

Abstract
Understanding the consequences of miR-145 reintroduction in human breast cancer (BC) could reveal its tumor-suppressive functions and may disclose new aspects of BC biology. Therefore, we characterized the effects of miR-145 re-expression in BC cell lines by using proliferation and apoptosis assays. As a result, we found that miR-145 exhibited a pro-apoptotic effect, which is dependent on TP53 activation, and that TP53 activation can, in turn, stimulate miR-145 expression, thus establishing a death-promoting loop between miR-145 and TP53. We also found that miR-145 can downregulate estrogen receptor-α (ER-α) protein expression through direct interaction with two complementary sites within its coding sequence. In conclusion, we described a tumor suppression function of miR-145 in BC cell lines, and we linked miR-145 to TP53 and ER-α. Moreover, our findings support a view that miR-145 re-expression therapy could be mainly envisioned in the specific group of patients with ER-α-positive and/or TP53 wild-type tumors.
Keywords: miR-145, ER-α, TP53, human BC, apoptosis, cell proliferation
Introduction
Breast cancer (BC) is one of the most commonly diagnosed cancers and is the second leading cause of cancer deaths.1 Although the overall survival rate for BC is increasing, an effective therapy for it has yet to be found. To meet this need, a better understanding of BC biology is required.
MicroRNAs (miRNAs) are short (19–24 nucleotides) noncoding RNAs that are generated from longer transcripts (pri-miRNA and pre-miRNA) in sequential maturation steps.2 The main known function of miRNAs is the regulation of gene expression at the post-transcriptional level either by protein translation inhibition or messenger RNA (mRNA) degradation through binding to imperfect sequence homology sites of target mRNAs and recruitment of the RNA-induced silencing complex (RISC).3 miRNAs are involved in fundamental processes, such as embryonic development and cell differentiation,4 in which fine regulation of gene expression in time and space is required for the correct execution of these processes. Consequently, the fact that researchers have found alterations of miRNA expression in many human diseases, including cancer,5 is not surprising. In fact, all human tumors analyzed so far have abnormalities in miRNA expression, and studies have identified signatures of deregulated miRNAs in different tumors.6, 7 miR-145 is one of the miRNAs whose expression is most commonly reduced in various human cancers, including BC,8 colon9 and lung10 cancers. In particular, the role of miR-145 in BC merits special attention, because several lines of evidence have shown that miR-145 expression is inversely correlated with BC tumor grade,11 tumor size11 and Ki-67 proliferation index.8
Researchers previously studied the function of miR-145 in colon and cervical cancer cells12, 13 and found miR-145 targets, such as insulin receptor substrate 1 (IRS-1) in colon cancer cells,14 homeobox A9 (HOXA9) in immortalized bone marrow cells,15 and octamer-binding transcription factor 4 (OCT4), sex determining region Y box 2 (SOX2) and Kruppel-like factor 4 (KLF4) in human embryonic stem cells.16 However, to our knowledge, no authors have reported on the effects of miR-145 re-expression in BC cells. Discovering the functions of miR-145 in BC may disclose new aspects of BC biology that can eventually generate new therapeutic approaches. Therefore, the purpose of our study was to investigate the effects of miR-145 reintroduction and identify new miR-145 targets in BC. Our strategy consisted in the use of proliferation and apoptosis assays after transfection of miR-145. As a result, we characterized the molecular pathways involved in miR-145-dependent apoptosis and identified new targets of miR-145 in BC cell lines. Finally, we found that the status of molecular markers, such as estrogen receptor-α (ER-α) and TP53, predicted the BC response to miR-145 reintroduction.
Results
Breast cancer miR-145 impairs cell proliferation in BC cell lines through apoptosis
We previously showed that miR-145 expression levels were consistently lower in primary human breast tumors and BC cell lines than in normal breast tissues.8 Herein, we extended these findings by measuring miR-145 expression levels in 14 breast tumors and in 10 BC cell lines using quantitative real-time PCR (qRT-PCR). As a result, we found lower expression of miR-145 in breast tumors (13-fold) and BC cell lines (150- to 700-fold) than in normal breast tissues (Supplementary Figures 1a and b).
To study the effects of miR-145 re-expression in BC cell lines, we first carried out an in vitro proliferation assay using tetrazolium staining (MTT assay) on six BC cell lines after transfection with a synthetic miR-145 precursor (Ambion, Austin, TX, USA). Four cell lines (MCF-7, CAMA-1, MCF-10A and T47D) showed a significant reduction in cell number, whereas the remaining two (MDA-MB-231 and MDA-MB-436) exhibited no effect on cell number after miR-145 transfection (Figure 1a). To further confirm the specificity of miR-145 effects, we transfected scalar amounts of this miRNA in MCF-7 and MDA-MB-231 cells and assessed proliferation by MTT assay. MCF-7 cells had a dose-dependent response to miR-145, whereas MDA-MB-231 cells did not have any significant response (Figure 1b). We assayed the transfection efficiency of miR-145 in these cell lines using qRT-PCR, and we confirmed a similar fold increase of miR-145 expression both in MCF-7 and in MDA-MB-231 (Supplementary Figures 3a). Interestingly, these experiments suggested that the anti-proliferative effect of miR-145 was occurring in cells expressing wild-type (wt) TP53 or ER-α.
