

Abstract
AIM: To determine hepatic expression of apurinic/apyrimidinic endonuclease 1 (APE-1) and 8-hydroxydeoxyguanosine (8-OHdG) in patients with chronic hepatitis B and C.
METHODS: Liver biopsies were obtained from 27 patients with chronic hepatitis B virus (HBV), 30 with chronic hepatitis C virus (HCV), 6 with autoimmune hepatitis (AIH), and 6 with primary biliary cirrhosis (PBC). Normal liver tissue was obtained from surgical resection specimens of four patients. Hepatic APE-1 protein and mRNA expression were assayed by Western blot and by real-time polymerase chain reaction, respectively. Hepatocellular APE-1 and 8-OHdG expression were determined by immunohistochemistry.
RESULTS: The staining intensity of hepatocellular nuclear APE-1 was lower in the HBV group than in the other groups (P < 0.05). Hepatic APE-1 protein levels were reduced in the HBV group relative to the other groups. Hepatic APE-1 mRNA levels were also lower in the HBV group. The proportion of hepatocytes with 8-OHdG-positive nuclei was increased in the HCV, AIH and PBC groups (P < 0.05), but not in the HBV group. Hepatocellular nuclear APE-1 levels were positively correlated with hepatocellular 8-OHdG levels in both the HBV and HCV groups (HBV, r = 0.34, P < 0.05; HCV, r = 0.54, P < 0.01).
CONCLUSION: An imbalance between oxidative DNA damage and APE-1 expression may contribute to hepatocarcinogenesis in chronic viral hepatitis.
Keywords: Apurinic/apyrimidinic endonuclease 1, 8-hydroxydeoxyguanosine, Oxidative stress, Viral hepatitis
INTRODUCTION
Reactive oxygen species (ROS) generated by chronic inflammation are closely linked to hepatocellular oxidative DNA damage and may be involved in hepatocarcinogenesis[1]. During oxidative stress, ROS attack DNA leading to oxidative DNA damage such as 8-hydroxydeoxyguanosine (8-OHdG). Cellular DNA damage can lead to mutation induction and subsequent carcinogenesis if DNA repair processes are not completely effective[2]. Recently, enhanced oxidative DNA damage has been reported in the livers of patients with chronic hepatitis B virus (HBV) or chronic hepatitis C virus (HCV) infection[3,4]. This raises the possibility that oxidative DNA damage participates in the pathogenesis of hepatocarcinogenesis during chronic HBV or HCV infection.
Oxidative DNA damage can generate apurinic/apyrimidinic (AP) sites resulting from loss of bases in DNA, either spontaneously through free radical attack or by the action of DNA glycosylases that remove bases modified by ROS. AP sites must be repaired efficiently because of their potential mutagenicity; AP sites result in base substitution mutations and loss of genetic integrity[5]. Human AP endonuclease 1 (APE-1) (also designated reduction-oxidation factor-1) is a key enzyme of DNA repair that is distributed in a wide range of normal tissues including the liver[4]. This enzyme catalyzes the initial step in AP site repair by rapidly introducing DNA strand breaks on the 5’ side of the AP site. APE-1 is also known to be a potent reduction-oxidation (redox) factor, regulating the DNA-binding activity of several transcription factors involved in cell differentiation, proliferation and apoptosis [e.g. activator protein-1, nuclear factor-κB (NF-κB) and p53], independently of its DNA repair function[6]. ROS mediate and enhance APE-1 expression and activity, while APE-1 controls intracellular ROS production by negatively regulating the activity of the ROS-related guanosine triphosphate hydrolase.
In view of its role in DNA repair and redox regulation, APE-1 is likely to protect DNA and transcription factors from oxidative damage and to repair damaged DNA in hepatocytes under conditions of enhanced oxidative stress in chronic viral hepatitis. Thus APE-1 may play an important role in the prevention of hepatocarcinogenesis. The aim of the present study was to compare hepatic APE-1 and 8-OHdG expression among patients with chronic HBV or HCV infection, patients with autoimmune liver diseases and normal controls, in whom hepatocellular carcinoma (HCC) is rarely encountered. This is a first step toward understanding the possible role of APE-1 in the pathogenesis of hepatocarcinogenesis in chronic viral hepatitis.
