidal neurons are characterized by higher-frequency firings and higher input resistance than pyramidal neurons in the same cortical layer (Compare with Fig. 4A). This neuron fired tonic spike discharges of ~33 s followed by phasic burst firings in Mg2+-free condition (Mg2+-free). B, Recording from a layer II/III non-pyramidal neuron. This neuron was silent in normal ACSF containing 1.2mM Mg2+ (Normal ACSF), but showed spontaneous depolarization with spike discharges in Mg2+-free condition (Mg2+-free). Bath-application of d-APV (50 μM), an NMDA-R antagonist abolished spontaneous depolarization with spike discharges (APV).
On the other hand, in a condition where picrotoxin (100 μM) was included in Mg2+-free ACSF, a repetitive appearance of longer depolarization with more spike firings was observed in layer V pyramidal neurons (8 cells from 8 slices) (Fig. 6A, Compare Mg2+-free + PTX with Mg2+-free). The duration of depolarization (a size of more than 2 mV) was significantly longer in the presence (6.1 ± 1.5 s) than in the absence (1.1 ± 0.7 s) of picrotoxin in Mg2+-free condition (p<0.01, paired t-test, n=8) (Fig. 6A, See the traces with fast time scales). Such prolonged depolarization and increased burst spike firings were observed in non-pyramidal neurons as well (n=2) (Fig. 6B, Mg2+-free + PTX), which became much less when picrotoxin was removed from Mg2+-free ACSF (Fig. 6B, Wash of PTX). Thus, the blockade of GABAA-R amplified glutamatergic excitatory synaptic transmission in both pyramidal and non-pyramidal neurons through suppression of the inhibitory cortical network.
Fig. 6. Enhancement of seizure activity in Mg2+-free condition with concurrent GABAA-R blockade in cortical slices.
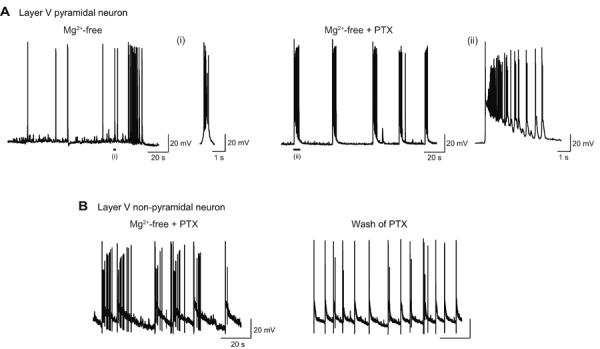
A, Recording from a layer V pyramidal neuron. Incubation in Mg2+-free condition induced spontaneous depolarization and burst spike firings (Mg2+-free). Application of picrotoxin (PTX, 100 μM) caused prolonged depolarization and increased burst spike firings (Mg2+-free + PTX). Two typical depolarization events are shown with faster time scales [(i) and (ii)]. The duration of these depolarization events (a size of more than 2 mV) was 0.89 s [(i)] and 5.8 s [(ii)], respectively. B, Recording from a layer V non-pyramidal neuron. Prolonged depolarization and increased burst spike firings observed in Mg2+-free condition with picrotoxin (Mg2+-free + PTX) became much less when picrotoxin was washed out of the bathing medium (Wash of PTX).