Figure 1.

miR-145 inhibits breast cancer cell growth. (a) Assessment of cell number by tetrazolium staining (MTT assay) in six BC cell lines. MiR-145 effects on cell counting were measured up to 96 h after miR-145 transfection (time 0). Each time point was expressed as a relative value (fold change, Y axis) to time 0. Values represent the average and bars represent S.D. of three (MCF7 and MDA-MB-231) or two (all other cell lines) independent experiments carried out each time in quadruplicates. For each cell line, TP53 and ER-α protein status are shown according to Neve et al.42 For TP53, − indicates no expression, +/− low expression, + medium expression, ++ high expression, mut mutated and wt wild type. For ER-α, protein levels are indicated as + (positive) or − (negative). (b) Assessment of cell number by tetrazolium staining (MTT assay) of MCF-7 and MDA-MB-231 cell lines transfected with scalar amounts of miR-145 and scrambled (from 100 nM to 3.125 nM). The cells were counted 48 h after miR-145 and scrambled transfection. For each concentration, miR-145 effect was expressed as a relative value (fold change, Y axis) to scrambled control. The symbol asterisk represents a significant difference (P<0.05) compared with scrambled by t-test. Null cells were treated only with lipofectamine.
The anti-proliferative effect that we observed after miR-145 re-expression in BC cell lines can be explained by an increase of cell death (that is, apoptosis) and/or a reduction of the number of cells that are cycling. First, we determined whether miR-145 effect on BC cell number was a result of apoptosis induction. Therefore, we analyzed early apoptotic events, such as exposure of phosphatidylserine outside the cellular membrane, by annexin V and propidium iodide (PI) staining of living cells. After transfection with miR-145, we observed higher levels of early apoptotic cells (annexin V-positive and PI-negative) in CAMA-1 (up to six times) and in MCF-7 (up to three times) compared with scrambled control, whereas, we did not observe any effect in MDA-MB-231 cells (Figure 2a). Furthermore, the analysis of caspase-3/7 activity and PARP cleavage after miR-145 transfection showed an induction of apoptosis in CAMA-1, MCF-7 and MCF-10A, but not in MDA-MB-231 (Figures 2b–e; Supplementary Figures 2a and b).
Figure 2.

miR-145 induces apoptosis in BC cell lines. (a) Annexin V staining of BC cell lines at different time points after miR-145 transfection (time 0). For each time point, we measured the percentage of annexin V-positive and propidium iodide-negative cells (annexin V+/PI−) (Y axis). Values represent averages and bars represent S.D. of two independent experiments. (b) Detection of caspase 3/7 activity by luminescent assay (luminescence, Y axis) 24 and 48 h after miR-145 transfection. Cisplatin treatment (40 μM) was used as a control of apoptosis in MDA-MB-231. Values represent average and bars represent S.D. of three replicates. (c) Western blotting of PARP protein in four BC cell lines 24 and 48 h after miR-145 transfection. Two bands are shown, full-length PARP (116 kDa) and cleaved PARP (89 kDa). The cleaved form is the marker of apoptosis. An increased ratio between cleaved and total PARP indicates an induction of apoptosis. (d) miR-145 effects on caspase 3 and 7 activity were measured by luminescent assay (luminescence, Y axis) in three BC cell lines that were transfected with scalar concentrations of miR-145 (100 nM, 50 nM and 5 nM). (e) Western blotting of full-length and cleaved PARP in three BC cell lines after transfection with different concentrations of miR-145 (100 nM, 50 nM and 5 nM). The asterisk represents a statistically significant difference (P<0.05) compared with scrambled by t-test. Null cells were treated only with lipofectamine.
Then, we determined whether miR-145 effects on BC cell proliferation were a result of changes in cell cycle distribution. Specifically, we assessed cell cycle phases in MCF-10A, MCF-7 and MDA-MB-231 cells by PI staining coupled with FACS analysis 24, 48 and 72 h after transfection with miR-145. As a result, we did not observe any consistent changes in the cell cycle profile of BC cell transfected with miR-145 cells versus cells transfected with scrambled control, except for an increase in the percentage of cells in sub-G1 phase, which is an indirect sign of apoptosis (Supplementary Figure 2c).