MATERIALS AND METHODS
Patients
This study included 27 consecutive patients with chronic HBV infection, 30 with chronic HCV infection, 6 with autoimmune hepatitis (AIH), 6 with primary biliary cirrhosis (PBC), who were evaluated and underwent liver biopsies at the University Hospital of Hamamatsu University School of Medicine. Inclusion of the 69 patients in this study was dependent upon the availability of sufficient biopsy material for histological and immunohistochemical assessment. Diagnosis of chronic HBV or HCV infection was based on elevated serum transaminase levels over at least 6 mo in the presence of HBV surface antigen or anti-HCV antibodies in serum. The sera of all 27 HBV and 30 HCV patients were HBV DNA-positive or HCV RNA-positive by polymerase chain reaction (PCR), respectively. Diagnosis of AIH or PBC was based on clinical, laboratory and histological findings. The sera of all six AIH patients were positive for anti-nuclear antibodies, and all 6 PBC patients had anti-mitochondrial antibodies in their serum. HBV surface antigen or anti-HCV antibodies were not detected in the sera of AIH and PBC patients. None of the patients received any specific treatment prior to liver biopsy. Normal liver tissue was obtained from surgical resection specimens of four patients who underwent hepatectomy for liver metastasis of colon cancer. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki (6th revision, 2008) as reflected in a priori approval by the institution’s human research committee.
Liver biopsies
Liver tissue was obtained by percutaneous needle biopsy in all 69 cases. All liver biopsy specimens were fixed in formalin and paraffin-embedded for hematoxylin and eosin, and Azan-Mallory staining, and for subsequent immunohistochemical analysis. A portion of each sample from 50 patients (HBV, 21; HCV, 23; AIH, 3; PBC, 3) was immediately frozen in liquid nitrogen and stored at -80 °C until use for protein or RNA extraction. Histological characteristics of chronic viral hepatitis and AIH were evaluated using the standard criteria proposed by Desmet[7]. PBC liver specimens were staged according to the method proposed by Ludwig et al[8].
Immunohistochemical analysis
Immunohistochemical detection of APE-1 protein and 8-OHdG were performed using a streptavidin-biotin complex peroxidase kit according to the manufacturer’s instructions (Nichirei, Tokyo, Japan). Briefly, deparaffinized sections (4 mm thick) were subjected to autoclave heating treatment in 10 mmol/L citrate buffer (pH = 6.0) for antigen retrieval. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol. The sections were treated with 10% normal goat serum to block nonspecific binding of antibodies, and incubated with rabbit polyclonal anti-APE-1 (Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:100) or mouse monoclonal anti-8-OHdG (Santa Cruz Biotechnology; dilution 1:50) antibodies at 4 °C overnight. After the incubation, biotinylated secondary antibody, and streptavidin conjugated with peroxidase were added sequentially, followed by color development with 3,3’-diaminobenzidine tetrahydrochloride and hydrogen peroxide. Nuclear staining was carried out with Mayer’s hematoxylin. The specificity of APE-1 staining was confirmed by preincubating the antibody with an excess of specific antigen peptide. As shown in Figure 1, the intensity of hepatocytic nuclear APE-1 immunoreactivity was graded into four categories (none, faint, weak, or strong staining). This evaluation assessed the mean signal intensity of the entire slide.
Figure 1.
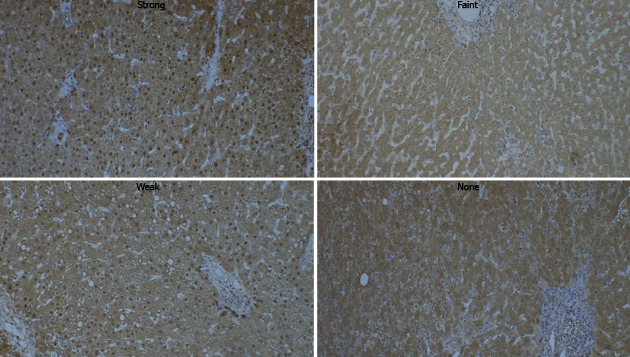
Immunohistochemical detection of hepatic apurinic/apyrimidinic endonuclease 1 protein in patients with chronic liver disease. The intensity of hepatocytic nuclear apurinic/apyrimidinic endonuclease 1 immunoreactivity was classified into four grades.
For semi-quantitative assessment of 8-OHdG expression, the number of nuclei positive for 8-OHdG among 300 hepatocytes was counted in each section, and the percentage of positive cells was calculated.
Western blot analysis
Frozen liver tissue from each patient (HBV, 16; HCV, 18; AIH, 3; PBC, 3; normal, 4) was homogenized in a radioimmunoreactive protein extraction assay lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, United States) containing a complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The lysates were diluted 1:1 with ×2 Laemmli sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Protein concentration of samples in SDS sample buffer was determined using Peterson’s modification of the micro-Lowry method. The protein extracts were subjected to SDS-PAGE on 12.5% polyacrylamide gels. The resolved proteins were electrophoretically transferred to polyvinylidene difluoride membranes. The blots were blocked overnight at 4 °C with TBS-T buffer (20 mmol/L Tris-HCl, pH 7.6, 137 mmol/L NaCl, 0.5% Tween 20) containing 10% nonfat dry milk, and were probed with rabbit polyclonal anti-APE-1 (1:100 dilution) or anti-glyceraldehyde-3-phosphate dehydrogenase (G3PDH) antibodies (Trevigen, Gaithersburg, MD, United States). Bound primary antibody was detected using anti-rabbit IgG horseradish peroxidase-conjugate (Santa Cruz Biotechnology; 1:5000 dilution) and the blots were visualized by chemiluminescence. Band signal intensities were determined with a densitometer. The levels of APE-1 were normalized to those of G3PDH.