3. Discussion
In this study, we employed two different conditions: (1) omission of extracellular Mg2+ alone and (2) with concurrent application of picrotoxin, to elicit NMDA-R-induced seizure activity in slice preparations from rat somatosensory cortex, and analyzed ERK1/2 activity biochemically and neuronal activity electrophysiologically. Incubation of cortical slices in Mg2+-free medium, which caused phasic spontaneous depolarization with spike firings in both pyramidal and non-pyramidal neurons in different cortical layers (Figs. 4 and 5), did not induce a detectable level of ERK1/2 activation (Fig. 1). On the other hand, in a condition where picrotoxin was included in Mg2+-free medium to suppress GABAA-R-mediated inhibition, which resulted in more prolonged membrane depolarization and enhanced burst action potential firings in both pyramidal and non-pyramidal neurons (Fig. 6), robust ERK1/2 activation occurred (Fig. 1). Such ERK1/2 activation was dependent on NMDA-Rs, as well as on non-NMDA-Rs. In Mg2+-free condition with picrotoxin, phospho-ERK1/2-positive neurons were markedly increased in the superficial and deep cortical layers, and among them, pyramidal neurons were distinct, which are characterized by a pyramidal-shaped soma and a prominent apical dendrite (Fig. 2). We cannot rule out the possibility that non-pyramidal neurons that lack apparent apical dendrites may also be positive with phospho-ERK1/2, but they are difficult to be identified by morphological analysis. Pronounced phospho-ERK1/2-staining in the soma and dendrites (Fig. 2) clearly indicates activation of ERK1/2 on the postsynaptic side. Thus, our study demonstrated that NMDA-R activation through a release from Mg2+-blockade, did not cause detectable ERK1/2 activation, but concurrent suppression of GABAergic inhibition that further enhances excitatory glutamatergic transmission did cause robust activation of ERK1/2 in cortical slices.
Such differential ERK1/2 activation was supported by a selective increase in the phospho-site 4/5 level of synapsin I in Mg2+-free condition exclusively with picrotoxin (Fig. 3). Synapsin I is a presynaptic vesicle-associated protein and the presynaptic pool of ERK1/2 is much smaller than its postsynaptic pool. However, seizure activity is considered to involve the entire cortical network that includes both pre- and post-synaptic compartments, and we previously demonstrated that phospho-site 4/5 of synapsin I was indeed regulated by ERK1/2 activity during acute seizure activity in the brain in vivo (Yamagata et al., 2002). Therefore, we expected that it could be a good marker for ERK1/2 activation when its activation was so robust as in the current study, and it turned out to be the case. On the other hand, a large decrease in the phospho-site 4/5 level in Mg2+-free condition (Fig. 3) seems to be caused by Ca2+/calmodulin-dependent protein phosphatase 2B, i.e., calcineurin, as this site was preferentially dephosphorylated by calcineurin not only in vitro, but also in synaptosome preparations in a depolarizing condition (Jovanovic et al., 2001). It should also be noted that the level of phospho-site 3 that is dependent on CaMKII, but not on ERK1/2, showed a profound decrease in Mg2+-free condition with picrotoxin (Fig. 3), demonstrating the selectivity of the changes in phospho-site 4/5 in that condition. The decrease in the level of CaMKII-dependent phospho-site 3 is in agreement with a previous observation showing a similar decrease in the phospho-site 3 level, along with a decrease in the Ca2+/calmodulin-independent, autonomous activity of CaMKII in the brain during acute seizure activity in rats in vivo (Yamagata and Obata, 1998; Yamagata et al., 2002; Yamagata, 2003).
Many classical studies in cultured neurons showed activation of ERK1/2 by NMDA-R-dependent and non-NMDA-R-dependent glutamatergic stimulation (Bading and Greenberg, 1991; Fiore et al., 1993; Murphy et al., 1994; Kurino et al., 1995), and pronounced ERK1/2 activation has been observed in various seizure models in vivo and in vitro (Baraban et al., 1993; de Lemos et al., 2010; Gass et al., 1993; Kim et al., 1994; Merlo et al., 2004; Murray et al., 1998; Yamagata et al., 2002). Thus, there seems to be a close relationship between neuronal excitation and ERK1/2 activation. On the other hand, recent studies in cultured neurons revealed that NMDA-R stimulation could result in either ERK1/2 activation or non-activation depending on the level of NMDA-R activation or on the location of activated NMDA-Rs on neuronal cell surface (Chandler et al., 2001; Ivanov et al., 2006; Léveillé et al., 2008). Chandler et al. (2001) reported that after a blockade of a high basal level of phospho-ERK1/2 in cortical neuronal cultures, application of NMDA produced a bell-shaped dose-response curve for stimulation of phospho-ERK1/2. Ivanov et al. (2006) and Léveillé et al. (2008) reported in hippocampal and cortical neuronal cultures, respectively, that pharmacological manipulation that stimulated synaptic NMDA-Rs resulted in activation of ERK1/2, whereas one that stimulated extrasynaptic NMDA-Rs did not or may even inactivate ERK1/2. According to these previous studies, it was expected that relatively mild NMDA-R activation would stimulate ERK1/2 activation, and that strong NMDA-R activation would trigger extrasynaptic NMDA-R activation and might suppress ERK1/2 activation (Hardingham and Bading, 2010). However, that was not the case in our present study using cortical slice preparations.