Taken together, these results indicated that miR-145 reintroduction reduced the number of BC cells by inducing apoptosis in cells either expressing wt TP53 or ER-α
miR-145 activates TP53 pathway
As we observed that miR-145 inhibited cell growth in TP53 wt cell lines, we hypothesized that this effect may depend on TP53 activation. To verify this, first, we tested whether miR-145 induced the expression of TP53 transcriptional targets, such as p53 upregulated modulator of apoptosis (PUMA)17 and cyclin-dependent kinase inhibitor 1A (CDKN1A/P21), which is an inhibitor of cell cycle progression.18 Therefore, we transfected miR-145 and scrambled control in MCF-7 in TP53 wt MCF-7, and 24 h later, we measured mRNA levels of PUMA and P21. As a result, we observed higher levels (up to three times) of PUMA and P21 mRNA after miR-145 transfection compared with scrambled control in MCF-7. Instead, neither PUMA nor P21 increased after miR-145 transfection in TP53-mutated MDA-MB-231 (Figure 3a). At the same time, we observed an increase in TP53-acetylated levels and PUMA protein expression and a slight decrease in total TP53 and p21 protein expression after miR-145 transfection in MCF-10A (Figure 3b). To confirm a dose-dependent increase in PUMA protein expression after miR-145 expression, we also transfected scalar amounts of this miRNA in MCF-10A (Supplementary Figure 3b). At the same time, to verify whether miR-145 induction of PUMA and P21 mRNA was dependent on TP53, we co-transfected miR-145 and small interfering RNA (siRNA) targeting TP53 in MCF-10A (TP53 wt), and 24 h later, we measured mRNA levels of PUMA and P21. This experiment confirmed the induction of PUMA and P21 mRNA after miR-145 transfection in MCF-10A compared with scrambled control (Figure 3c), and most importantly it showed that this effect was completely impaired if TP53 was silenced during miR-145 reintroduction (Figure 3c).
Figure 3.

miR-145 enhances TP53 transcriptional activity and functions in part through TP53 pathway activation. (a) mRNA levels of PUMA and p21 measured by qRT-PCR 48 h after miR-145 or scrambled transfection in two cell lines, MCF7 (TP53 wt) and MDA-MB-231 (TP53 mutated). Fold changes of P21 and PUMA have been calculated using 2−ΔCt method. GAPDH mRNA levels were used as an internal normalization control. Values represent averages and bars represent S.D. of three independent experiments. Samples treated with miR-145 and scrambled have been normalized to null samples. (b) Western blotting for the indicated proteins in MCF-10 samples 24 h after transfection with miR-145 (100 nM) or scrambled (100 nM). GAPDH was used as loading controls. (c) P21 and PUMA mRNA levels measured by qRT-PCR after transfection of miR-145 and siRNA anti-TP53 or miR-145 alone in MCF-10A. Fold changes of P21 and PUMA have been calculated using 2−ΔCt method. GAPDH mRNA levels were used as an internal normalization control. The cells were first transfected with siRNA anti-TP53, and 36 h later the cells were transfected with miR-145, scrambled or with lipofectamine only (null). (d) Assessment of cell number by MTT assay in MCF-7 48 h after miR-145 transfection in the presence or absence (by siRNA) of TP53 or of PUMA. Y axis represent fold change relative to null cells. Values represent averages and bars represent S.D. of three experiments. At the bottom of the graph, western blottings of PUMA and β-actin are shown. The asterisk represents a statistically significant difference (P<0.05) compared with scrambled by t-test allowing unequal variances between samples. Null cells were treated only with lipofectamine.
To verify that BC cell growth inhibition after miR-145 transfection was dependent on TP53-mediated transactivation of PUMA, we tested the hypothesis whether inhibition of TP53 or PUMA expression by siRNA was able to reduce the anti-growth effect of miR-145. Therefore, we co-transfected MCF-7 cells with miR-145 and siRNAs targeting TP53 or PUMA, or with miR-145 alone. Twenty-four hours later, we observed a 20% increase in the number of cells when we transfected miR-145 together with siRNA against PUMA or against TP53 versus when we transfected miR-145 alone (Figure 3d).
Furthermore, Sachdeva et al.19 recently reported that TP53 can activate miR-145 transcription in a colon cancer cell model. To confirm that TP53 induces miR-145 expression in our BC model as well, we treated TP53 wt MCF-10A and TP53-mutated MDA-MB-231 cells with two different TP53-activating drugs, that is, Adriamycin (1.72 μM) and Nutlin-3 (10 μM), and 24 h later, we measured miR-145 expression levels by qRT-PCR. We also measured miR-34c expression levels, and we used it as positive control, because He et al.20 previously proved that TP53 transactivates miR-34c transcription. After TP53 activation with both Adriamycin or Nutlin-3 treatment, we observed an increase in miR-145 and miR-34c levels in TP53 wt MCF-10A cells, whereas neither miR-34c nor miR-145 increased in TP53-mutated MDA–MB-2321 cells (Figure 4).
Figure 4.