Quantitative real-time PCR analysis
Total RNA was extracted from liver biopsy samples (HBV, 5; HCV, 5) using TRIzol reagent (Invitrogen, Carlsbad, CA, United States), according to the manufacturer’s instructions. A total of 1 μg of RNA was reverse-transcribed per reaction using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany). SYBR green-based quantitative real-time PCR was performed in a LightCycler (Roche Diagnostics GmbH, Mannheim, Germany) and the optimal reaction conditions were determined for each template. The cDNA for the human APE-1 gene was amplified with the forward primer, 5’-AGTTTCTTACGGCATAGGCG-3’ and the reverse primer 5’-ACTTCGAAAGGCTTCATCC-3’ to generate a 161 bp product. β-actin cDNA was amplified with the forward primer 5’-CAGGGCGTGATGGTGGGCATG-3’ and the reverse primer 5’-GGCGACGTAGCACAGCTTCTCC-3’ (540 bp product) as an internal control.
Statistical analysis
Data are presented as mean ± SE unless otherwise stated. Comparisons of mean values between groups were performed using Mann-Whitney U tests. Comparisons between the frequencies of observations were performed using Fisher’s exact test. The correlations between various parameters were calculated by univariate linear regression analysis and expressed as Pearson’s correlation coefficients. A P value of < 0.05 was considered to indicate significance.
RESULTS
Clinical characteristics of patients
The clinical characteristics of 27 patients with chronic HBV infection, 30 with chronic HCV infection, 6 with AIH and 6 with PBC are shown in Table 1. There were no significant differences in gender or mean age among the chronic liver disease groups. Serum alanine aminotransferase (ALT), aspartate aminotransferase and γ glutamyl transpeptidase levels were significantly higher in the HBV group than in the HCV group (P < 0.001), but there was no significant difference in the degree of hepatic necroinflammation between these groups.
Table 1.
Clinical characteristics in patients with chronic liver disease
| Characteristics | HCV (n = 30) | HBV (n = 27) | AIH (n = 6) | PBC (n = 6) | HCV vs HBV |
| Gender (male/female) | 18/12 | 18/9 | 0/6 | 1/5 | NS |
| Age (yr) | 56.8 ± 9.1 (39-73) | 52.6 ± 12.9 (22-69) | 58.5 ± 12.2 (47-74) | 56.3 ± 7.0 (44-64) | NS |
| Platelet count (× 104/mm3) | 15.7 ± 5.9 (9.3-32.3) | 14.6 ± 6.2 (8.2-28.4) | 14.9 ± 7.6 (12.7-26.0) | 16.0 ± 4.4 (13.4-23.6) | NS |
| AST (IU/L) | 55 ± 36 (13-191) | 135 ± 84 (59-341) | 118 ± 32 (47-120) | 32 ± 6 (21-35) | P < 0.001 |
| ALT (IU/L) | 71 ± 60 (16-307) | 180 ± 153 (79-554) | 138 ± 37 (54-144) | 45 ± 18 (11-53) | P < 0.001 |
| γGTP (IU/L) | 45 ± 36 (14-175) | 84 ± 43 (41-135) | 147 ± 53 (35-228) | 114 ± 63 (23-179) | P < 0.01 |
| Viral load | > 500 KIU/mL (n = 16) | ||||
| 100-500 KIU/mL (n = 4) | > 7.6 log copies/mL (n = 11) | ||||
| < 100 KIU/mL (n = 10) | ≤ 7.6 log copies/mL (n = 16) | ||||
| Liver histopathology | Ludwig’s stage | ||||
| Necroinflammation (mild/moderate/severe) | 5/20/5 | 7/15/5 | 2/3/1 | Portal stage (n = 2) | NS |
| Periportal stage (n = 4) | |||||
| Fibrosis (none/mild/moderate/severe/cirrhosis) | 0/17/8/4/1 | 3/10/8/4/2 | 1/4/1/0/0 | Septal stage (n = 0) | |
| Cirrhotic stage (n = 0) | NS |
Values represent mean ± SE unless otherwise stated. HCV: Hepatitis C virus; HBV: Hepatitis B virus; AIH: Autoimmune hepatitis; PBC: Primary biliary cirrhosis; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; γGTP: γ glutamyl transpeptidase.