Using a cortical slice model of seizure activity, we examined how NMDA-R activation regulates ERK1/2 activity at a cortical network level. It should be noted that in cortical slices in normal Mg2+ condition, spontaneous neuronal firing was rare, and the network activities were almost absent (Fig. 4C, E; Fig. 5B, Normal ACSF), as reported in previous studies (Kawaguchi, 2001; Luhmann and Prince, 1990). In addition, a very low basal level of phospho-ERK1/2 was detected in this control condition (Fig. 1B, Cont). By omission of extracellular Mg2+, NMDA-R-dependent seizure activity could be readily induced, and our electrophysiological recordings showed that NMDA-R activation enhanced not only excitatory glutamatergic, but also inhibitory GABAergic transmission (Figs. 4 and 5). The enhanced GABAergic inhibition apparently suppressed NMDA-R-mediated inward currents postsynaptically and/or glutamate release presynaptically, and thereby limiting the duration of depolarization of excitatory neurons. With both excitatory glutamatergic and inhibitory GABAergic transmission enhanced, long but limited depolarization may not be enough to cause activation of ERK1/2 in neurons. With concurrent blockade of GABAA-R-mediated inhibition, however, the duration of depolarization became prolonged (Fig. 6), leading to activation of ERK1/2 probably through increased Ca2+-influx into neurons. Such ERK1/2 activation was suppressed not only by NMDA-R blockade, but also by non-NMDA-R blockade (Fig. 1C), probably because non-NMDA-R activation further enhances excitability of neurons, which in turn increases glutamate release and induces more activation of NMDA-Rs. Our results indicate that moderate stimulation of synaptic NMDA-Rs is not enough, but profound glutamatergic excitation that involves both NMDA-R and non-NMDA-R activation is necessary for robust ERK1/2 activation to occur at a cortical network level.
We cannot rule out the possibility that enhanced glutamatergic transmission in Mg2+-free condition with GABAA-R blockade may have caused spillover of glutamate to extrasynaptic sites, leading to activation of extrasynaptic NMDA-Rs. However, that is unlikely, given that extrasynaptic NMDA-R activation would be to suppress ERK1/2 activation (Ivanov et al., 2006; Léveillé et al., 2008; Hardingham and Bading, 2010). Rather, activation of synaptic NMDA-Rs is strongly suggested in the current seizure models, based on our electrophysiological observations showing enhanced synaptic activity in Mg2+-free condition and more profound activity with concurrent GABAA-R blockade (Figs. 4–6). Our results implicate that robust ERK1/2 activation that had been observed in various seizure models in vivo (Baraban et al., 1993; de Lemos et al., 2010; Gass et al., 1993; Kim et al., 1994; Yamagata et al., 2002) may also have been caused by profound synaptic NMDA-R and non-NMDA-R activation.
In conclusion, our study clearly demonstrated that: (1) NMDA-R activation by omission of extracellular Mg2+ was not enough, but (2) additional strong glutamatergic excitation by concurrent GABAA-R blockade, which involves both NMDA-R and non-NMDA-R activation, was necessary for ERK1/2 activation to occur at a cortical network level. Our results indicate the importance of the network-level analysis toward the understanding of activity-dependent regulation of ERK1/2 in vivo.