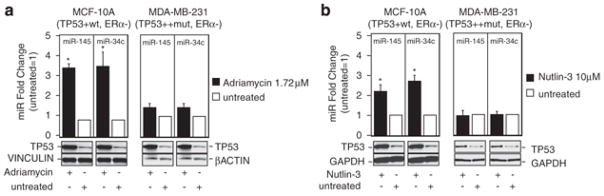
TP53 induces miR-145 expression levels. (a and b) miR-145 and miR-34c levels were measured by qRT-PCR 24 h after treatment with Adriamycin and Nutlin-3, respectively. Fold changes of miR-145 and miR-34c have been calculated using 2−ΔCt method. U6 mRNA levels were used as an internal normalization control. Values represent average and bars represent S.D. of three independent experiments. At the bottom of the graph, western blotting of TP53 protein levels is shown to prove induction of TP53 protein levels after Adriamycin and Nutlin-3 treatment. GAPDH and β-actin protein levels were used as loading controls. The asterisk represents a significant difference (P<0.05) compared with untreated samples by t-test.
Taken together, the miR-145-dependent activation of TP53 and PUMA and the TP53-dependent transactivation of miR-145 expression showed the existence of a death-promoting loop between miR-145 and TP53 in BC cells.
miR-145 directly targets the coding sequence of estrogen receptor α
As the response of BC cell lines to miR-145 overexpression correlated not only with TP53 status but also with ER-α (Figure 1), we hypothesized that miR-145 could exert its anti-proliferative effect through the regulation of ER-α protein expression as well. We first determined whether miR-145 decreased ER-α protein expression; therefore, we transfected ER-α-positive MCF-7 cells with miR-145 and measured ER-α protein expression levels by western blotting. As a result, we detected consistent reduction in ER-α protein expression 48 h after miR-145 transfection, which was also evident after transfection of scalar amounts of miR-145 (75% lower than in scrambled control) (Figure 5a–c and Supplementary Figure 4). We also evaluated cyclin D1 protein expression, a downstream target of ER-α, and we detected a parallel reduction in cyclin D1 levels (Figure 5a–b; Supplementary Figure 4). The decrease in ER-α and cyclin D1 protein levels was also confirmed by reverse-phase proteomic array (data not shown).
Figure 5.

miR-145 represses ER-α protein in MCF-7 by direct binding within the ESR1-coding sequence. (a) Western blotting analysis of ER-α, cyclin D1, and phosphorylated ER-α in serine 118 (pS118) protein levels in MCF7 48 h after miR-145 transfection. GAPDH was used as loading control. (b) ER-α and cyclin D1 protein levels were measured by western blotting 48 h after miR-145 (100 nM) or scrambled transfection (100 nM). Values represent average and bars represent S.D. of three independent experiments, one of which is shown in panel a. GAPDH was used as internal normalization control. (c) ER-α protein levels were measured 48 h after transfection of scalar doses of miR-145 (100 nM, 50 nM and 5 nM). Vinculin was used as loading control. (d) Top part of the panel represents the isoform1 of ESR1 mRNA. The miR-145-predicted target sites are shown as triangles (white triangles if they are located in the 3′ UTR, black if they are located in the CDS), and they are labeled from 1 to 5 starting from the 5′ of the mRNA. The middle cartoon represents the luciferase construct that has been used for in vitro assay. The bottom panel represents the results of in vitro experiments with luciferase constructs that contain miR-145-predicted targets. Mutated constructs have been generated by deletion of seven nucleotides in the 3′ of the target sites. Values represent averages and bars represent S.D. of three independent experiments. Luciferase activity was normalized to that of scrambled control. The asterisk represents a statistically significant difference (P<0.05) compared with scrambled by t-test. Null cells were treated only with lipofectamine.
Next, we sought to clarify how miR-145 reduced ER-α protein expression in BC cells. We tested whether miR-145 regulated ER-α expression at the post-transcriptional level by directly targeting ER-α mRNA (ESR-1).2 Hence, we interrogated miRNA target prediction programs for miR-145 target sites in the 3′UTR of ESR1 mRNA (Pictar,21 Miranda,22 Diana-microT23 and TargetScan24), and we also interrogated the RNA22 prediction program25 for miR-145 target sites in the full-length sequence of ESR1 mRNA. Five putative miR-145-binding sites were predicted, three located inside the coding sequence (CDS) of ESR1 and two in the 3′UTR of ESR1 (Figure 5d, Supplementary Table 1). Therefore, we cloned each of them into pGL3 luciferase reporter vectors downstream of the luciferase reporter gene (Figure 5d). In parallel, we generated pGL3-mutated constructs by deleting the predicted target sites of miR-145 in ESR1 (Supplementary Table 1). Two out of the five sites, named target #2 and #3 in Figure 5d, caused a significant reduction in luciferase activity in MCF-7 after miR-145 re-expression compared with scrambled control; at the same time, these two sites did not cause a significant decrease in luciferase activity when their mutated version was used (Figure 5d). Both of these target sites were located in the CDS of ESR1 and have a 5′ seed region of perfect complementarity (Supplementary Table 1).