Hepatic APE-1 protein and mRNA expression
In liver specimens from patients with chronic liver disease, positive staining for APE-1 protein was detected preferentially in hepatocytes and bile duct cells, and occasionally in sinusoidal cells and portal inflammatory cells. In hepatocytes, the nucleus was preferentially stained in a mottled pattern. When nuclear staining was evaluated, the percentage of positive hepatocellular nuclei throughout the slide was fairly constant at 80%-100%, regardless of the etiology of chronic liver disease, while the staining intensity was lower in HBV than in HCV, AIH/PBC, or normal samples (Figure 2A). With regard to hepatocellular cytoplasmic staining, the staining pattern was uniform within individual cells, and homogeneous throughout slides in most specimens; no significant differences in staining intensity were observed among the four groups of patients. In addition, the staining intensity of hepatocytic nuclear APE-1 protein was positively correlated with serum ALT levels in HBV (r = 0.42, P < 0.05) and HCV (r = 0.38, P < 0.05) groups (Figure 2B); there were no other correlations between laboratory values and histological findings.
Figure 2.
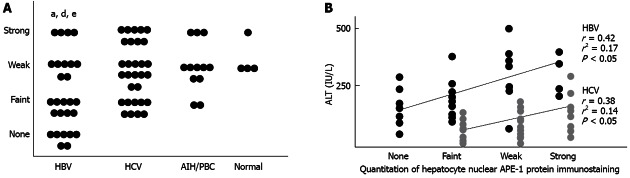
Quantitation of hepatocyte nuclear apurinic/apyrimidinic endonuclease 1 protein immunostaining. A: The percentage of positive hepatocellular nuclei throughout the slide was consistently 80%-100%, regardless of the etiology of chronic liver disease, while the staining intensity was lower in hepatitis B virus (HBV) group than in hepatitis C virus (HCV), autoimmune hepatitis (AIH)/primary biliary cirrhosis (PBC) and normal groups (aP < 0.05 vs HCV, dP < 0.01 vs AIH/PBC, eP < 0.05 vs normal); B: The staining intensity of hepatocytic nuclear apurinic/apyrimidinic endonuclease 1 (APE-1) protein was positively correlated with serum alanine aminotransferase (ALT) levels in the HBV (r = 0.42, P < 0.05) and HCV groups (r = 0.38, P < 0.05).
Western blot analysis showed that APE-1 protein levels were lower in HBV livers compared with normal livers, while there were no significant differences in APE-1 protein levels among HCV, PBC/AIH, and normal livers (Figure 3A and B). Hepatic APE-1 protein levels were reduced by 64% in the HBV group compared with the HCV group. There was a positive correlation between APE-1 protein levels by Western blot analysis and by immunohistochemistry in the livers of patients with chronic liver disease (Figure 3C).
Figure 3.
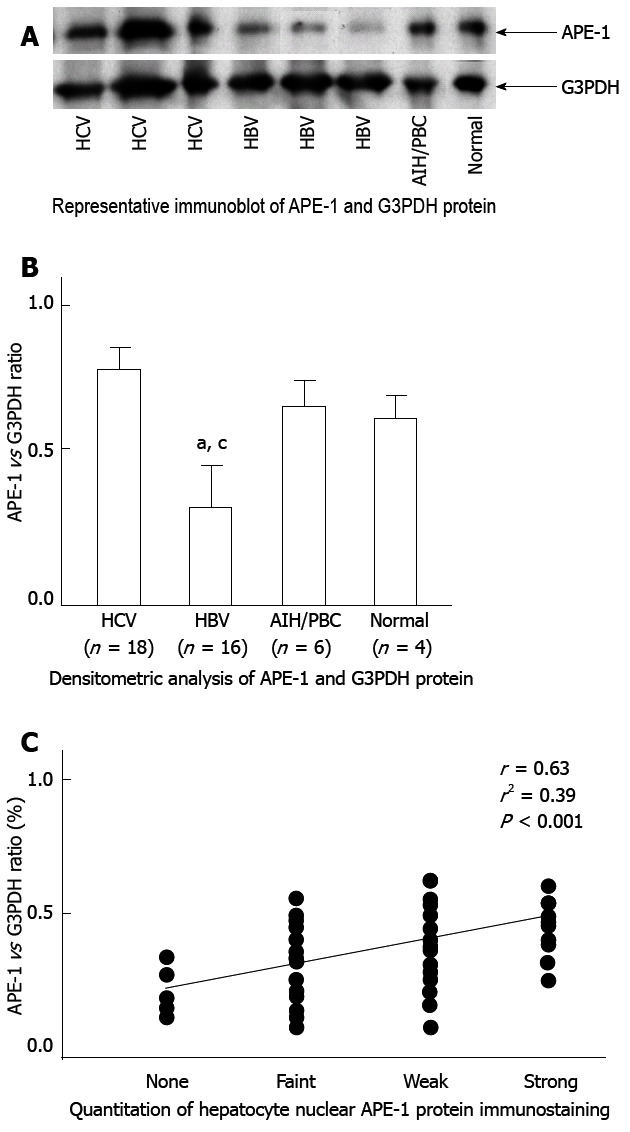
Hepatic apurinic/apyrimidinic endonuclease 1 protein expression in patients with chronic liver disease. A, B: Western blot analysis showed that apurinic/apyrimidinic endonuclease 1 (APE-1) protein levels were reduced in HBV livers compared with normal and HCV livers; HCV livers showed no significant difference in APE-1 protein level compared with normal livers (aP < 0.05 vs HCV, cP < 0.05 vs normal); C: Correlation between hepatocytic APE-1 protein determined by Western blot and immunohistochemistry in patients with chronic hepatitis. G3PDH: Anti-glyceraldehyde-3-phosphate dehydrogenase.