4. Experimental Procedures
4.1. Animal experiments
Male Wistar rats (5 weeks old) (Japan SLC, Hamamatsu, Japan) were used for experiments. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of National Institutes of Natural Sciences. All experiments were conducted in accordance with the Guide for Animal Experimentation in the Institute. Animals were housed in cages with ad libitum access to water and food and maintained on a 12 h light/dark cycle.
4.2. Neocortical slice preparation
Rats were deeply anesthetized with pentobarbital (50 mg/kg i.p.; Dainippon Sumitomo Pharma, Osaka, Japan) and then decapitated. The brains were rapidly removed and placed in ice-cold modified artificial cerebrospinal fluid (modified ACSF) containing (in mM): choline chloride 120, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, glucose 15, MgCl2 7, CaCl2 0.5 (Kaneko et al., 2008). Coronal slices (400-μm-thick) containing somatosensory cortex were cut from the bilateral hemispheres using a vibratome (Series 1000, Technical Products International, St. Louis, MO, USA) in modified ACSF, and subcortical structures were removed. These slices were placed alternately in two holding interface chambers (ca. 10 slices in each chamber) filled with normal ACSF composed of (in mM): NaCl 125, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, CaCl2 2.5, MgCl2 1.2, glucose 15, and bubbled continuously with a mixture of 95% O2 - 5% CO2 (pH 7.4). Slices in both chambers were allowed to recover in normal ACSF for at least one hour at room temperature under humid gas of 95% O2-5% CO2.
After that, slices in one chamber were exposed to either one of the following seizure-inducing conditions: (1) Mg2+-free condition, in which Mg2+ concentration in ACSF was reduced to nominally zero to enhance the activation of NMDA-Rs; and (2) Mg2+-free condition with picrotoxin, in which picrotoxin (100 μM; Sigma, St. Louis, MO, USA), a GABAA-R blocker was added to Mg2+-free condition to further increase the excitability of neuronal circuits within the slices. On the other hand, slices in the other chamber were incubated in parallel in normal ACSF containing 1.2 mM Mg2+ to serve as controls. After 40–60 min of incubation at around body temperature, eight slices from each chamber were processed for biochemical studies, and the remaining one or two slices were used for electrophysiological recording or immunohistochemistry.
To assess the effect of NMDA-R or non-NMDA-R inhibition in Mg2+-free condition with picrotoxin, D-APV (50 μM; Tocris, Bristol, UK), an NMDA-R antagonist or CNQX (10 μM; Tocris), a non-NMDA-R antagonist was included in Mg2+-free ACSF in addition to picrotoxin. Control condition remained the same (normal ACSF).
4.3. Sample preparation for biochemical analyses
Eight slices from each chamber were transferred separately to a 1 ml-Teflon-glass homogenizer on ice and immediately homogenized in 0.5 ml of homogenization buffer consisting of 20 mM Tris/HCl, pH 7.5, 5 mM EDTA, 1 mM EGTA, 10 mM sodium pyrophosphate, 50 mM NaF, 1 mM Na3VO4 (ortho), 1 mM dithiothreitol, 10 μg/ml each of leupeptin, antipain, pepstatin, and chymostatin, and 0.1 mM phenylmethylsulfonyl fluoride. Each homogenate was immediately separated into the soluble and particulate fractions by ultracentrifugation at 175,000 g for 35 min at 4°C. The soluble and particulate fractions were recovered, and aliquots were saved for measurement of protein concentration, sodium dodecyl sulfate-polyacrylamide gel electrophoresis and kinase activity assay as described (Yamagata et al., 2006).
4.4. ERK1/2 activity assay
ERK1/2 activity was determined by using a p42/p44 MAP kinase enzyme assay system (Amersham, Buckinghamshire, UK), according to manufacturer's procedures with some modifications as described (Yamagata et al., 2002). The amount of protein used was 0.8 μg from the soluble fraction where ERK1/2 was enriched. The reaction was linear in terms of both protein concentration and incubation time.