Previous studies showed that miRNAs could regulate mRNA expression by inhibiting mRNA translation or by inducing mRNA degradation. Moreover, authors proved that the luciferase assay that we used (Figure 5d) is not useful to discriminate how miRNAs repress their targets (that is, inhibition of mRNA translation or degradation of mRNA).26 Therefore, to understand how miR-145 decreases ER-α protein expression, we measured ESR1 mRNA expression levels in the same samples in which we previously observed a reduction in ER-α protein expression levels after miR-145 transfection (Figure 5a). As a result, we observed an increase in ESR1 mRNA expression after miR-145 transfection (Figure 6a). Although all of the evidences that we have collected so far suggested that miR-145 directly targeted ESR1 only by inhibition of protein translation, still we sought to exclude the possibility that miR-145 could indirectly reduce ER-α protein expression by reducing ER-α protein stability.
Figure 6.
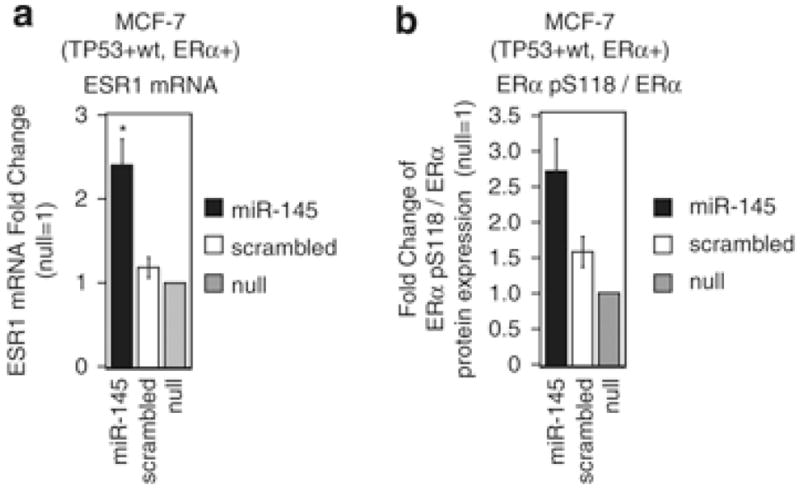
miR-145 does not decrease ESR1 mRNA or ER-α protein stability. (a) ESR1 mRNA and (b) ER-α pS118/ER-α protein levels were measured 48 h after miR-145 transfection. Values represent average and bars represent S.D. of three independent experiments. Fold changes of ESR1 mRNA levels were calculated using 2−ΔCt method. GAPDH mRNA levels were used as an internal normalization control. Fold changes of ER-α pS118/ER-α ratios were calculated by western blotting (see Figure 5a). The asterisk represents a significant difference (P<0.05) compared with scrambled by t-test. Null cells were treated only with lipofectamine.
To measure ER-α stability, we used western blotting to quantify the phosphorylation of ER-α at serine 118 (ER-α-pS118), which is known to inhibit proteosomal degradation of ER-α,27 and we expressed stability as the ratio of ER-α-pS118 to total ER-α. The ER-α-pS118/total ER-α ratio was even higher in MCF7 cells after treatment with miR-145 than with scrambled control (Figure 6b). Therefore, we ruled out an indirect effect of miR-145 on ER-α protein stability. In conclusion, these data indicated that miR-145 inhibited ER-α protein expression exclusively by repressing ESR1 mRNA translation through interaction with two target sites within the ESR1 CDS.
Discussion
Our findings indicated that miR-145 inhibited proliferation and induced apoptosis of BC cells through activation of the TP53 pathway and reduction in ER-α expression. To the best of our knowledge, we are the first to describe a tumor-suppressor function of miR-145 in BC cells and to directly link miR-145 with ER-α and TP53, both of which have important roles in breast tumor biology.28, 29
Previous studies showed the anti-proliferative properties of miR-145 in colon cancer cell models and reported various targets. For example, miR-145 could inhibit the growth of HCT-116 colon cancer cells through the targeting of IRS-1.14 As HCT116 colon cancer cells carry a wt TP53 gene, it is possible that the activation of TP53 could have also been involved in the observed miR-145 effect in these cells. However, apoptosis may not be the only response to miR-145 upregulation. For example, it has been recently shown that miR-145 is low in self-renewing human embryonic stem cells and increases during differentiation. In these cells, miR-145 may facilitate differentiation by repressing the core pluripotency factors, OCT4, SOX2 and KLF4, thereby silencing the self-renewal program.16 Therefore, it seems plausible that miR-145 function may depend on different target genes that are operating in specific cell types. At the same time, a general mechanism appears to be the activation of TP53, and this mechanism would lead to apoptosis in wt TP53 cancer cells. Hence, similarly to TP53 loss, the loss of miR-145 expression in most human tumors and cancer cell lines may provide a favorable environment for cell survival.