Real-time PCR analysis showed that APE-1 mRNA levels were 58% lower in HBV livers than in HCV livers (Figure 4).
Figure 4.
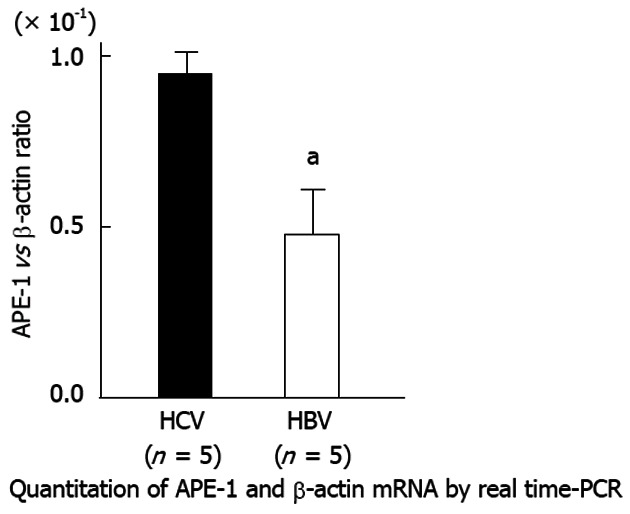
Hepatic apurinic/apyrimidinic endonuclease 1 mRNA expression in patients with chronic liver disease. Real-time polymerase chain reaction (PCR) analysis showed that apurinic/apyrimidinic endonuclease 1 (APE-1) mRNA levels were reduced by 58% in HBV livers compared with HCV livers (aP < 0.05 vs HCV).
Hepatic oxidative DNA damage in patients with chronic liver disease
In liver specimens from patients with chronic liver disease, positive staining for 8-OHdG was detected preferentially in the nuclei of hepatocytes, bile duct cells, and portal inflammatory cells, and occasionally in the nuclei of sinusoidal cells. As shown in Figure 5A, hepatocytic 8-OHdG labeling index was increased in HCV, AIH, and PBC groups (HCV, AIH or PBC vs normal; 64%, 64% or 68% vs 35%, respectively, P < 0.05). This was not seen in the HBV group, and the labeling index was lower in HBV than in HCV samples (HBV vs HCV; 41% vs 64%, respectively, P < 0.001). In both the HBV and HCV groups, the labeling index was positively correlated with the staining intensity of hepatocytic nuclear APE-1 protein (HBV, r = 0.34, P < 0.05; HCV, r = 0.54, P < 0.01) (Figure 5B and C).
Figure 5.
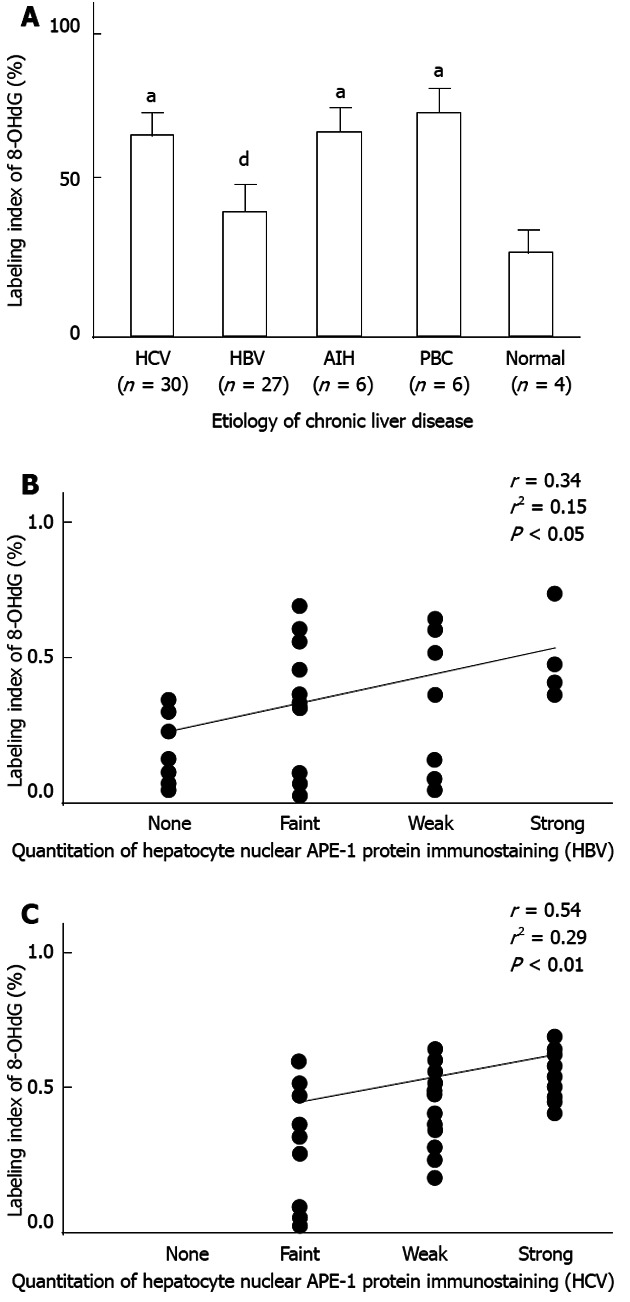
Hepatocytic 8-hydroxydeoxyguanosine expression in patients with chronic liver disease. A: Hepatocytic 8-hydroxydeoxyguanosine labeling index was increased in the hepatitis C virus (HCV), autoimmune hepatitis (AIH), and primary biliary cirrhosis (PBC) groups (HCV, AIH or PBC vs normal; 64%, 64% or 68% vs 35%, respectively, aP < 0.05 vs normal), but not in the hepatitis B virus (HBV) group, and the labeling index was lower in the HBV than in the HCV group (HBV vs HCV; 41% vs 64%, respectively, dP < 0.001 vs HCV); B, C: The labeling indices of HBV and HCV samples were positively correlated with the staining intensity of hepatocytic nuclear apurinic/apyrimidinic endonuclease 1 (APE-1) protein (HBV, r = 0.34, P < 0.05; HCV, r = 0.54, P < 0.01).
DISCUSSION
HCC is frequently encountered in chronic HBV or HCV infection. Hepatocarcinogenesis is most often a multistep process in which a number of genetic alterations accumulate in the hepatocyte[9]. In the case of HCC due to hepatitis virus infection, oncogenesis may be related to viral-induced chronic inflammation. Due to enhanced cell turnover, the repair of damaged DNA may be compromised, rendering the cells more susceptible to spontaneous or mutagen-induced alterations. In addition, with HBV, integration of viral DNA into the host cell genomic DNA may induce transformation. A third possible mechanism involves viral proteins [hepatitis B virus x (HBx) or HCV core] that