4.5. Immunoblot analyses
Immunoblot analysis to detect the phospho- and total ERK1/2 was performed by using monoclonal anti-phospho-p44/42 MAPK antibody (anti-phospho-ERK1/2; Cell Signaling, Beverly, MA, USA) directed to dually phosphorylated ERK1/2 at Thr202/Tyr204, and polyclonal anti-p44/42 MAPK antibody (anti-ERK1/2; Cell Signaling) as previously described (Yamagata et al., 2002). The amount of protein used was 10 μg from the soluble fraction where ERK1/2 was enriched.
Quantitative immunoblot analysis for synapsin I was performed by using anti-phospho-site 4/5 (Ser-62 and Ser-67) (G-526; Jovanovic et al., 1996), anti-phospho-site 3 (Ser-603) (RU20; Czernik et al., 1995; Yamagata et al., 1995) and anti-synapsin I (G-454/455; Czernik et al., 1995) as previously described (Yamagata et al., 2002). The amounts of protein used were 30 μg for anti-phospho-site 4/5 and 6.5 μg for anti-phospho-site 3 and anti-synapsin I from the particulate fraction where synapsin I was localized. The measured immunoreactivity was linear in terms of protein amounts used for each antibody.
4.6. Immunohistchemistry
A brain slice from each chamber was fixed by immersion in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) overnight at 4°C. Slices were resectioned to a thickness of 50 μm using a vibratome, and processed for fluorescence immunohistochemistry, except for one section that was subjected to Nissl staining. After blocking in 50 mM phosphate buffered saline (PBS) containing 5% BSA and 0.3% Triton X-100, sections were incubated with anti-phospho-ERK1/2 (1:1000) or anti-ERK1/2 (1:500) in the blocking buffer over three nights at 4°C. After washing in PBS, they were incubated in an Alexa 594-conjugated secondary antibody (Donkey anti-mouse IgG or Goat anti-rabbit IgG, 1:250; Molecular Probes, Eugene, OR) in PBS containing 2.5% BSA and 0.3% Triton X-100 for 2 h at 4°C. Sections were then mounted on gelatin-coated slides, coverslipped with Vectashield (Vector Laboratories, Burlingame, CA) and observed using epifluorescence (BX51WI, Olympus, Tokyo, Japan). When comparing the sections from control and stimulated slices, images were taken consecutively across the cortical layers using the same exposure time between control and stimulated slices, and were reconstructed afterwards. Cortical layers were determined based on Nissl staining of adjacent sections. For observation with a higher magnification, a confocal laser-scanning microscope (FV1000, Olympus) was used. The contrast and brightness of digital images were adjusted using Adobe Photoshop (Adobe Systems, San Jose, CA), and images were saved as TIFF files.
4.7. Electrophysiology
A brain slice from either one of the holding chambers (Normal ACSF, Mg2+-free ACSF or Mg2+-free ACSF with picrotoxin) was transferred to a submerged chamber mounted on an upright microscope (BX50WI, Olympus) with a ×40 water-immersion objective, and continuously perfused with the corresponding oxygenated ACSF at a flow rate of 2 ml/min at 30–32°C. Whole-cell recordings were made from pyramidal and non-pyramidal neurons in layers V and II/III in the somatosensory cortex using infrared differential interference contrast techniques. Pyramidal and non-pyramidal neurons were identified as such based on their locations, shape of the soma, input resistance and spike-firing patterns as previously described (Kawaguchi, 1993). Electrodes (3–5 MΩ) were filled with a pipette solution containing (in mM): KCH3SO3 133, KCl 7, HEPES/KOH 10, MgATP 5, and Na2GTP 0.4, EGTA 0.5 (pH 7.2 with KOH).