Our results indicated that miR-145 activated TP53 toward a prevalent apoptosis response more than toward a cell cycle arrest response. In fact, we observed an almost unique increase in PUMA protein levels instead of p21 protein levels after miR-145 transfection. Further study is needed to determine how miR-145 activates TP53, in other words, identify the miR-145 target(s) responsible for TP53 activation.
Besides showing that miR-145 activated TP53 pathway, we also found that TP53 itself induced miR-145 transcription in BC cells. It was previously reported that TP53 activates miR-145 transcription during starvation in a colon cancer cell line.19 Here, we showed that two drugs (that is, the MDM-2 inhibitor Nutlin-3 and the DNA-damage inducer Adriamycin) that activate the TP53 pathway increased miR-145 expression, and we also showed that this effect was missing in TP53-mutated MDA-MB-231 BC cell line. Thus, we conclude that a positive regulatory loop between miR-145 and TP53 exists, which leads to apoptotic death in cancer cells carrying a wt TP53 gene. The existence of regulatory loops between miRNAs and transcription factors was previously reported.30 For example, E2F1, E2F2 and E2F3 directly bind the promoter of the miR-17-92 cluster, activating its transcription, and miR-20a, a member of the miR-17-92 cluster, modulates the translation of the E2F2 and E2F3 mRNAs,31 and this negative feed back regulatory loop probably protects cells from apoptosis caused by excessive E2F expression. Furthermore, transcription of miR-34 family is regulated by TP5332 and miR-34a itself has been shown to regulate TP53 activity.33
Previous studies described a direct correlation of miR-145 with ER-α-positive tumor status11, 34 and an inverse correlation of it with the Ki-67 proliferation index.8 The inverse correlation of miR-145 with Ki-67 agrees with the anti-proliferative effect of miR-145 that we describe herein, whereas the direct correlation of miR-145 with ER-α expression is unexpected and more difficult to explain. It is possible that, as the expression of miR-145 was associated with cell differentiation,16 this direct correlation may be linked to the higher grade of differentiation associated with ER-α-positive BCs. Different groups have identified other miRNAs that directly target ESR1 (for example, miR-221, miR-222 and miR-206).35, 36 Kondo et al. 37 described an inverse correlation between the levels of miR-206 and the ER-α protein status of the tumors. At the same time, authors described a direct correlation of miR-221/miR-222 with ER-α protein status of the tumors.34 The direct correlations of miR-145 and miR-221/miR-222 with ER-α status are puzzling and contradictory and need further work.
It is noted that miR-145 is located in the 5q33.1 region of human genome and clusters with miR-143. In colon cancer cell lines, Akao et al.38 showed that both miR-143 and miR-145 impaired cell growth. Moreover, expression of both miR-145 and miR-143 is commonly reduced in several types of tumors.8, 39 Using Rapid Amplification of cDNA Ends (RACE), we were able to clone a single cDNA fragment containing both of these miRNAs (Supplementary Figure 5a) (GenBank accession number GQ292874). However, in the BC cell lines that we used in this study, expression of miR-143 was much lower than that of miR-145 (10- to 100-fold) (data not shown), making difficult to obtain consistent results regarding miR-143 regulation by TP53. Nevertheless, the common reduction in miR-143 and miR-145 expression in BC tumor samples and the common transcription of the two microRNAs (Supplementary Figure 5b) both suggest that miR-143 may have a tumor-suppressor function in BC, as miR-145 does.
In conclusion, miR-145 tumor-suppressor effect occurs through promotion of apoptosis in both ER-α-positive and wt TP53-expressing BC cells. miR-145 expression activates TP53 and suppresses ER-α, whereas TP53 activates miR-145, representing a positive regulatory death loop that may open the avenue for miR-145 re-expression therapy in particular in the specific group of BC patients carrying ER-α-positive and/or wt TP53 tumors.
Materials and Methods
Cell lines and patient samples
MCF-7, CAMA-1, MDA-MB-231, MCF-10A, T47D and MDA-MB-436 were obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and cultured according to the ATCC protocols. Normal breast tissue and breast tumor samples were collected in the University of Ferrara and processed as described previously.8
miRNA precursor molecules and siRNAs
Synthetic miRNA precursor molecules and negative control #1 (scrambled) were purchased form Ambion. These synthetic oligos were dissolved in nuclease-free water to a stock concentration of 50 μM. siRNAs against TP53 and PUMA were purchased from Dharmacon (Chicago, IL, USA). If not otherwise specified, miR-145 and scrambled were used at 100 nM concentration.
Hormones and drugs
Nutlin-3 was purchased from Cayman Chemical (Ann Arbor, MI, USA) and dissolved in dimethyl sulfoxide to a stock solution of 2 mM. Adriamycin was kindly provided by Dr. Waldemar Priebe (The University of Texas MD Anderson Cancer Center, Houston, TX, USA) and resuspended in water to a final concentration of 0.5 μM.