may act as trans-activators of cellular genes, or may induce cellular stress or oxidative stress leading to DNA damage. However, the mechanisms responsible for hepatocarcinogenesis have not been fully clarified. Therefore, we have focused on investigating hepatic expression of 8-OHdG, a useful marker of oxidative DNA damage, and APE-1, a rate-limiting enzyme in the repair of oxidative DNA damage[10], in patients with chronic HBV or HCV infection. Our findings can be summarized as follows: (1) hepatocellular nuclear APE-1 staining and hepatic APE-1 expression at the protein and mRNA levels are reduced in HBV patients, but not in HCV patients; and (2) the number of hepatocytes with 8-OHdG-positive nuclei is increased among HCV patients, but not HBV patients.
Our data suggest that hepatocellular nuclear expression of APE-1 protein was downregulated in patients with chronic HBV infection compared with normal controls. The downregulation of nuclear APE-1 may be due to altered subcellular translocation, decreased protein synthesis, and/or increased protein degradation of APE-1. The nuclear import of APE-1 is mediated by its nuclear localization signal with the involvement of an importin system[11], while the nuclear export of APE-1 is controlled by S-nitrosation of its nuclear export signal[12]. In addition, the interaction with specific nuclear proteins such as GADD45a appears to maintain APE-1 within the nucleus[13]. Decreased nuclear import, increased nuclear export of APE-1 and/or reduced stability of nuclear APE-1 may downregulate nuclear expression of the protein in HBV-infected hepatocytes, given the interaction of HBx protein with the Crm1-dependent nuclear export pathway[14]. APE-1 induction can be reduced at the transcriptional level. The activation of negative calcium response elements in the promoter region of the APE-1 gene represses APE-1 transcription[15]. In addition, activation of p53 downregulates APE-1 expression, interfering with Sp1 binding to the APE-1 promoter[16]. Similar mechanisms may be involved in the reduction of hepatic APE-1 mRNA levels in HBV-infected livers, since HBx can modify the transcriptional machinery[17]. The reduction of hepatocellular nuclear APE-1 expression may also be due to APE-1 degradation. APE-1 undergoes proteolysis by granzymes A and K, leading to enhanced cell death[18,19]. This may explain our observation that patients with chronic HBV infection had higher levels of serum transaminase and lower levels of hepatocellular nuclear APE-1 expression than those with chronic HCV infection. In addition, HBx may promote ubiquitin-mediated proteasomal degradation of APE-1, as it does of β-catenin[20], since ubiquitination of APE-1 by MDM2 leads to cytoplasmic translocation and proteasomal degradation of APE-1[21].