Whole cell current-clamp recordings were made with an EPC-9 amplifier (HEKA Elektronik, Lambrecht, Germany), filtered at 3 kHz, digitized at 4 kHz or 20 kHz, and analyzed offline with PULSE (HEKA Elektronik) and Igor Pro (WaveMetrics, Lake Oswego, OR) as described (Kaneko et al., 2008). Only cells with resting membrane potential more negative than −55 mV and with overshooting spikes were analyzed. To investigate the firing properties of neurons, seven current injection steps (400 ms) were applied from −150 to +300 pA in 75 pA increments. After examining the pattern of evoked firings, spontaneous activity was recorded in current-clamp mode at around resting membrane potential of the recorded cells.
In some electrophysiological recordings, superfusing solution was switched from normal ACSF to Mg2+-free ACSF, from Mg2+-free ACSF to Mg2+-free ACSF with d-APV (50 μM), from Mg2+-free ACSF to Mg2+-free ACSF with picrotoxin (100 μM), or vice versa to directly compare spontaneous activity of the same neurons in different conditions.
4.8. Data analysis
All data are expressed as mean ± SEM.
Highlights
Cortical slices were used for electrophysiological and ERK1/2 activation analyses.
NMDA-R activation by omission of extracellular Mg2+ did not cause ERK1/2 activation.
Additional strong excitation by GABAA-R blockade was necessary for ERK1/2 activation.
Network-level analysis is important to understand activity-dependent ERK regulation.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science (KAKENHI) (Y.Y.), and by NIH grant DA10044 (A.C.N.). We thank Dr. Paul Greengard (The Rockefeller University) for synapsin I antibodies, Ms. Miho Tanaka for technical assistance, and Drs. Yasuo Kawaguchi and Atsushi Nambu (National institute for Physiological Sciences) for valuable comments on the manuscript. We also thank the staffs in the Center for Radioisotope Facilities at National Institutes of Natural Sciences.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- anti-ERK1/2
anti-p44/42 MAPK antibody
- anti-phospho-ERK1/2
anti-phospho-p44/42 MAPK antibody
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- d-APV
d-2-amino-5-phosphonovaleric acid
- ERK1/2
extracellular signal-regulated kinase 1/2
- GABAA-R
γ-aminobutyric acid type A receptor
- MAPK
mitogen-activated protein kinase
- MEK
ERK kinase
- NMDA-R
N-methyl-d-aspartate-type glutamate receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nature Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- Baraban JM, Fiore RS, Sanghera JS, Paddon HB, Pelech SL. Identification of p42 mitogen-activated protein kinase as a tyrosine kinase substrate activated by maximal electroconvulsive shock in hippocampus. J. Neurochem. 1993;60:330–336. doi: 10.1111/j.1471-4159.1993.tb05855.x. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J. Biol. Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Czernik AJ, Mathers J, Tsou K, Greengard P, Mische SM. Phosphorylation state-specific antibodies: preparation and applications. Neuroprotocols. 1995;6:56–61. [Google Scholar]
- de Lemos L, Junyent F, Verdaguer E, Folch J, Romero R, Pallàs M, Ferrer I, Auladell C, Camins A. Differences in activation of ERK1/2 and p38 kinase in Jnk3 null mice following KA treatment. J. Neurochem. 2010;114:1315–1322. doi: 10.1111/j.1471-4159.2010.06853.x. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J. Biol. Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J. Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- Flint AC, Connors BW. Two types of network oscillations in neocortex mediated by distinct glutamate receptor subtypes and neuronal populations. J. Neurophysiol. 1996;75:951–957. doi: 10.1152/jn.1996.75.2.951. [DOI] [PubMed] [Google Scholar]
- Gass P, Kiessling M, Bading H. Regionally selective stimulation of mitogen activated protein (MAP) kinase tyrosine phosphorylation after generalized seizures in the rat brain. Neurosci. Lett. 1993;162:39–42. doi: 10.1016/0304-3940(93)90554-x. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Rev. Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao H-T, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Phil. Trans. R. Soc. Lond. B. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc. Natl. Acad. Sci. USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr., Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J. Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Tamamaki N, Owada H, Kakizaki T, Kume N, Totsuka M, Yamamoto T, Yawo H, Yagi T, Obata K, Yanagawa Y. Noradrenergic excitation of a subpopulation of GABAergic cells in the basolateral amygdala via both activation of nonselective cationic conductance and suppression of resting K+ conductance: A study using glutamate decarboxylase 67-green fluorescent protein knock-in mice. Neuroscience. 2008;157:781–797. doi: 10.1016/j.neuroscience.2008.09.029. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Groupings of nonpyramidal and pyramidal cells with specific physiological and morphological characteristics in rat frontal cortex. J. Neurophysiol. 1993;69:416–431. doi: 10.1152/jn.1993.69.2.416. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Distinct firing patterns of neuronal subtypes in cortical synchronized activities. J. Neurosci. 2001;21:7261–7272. doi: 10.1523/JNEUROSCI.21-18-07261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Hong KS, Seong YS, Park JB, Kuroda S, Kishi K, Kaibuchi K, Takai Y. Phosphorylation and activation of mitogen-activated protein kinase by kainic acid-induced seizure in rat hippocampus. Biochem. Biophy. Res. Comm. 1994;202:1163–1168. doi: 10.1006/bbrc.1994.2050. [DOI] [PubMed] [Google Scholar]
- Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J. Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- Léveillé F, El gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008;22:4258–4271. doi: 10.1096/fj.08-107268. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Prince DA. Control of NMDA receptor-mediated activity by GABAergic mechanisms in mature and developing rat neocortex. Dev. Brain Res. 1990;54:287–290. doi: 10.1016/0165-3806(90)90152-o. [DOI] [PubMed] [Google Scholar]
- Merlo D, Cifelli P, Cicconi S, Tancredi V, Avoli M. 4-Aminopyridine-induced epileptogenesis depends on activation of mitogen-activated protein kinase ERK. J. Neurochem. 2004;89:654–659. doi: 10.1111/j.1471-4159.2004.02382.x. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Blatter LA, Bhat RV, Fiore RS, Wier WG, Baraban JM. Differential regulation of calcium/calmodulin-dependent protein kinase II and p42 MAP kinase activity by synaptic transmission. J. Neurosci. 1994;14:1320–1331. doi: 10.1523/JNEUROSCI.14-03-01320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. USA. 1998;95:1975–1980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Unsicker K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience. 2006;138:1055–1065. doi: 10.1016/j.neuroscience.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. The neuronal MAPK kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opn. Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature Rev. Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC. N-methylaspartate receptors mediate epileptiform activity evoked in some, but not all, conditions in rat neocortical slices. Neuroscience. 1986;19:1161–1177. doi: 10.1016/0306-4522(86)90130-2. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Obata K, Greengard P, Czernik AJ. Increase in synapsin I phosphorylation implicates a presynaptic component in septal kindling. Neuroscience. 1995;64:1–4. doi: 10.1016/0306-4522(94)00492-n. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Obata K. Dynamic regulation of the activated, autophosphorylated state of Ca2+/calmodulin-dependent protein kinase II by acute neuronal excitation in vivo. J. Neurochem. 1998;71:427–439. doi: 10.1046/j.1471-4159.1998.71010427.x. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Jovanovic JN, Czernik AJ, Greengard P, Obata K. Bidirectional changes in synapsin I phosphorylation at MAP kinase-dependent sites by acute neuronal excitation in vivo. J. Neurochem. 2002;80:835–842. doi: 10.1046/j.0022-3042.2001.00753.x. [DOI] [PubMed] [Google Scholar]
- Yamagata Y. New aspects of neurotransmitter release and exocytosis: Dynamic and differential regulation of synapsin I phosphorylation by acute neuronal excitation in vivo. J. Pharmacol. Sci. 2003;93:22–29. doi: 10.1254/jphs.93.22. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Imoto K, Obata K. A mechanism for the inactivation of Ca2+/calmodulin-dependent protein kinase II during prolonged seizure activity and its consequence after the recovery from seizure activity in rats in vivo. Neuroscience. 2006;140:981–992. doi: 10.1016/j.neuroscience.2006.02.054. [DOI] [PubMed] [Google Scholar]