RNA isolation and qRT-PCR
Purification of total RNA using TRIzol reagent (Invitrogen, Carlsband, CA, USA) was carried out as described previously.8 miR-145 and RNU6B probes (Applied Biosystems, Foster City, CA, USA) were used for mature miRNA quantification according the manufacturer’s protocol. Five hundred nanograms of total RNA was used for the retrotranscription step. The 2−ΔCt method was used to calculate the relative abundance of miR-145 compared with RNU6B expression.40 To generate total cDNAs for protein-coding gene expression, 1 μg of total RNA was retrotranscribed using SuperScript II (Invitrogen) with random hexamers according to the manufacturer’s protocol. Real-time PCR analysis of the samples was carried out with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). The list of primers that we used herein is shown in Supplementary Table 1.
Protein isolation, western blotting and antibodies
Breast cancer cell were collected from 6-well plates using trypsin-ethylenediaminetetraacetic acid (EDTA) (Mediatech, Manassas, VA, USA) and dissolved in NP40 lysis buffer (0.5% NP40, 250 mM NaCl, 50 mM Hepes, 5 mM ethylenediaminetetraacetic acid, 0.5 mM egtazic acid) freshly supplemented with a complete protease inhibitor and phosphatase inhibitor cocktails 1 and 2 (Roche, Indianapolis, IN, USA). Proteins were purified as described previously.41 The complete list of antibodies is described in Supplementary Table 3.
In vitro proliferation assay
Breast cancer cells were plated in 96-well plates at a density of 10 000 cells/well. Transfection of the cells with either an miR-145 precursor molecule or a negative-control molecule (scrambled) at a final concentration of 100 nM was carried out using a Lipofectamine 2000 protocol (Invitrogen). Cells were also treated with Lipofectamine only (null cells). Both treatment and controls were performed in quadruplicate each time. Four hours after transfection (time 0), the absorbance of cells was estimated using a colorimetric MTT assay (CellTiter 96 AQueous One Solution; Promega, Madison, WI, USA), and this procedure was repeated every 24 h for 4 consecutive days.
Apoptotic assays
The number of early apoptotic of BC cells after miR-145 transfection was determined using an annexin V staining kit (BD Pharmingen, San Jose, CA, USA). Every 24 h, the cells were collected from 6-well plates, and annexin V staining of the cells was carried out according to the manufacturer’s protocol. The cells were counterstained with PI and immediately read using a FACSCalibur system (BD, Franklin Lakes, NJ, USA). Only annexin V-positive and PI-negative cells were counted at each time point. For further confirmation of apoptosis, MCF7 cells were analyzed using the caspase-3/7 assay according to the manufacturer’s protocol (Promega).
Cell cycle analysis
MCF7, MCF10A and MDA-MB-231 BC cell lines were seeded in 6-well plates and transfected with miR-145, scrambled control and with lipofectamine only (null). Every 24 h, the cells were collected from 6-well plate using trypsin-EDTA acid and washed twice with cold PBS. The cells were finally stained with PI solution containing RNase enzyme at 37°C for 30 min. The stained cells were read using a FACSCalibur system (BD), and the gates for sub-G1, G1, S and G2 were arbitrary chosen by the operator.
miR-145–TP53 loop identification
MCF7 cells were seeded into 24-well plates at a concentration of 75 × 103 cells/well in an antibiotic-free medium. Transfection of the cells was carried out with an miR-145 precursor molecule (Ambion), a negative-control molecule (scrambled) (Ambion), siRNA-PUMA (Dharmacon) and siRNA-p53 (Dharmacon) at a final concentration of 100 nM using Lipofectamine 2000 reagent (Invitrogen). Forty-eight hours after transfection, the cells were collected by trypsin-EDTA and were counted using a hemocytometer chamber.
Luciferase assay
Primer pairs were designed to amplify a region of about 200–300 bp around every predicted miR-145 target site within the ESR1 CDS and 3′UTR (Supplementary Table 2). The five amplicons (three in the CDS, two in the 3′UTR) were first mutated by sequence specific mutagenesis (Stratagene, La Jolla, CA, USA), and then both wt and mutated (mut) amplicons were cloned in pGL3 vector as described previously.41 MCF7 cells (0.2 × 106) were plated on 24-well plates; on the day after, the cells were transfected with miR-145 or scrambled control (10 nM), with each pGL3 ESR1 construct (wt and mut) (0.4 μg/well), and with pRLTK vector (0.05 μg/well). Twenty-four hours afterward, the cells were rinsed with phosphate-buffered saline and dissolved in 1 × Passive Lysis Buffer (Promega). Subsequently, the luciferase activity was measured using dual luciferase kit (Promega) using a Veritas luminometer (Turner Biosystems, Sunnyvale, CA, USA). Each combination of pGL3 (wt and mut) and pRLTK was tested in quadruplicate in three independent experiments.