Our findings indicate that HCV patients had enhanced hepatic 8-OHdG accumulation compared with HBV patients. This confirms the findings of previous studies[22,23]. In addition, in both HBV and HCV patients, the staining intensity of hepatocytic nuclear APE-1 protein was found to be positively correlated with the proportion of 8-OHdG-positive hepatocytes. This suggests that hepatocellular nuclear APE1 expression depends on oxidative DNA damage in HBV- and HCV-infected livers, and reflects the adaptive response of APE-1 to oxidative stress[10]. Furthermore, our data suggest an imbalance between oxidative DNA damage and APE-1 expression. In HBV-infected livers, APE-1 expression is reduced without an increase in oxidatively damaged DNA. In HCV-infected livers, APE-1 expression is not sufficiently induced in proportion to enhanced oxidative DNA damage. These imbalances between oxidative DNA damage and APE-1 expression may lead to the accumulation of AP sites, causing base-substitution mutations and subsequent hepatocarcinogenesis in chronic HBV and HCV infection, as described in a study of ulcerative colitis[24]. Recently, accumulation of AP sites and reduced APE-1 expression were reported in the livers of mice deficient in methionine adenosyltransferase 1a, one of the experimental animal models of HCC[25]. Previous experimental studies showed enhanced spontaneous mutagenesis and cancer predisposition in APE-1 heterozygous mice[26,27].
In conclusion, hepatic APE-1 expression is reduced without enhanced accumulation of hepatic 8-OHdG in chronic HBV infection, while enhanced accumulation of hepatic 8-OHdG exists without an increase in hepatic APE-1 expression in chronic HCV infection. The imbalance between oxidative DNA damage and APE-1 expression may contribute to hepatocarcinogenesis in chronic viral hepatitis.
COMMENTS
Background
Increased production of reactive oxygen species (ROS), which cause oxidative DNA damage, is considered to be related to hepatocarcinogenesis. Apurinic/apyrimidinic endonuclease-1 (APE-1) and 8-hydroxydeoxyguanosine (8-OHdG) are useful markers of DNA damage induced by oxidative stress. The aim of the present study was to examine hepatic expression of APE-1 and 8-OHdG in patients with chronic hepatitis.
Research frontiers
Hepatic APE-1 and 8-OHdG expression may undergo differential regulation in chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infection. Downregulation of hepatic APE-1 expression may contribute to hepatocarcinogenesis in chronic HBV infection.
Innovations and breakthroughs
Livers of patients with chronic HBV infection, but not chronic HCV infection, showed reduced expression of hepatic APE-1 compared with normal livers and other chronic liver diseases.
Applications
Future studies should evaluate the ROS increase generated by chronic inflammation, which is closely linked to the occurrence of hepatocellular oxidative damage and may be related to hepatocarcinogenesis.
Terminology
APE-1: APE-1 is one of the major DNA repair enzymes, and its altered expression is associated with tumorigenesis; 8-OHdG: 8-OHdG is a pro-mutagenic DNA lesion produced by oxygen radicals, and is recognized as a useful marker for estimation of DNA damage induced by oxidative stress.
Peer review
The authors have shown differential expression of hepatic APE-1, a DNA repair enzyme, in chronic hepatitis. By evaluating hepatic APE-1 and 8-OHdG expression in liver disease of various etiologies, they concluded that downregulation of hepatic expression may contribute to the development of hepatocellular carcinoma in chronic HBV infection. This study should be of value in elucidating the mechanisms that contribute to hepatic carcinogenesis.