Statistical analysis
The t-test was used to compare mean values in groups of samples (for example, miRNA expression data, number of proliferating cells). All reported P-values were calculated for groups with unequal variance using the Excel software program (Microsoft, Redmond, WA, USA). P<0.05 was considered significant.
Supplementary Material
Abbreviations
- miRNA
microRNA
- mRNA
messenger RNA
- 3′UTR
3′ untranslated region
- BC
breast cancer
- ER-α
estrogen receptor-α
- PCR
polymerase chain reaction
- wt
wild-type
- PI
propidium iodide
- PARP
poly(ADP-ribose) polymerase
- PUMA
p53 upregulated modulator of apoptosis
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- siRNA
small interfering RNA
- ESR1
estrogen receptor 1
- CDS
coding sequence
References
- 1.Jemal A, Ward E, Thun MJ. Recent trends in breast cancer incidence rates by age and tumor characteristics among US women. Breast Cancer Res. 2007;9:R28. doi: 10.1186/bcr1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 4.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 5.Voorhoeve PM, Agami R. Classifying microRNAs in cancer: the good, the bad and the ugly. Biochim Biophys Acta. 2007;1775:274–282. doi: 10.1016/j.bbcan.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 8.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 9.Michael MZ, O’Connor SM, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 10.Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer cell. 2006;9:189–198. doi: 10.1016/j.ccr.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 11.Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, et al. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105:13021–13026. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Tang S, Le SY, Lu R, Rader JS, Meyers C, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS ONE. 2008;3:e2557. doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akao Y, Nakagawa Y, Naoe T. MicroRNAs 143 and 145 are possible common onco-microRNAs in human cancers. Oncol Rep. 2006;16:845–850. [PubMed] [Google Scholar]
- 14.Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, deAngelis T, Baserga R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. JBiol Chem. 2007;282:32582–32590. doi: 10.1074/jbc.M702806200. [DOI] [PubMed] [Google Scholar]
- 15.Shen WF, Hu YL, Uttarwar L, Passegue E, Largman C. MicroRNA-126 regulates HOXA9 by binding to the homeobox. MolCell Biol. 2008;28:4609–4619. doi: 10.1128/MCB.01652-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 17.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Jenkins CW, Nichols MA, Xiong Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene. 1994;9:2261–2268. [PubMed] [Google Scholar]
- 19.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 22.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Devel. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 25.Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell. 2006;126:1203–1217. doi: 10.1016/j.cell.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 26.Kong YW, Cannell IG, de Moor CH, Hill K, Garside PG, Hamilton TL, et al. The mechanism of micro-RNA-mediated translation repression is determined by the promoter of the target gene. Proc Natl Acad Sci USA. 2008;105:8866–8871. doi: 10.1073/pnas.0800650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grisouard J, Medunjanin S, Hermani A, Shukla A, Mayer D. Glycogen synthase kinase-3 protects estrogen receptor alpha from proteasomal degradation and is required for full transcriptional activity of the receptor. Mol Endocrinol. 2007;21:2427–2439. doi: 10.1210/me.2007-0129. [DOI] [PubMed] [Google Scholar]
- 28.Jordan VC, O’Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol. 2007;25:5815–5824. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- 29.Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 30.Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282:2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 32.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network – another piece in the tumour-suppression puzzle. Nat Rev. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, Scott GK, et al. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer. 2006;5:24. doi: 10.1186/1476-4598-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams BD, Furneaux H, White BA. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007;21:1132–1147. doi: 10.1210/me.2007-0022. [DOI] [PubMed] [Google Scholar]
- 36.Zhao JJ, Lin J, Yang H, Kong W, He L, Ma X, et al. MicroRNA-221/222 negatively regulates estrogen receptor alpha and is associated with tamoxifen resistance in breast cancer. J Biol Chem. 2008;283:31079–31086. doi: 10.1074/jbc.M806041200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Kondo N, Toyama T, Sugiura H, Fujii Y, Yamashita H. miR-206 Expression is down-regulated in estrogen receptor alpha-positive human breast cancer. Cancer Res. 2008;68:5004–5008. doi: 10.1158/0008-5472.CAN-08-0180. [DOI] [PubMed] [Google Scholar]
- 38.Akao Y, Nakagawa Y, Naoe T. MicroRNA-143 and -145 in colon cancer. DNA Cell Biol. 2007;26:311–320. doi: 10.1089/dna.2006.0550. [DOI] [PubMed] [Google Scholar]
- 39.Michael MZ, SMOC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 40.Schmittgen TD, Lee EJ, Jiang J. High-throughput real-time PCR. Methods Mol Biol. 2008;429:89–98. doi: 10.1007/978-1-60327-040-3_7. [DOI] [PubMed] [Google Scholar]
- 41.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.