Footnotes
P- Reviewer Hyogo H S- Editor Wen LL L- Editor A E- Editor Li JY
References
- 1.Jüngst C, Cheng B, Gehrke R, Schmitz V, Nischalke HD, Ramakers J, Schramel P, Schirmacher P, Sauerbruch T, Caselmann WH. Oxidative damage is increased in human liver tissue adjacent to hepatocellular carcinoma. Hepatology. 2004;39:1663–1672. doi: 10.1002/hep.20241. [DOI] [PubMed] [Google Scholar]
- 2.Feitelson MA, Sun B, Satiroglu Tufan NL, Liu J, Pan J, Lian Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene. 2002;21:2593–2604. doi: 10.1038/sj.onc.1205434. [DOI] [PubMed] [Google Scholar]
- 3.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–372. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 4.Di Maso V, Avellini C, Crocè LS, Rosso N, Quadrifoglio F, Cesaratto L, Codarin E, Bedogni G, Beltrami CA, Tell G, et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med. 2007;13:89–96. doi: 10.2119/2006-00084.DiMaso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair (Amst) 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18:5609–5621. doi: 10.1093/emboj/18.20.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19:1513–1520. [PubMed] [Google Scholar]
- 8.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–112. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 9.Pang RW, Lee TK, Man K, Poon RT, Fan ST, Kwong YL, Tse E. PIN1 expression contributes to hepatic carcinogenesis. J Pathol. 2006;210:19–25. doi: 10.1002/path.2024. [DOI] [PubMed] [Google Scholar]
- 10.Ramana CV, Boldogh I, Izumi T, Mitra S. Activation of apurinic/apyrimidinic endonuclease in human cells by reactive oxygen species and its correlation with their adaptive response to genotoxicity of free radicals. Proc Natl Acad Sci USA. 1998;95:5061–5066. doi: 10.1073/pnas.95.9.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson EB, Theriot CA, Chattopadhyay R, Mitra S, Izumi T. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1) Nucleic Acids Res. 2005;33:3303–3312. doi: 10.1093/nar/gki641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson J, Smith R, Kudler H. Familial psychiatric illness in chronic posttraumatic stress disorder. Compr Psychiatry. 1989;30:339–345. doi: 10.1016/0010-440x(89)90059-x. [DOI] [PubMed] [Google Scholar]
- 13.Jung HJ, Kim EH, Mun JY, Park S, Smith ML, Han SS, Seo YR. Base excision DNA repair defect in Gadd45a-deficient cells. Oncogene. 2007;26:7517–7525. doi: 10.1038/sj.onc.1210557. [DOI] [PubMed] [Google Scholar]
- 14.Forgues M, Marrogi AJ, Spillare EA, Wu CG, Yang Q, Yoshida M, Wang XW. Interaction of the hepatitis B virus X protein with the Crm1-dependent nuclear export pathway. J Biol Chem. 2001;276:22797–22803. doi: 10.1074/jbc.M101259200. [DOI] [PubMed] [Google Scholar]
- 15.Izumi T, Henner WD, Mitra S. Negative regulation of the major human AP-endonuclease, a multifunctional protein. Biochemistry. 1996;35:14679–14683. doi: 10.1021/bi961995u. [DOI] [PubMed] [Google Scholar]
- 16.Zaky A, Busso C, Izumi T, Chattopadhyay R, Bassiouny A, Mitra S, Bhakat KK. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008;36:1555–1566. doi: 10.1093/nar/gkm1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arbuthnot P, Capovilla A, Kew M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J Gastroenterol Hepatol. 2000;15:357–368. doi: 10.1046/j.1440-1746.2000.02069.x. [DOI] [PubMed] [Google Scholar]
- 18.Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, Lieberman J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol. 2003;4:145–153. doi: 10.1038/ni885. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y, Chen J, Zhao T, Fan Z. Granzyme K degrades the redox/DNA repair enzyme Ape1 to trigger oxidative stress of target cells leading to cytotoxicity. Mol Immunol. 2008;45:2225–2235. doi: 10.1016/j.molimm.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 20.Jung JK, Kwun HJ, Lee JO, Arora P, Jang KL. Hepatitis B virus X protein differentially affects the ubiquitin-mediated proteasomal degradation of beta-catenin depending on the status of cellular p53. J Gen Virol. 2007;88:2144–2154. doi: 10.1099/vir.0.82836-0. [DOI] [PubMed] [Google Scholar]
- 21.Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009;28:1616–1625. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitada T, Seki S, Iwai S, Yamada T, Sakaguchi H, Wakasa K. In situ detection of oxidative DNA damage, 8-hydroxydeoxyguanosine, in chronic human liver disease. J Hepatol. 2001;35:613–618. doi: 10.1016/s0168-8278(01)00171-4. [DOI] [PubMed] [Google Scholar]
- 23.Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, Kawanishi S, Watanabe S, Kaito M, Takei Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat. 2008;15:498–507. doi: 10.1111/j.1365-2893.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 24.Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, Hussain SP, Roth RB, Zhou X, Mechanic LE, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Invest. 2003;112:1887–1894. doi: 10.1172/JCI19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomasi ML, Iglesias-Ara A, Yang H, Ramani K, Feo F, Pascale MR, Martínez-Chantar ML, Mato JM, Lu SC. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: implication in hepatocarcinogenesis. Gastroenterology. 2009;136:1025–1036. doi: 10.1053/j.gastro.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huamani J, McMahan CA, Herbert DC, Reddick R, McCarrey JR, MacInnes MI, Chen DJ, Walter CA. Spontaneous mutagenesis is enhanced in Apex heterozygous mice. Mol Cell Biol. 2004;24:8145–8153. doi: 10.1128/MCB.24.18.8145-8153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meira LB, Cheo DL, Hammer RE, Burns DK, Reis A, Friedberg EC. Genetic interaction between HAP1/REF-1 and p53. Nat Genet. 1997;17:145. doi: 10.1038/ng1097-145. [DOI] [PubMed] [Google Scholar]