

Abstract
A field defect is a field of pre-malignant tissue in which a new cancer is likely to arise. Field defects often appear to be histologically normal under the microscope. Recent research indicates that cells within a field defect characteristically have an increased frequency of epigenetic alterations and these may be fundamentally important as underlying factors in progression to cancer. However, understanding of epigenetic field defects is at an early stage, and the work of Katsurano et al published this year, is a key contribution to this field. One question examined by Katsurano et al was how early could the formation of an epigenetic field defect be detected in a mouse colitis model of tumorigenesis. They highlighted a number of measurable epigenetic alterations, detected very early in normal appearing tissue undergoing histologically invisible tumorigenesis. They also documented the increasing presence of the epigenetic alterations at successive times during progression to cancer. In this commentary, we offer a perspective on the changes they observed within a broader sequence of epigenetic events that occur in progression to cancer. In particular, we highlight the likely central role of epigenetic deficiencies in DNA repair gene expression that arise during progression to cancer.
Keywords: Field defect, Epigenetics, Tumorigenesis, Carcinogenesis, DNA damage, DNA repair, Colon cancer, Mouse, Human
COMMENTARY ON HOT TOPICS
We read with great interest the recent article by Katsurano et al[1] describing a mouse colitis model leading to tumor formation. They report that an epigenetic field defect forms early after treatment with dextran sulfate sodium (DSS) and that the epigenetic alterations continue to increase even with diminishing stimulation. Their study was unique in showing that particular epigenetic alterations, involving DNA methylation, increased even while inflammation was diminishing.
The term “field cancerization” was first used in 1953 to describe an area or “field” of epithelium that has been preconditioned by (at that time) largely unknown processes so as to predispose it towards development of cancer[2]. Since then, the terms “field cancerization” and “field defect” have been used to describe pre-malignant tissue in which new cancers are likely to arise.
Field defects are of crucial importance in progression to cancer, though they have not received a great deal of attention thus far. As pointed out by Rubin[3], “The vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro. Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion[4].” Field defects with mutations are precursors of cancers with those same mutations. Likewise, epigenetic alterations in field defects are precursors of cancers with those same epigenetic alterations.
Colon cancers contain a median of 76 non-silent sequence mutations, of which about 15 are “driver mutations” (the rest are “passenger mutations”)[5], as well as about 55 aneuploidy events[6]. By comparison, some frequent epigenetic alterations in colon cancers affect hundreds of genes. For example, CpG island methylation of the DNA sequence encoding microRNA miR-137 reduces its expression, and this is a frequent early epigenetic event in colorectal carcinogenesis, occurring in 81% of colon cancers and in 14% of the normal appearing colonic mucosa of the field defects associated with these cancers[7]. Silencing of miR-137 can affect expression of 491 genes, the targets of this miRNA[7]. Changes in the level of miR-137 expression result in changed mRNA expression of the target genes by 2 to 20-fold and corresponding, though often smaller, changes in expression of the protein products of the genes. Other microRNAs, with likely comparable numbers of target genes, are even more frequently epigenetically altered in colonic field defects and in the colon cancers that arise from them. These include miR-124a, miR-34b/c and miR-342 which are silenced by CpG island methylation of their encoding DNA sequences in primary tumors at rates of 99%, 93% and 86%, respectively, and in the adjacent normal appearing mucosa at rates of 59%, 26% and 56%, respectively[8,9]. In addition to epigenetic alteration of expression of miRNAs, other common types of epigenetic alterations in cancers that change gene expression levels include direct hypermethylation or hypomethlyation of CpG islands of protein-encoding genes and alterations in histones and chromosomal architecture that influence gene expression[10,11].
The specific epigenetic alterations studied by Katsurano et al[1], as well as epigenetic field defects in general, can be placed in a broad explanatory framework starting with the occurrence of DNA damage, a major primary event in progression to cancer as shown in Figure 1. In Figure 1 italicized capitalized abbreviations are symbols of epigenetically altered genes. ERCC1, WRN, MSH2, PMS2, MLH1 and MGMT are symbols for specific DNA repair genes. FOSb, FUT4, HOXa5, MEX3a, MSX1, SOX11 are symbols for epigenetically altered genes described by Katsurano et al[1]. These symbols for the genes (in the order listed above) are: FBJ osteosarcoma oncogene B, fucosyltransferase 4, homeobox A5, mex3 homolog A, homeobox msh-like 1, and SRY-box-containing gene 1, respectively.
Figure 1.
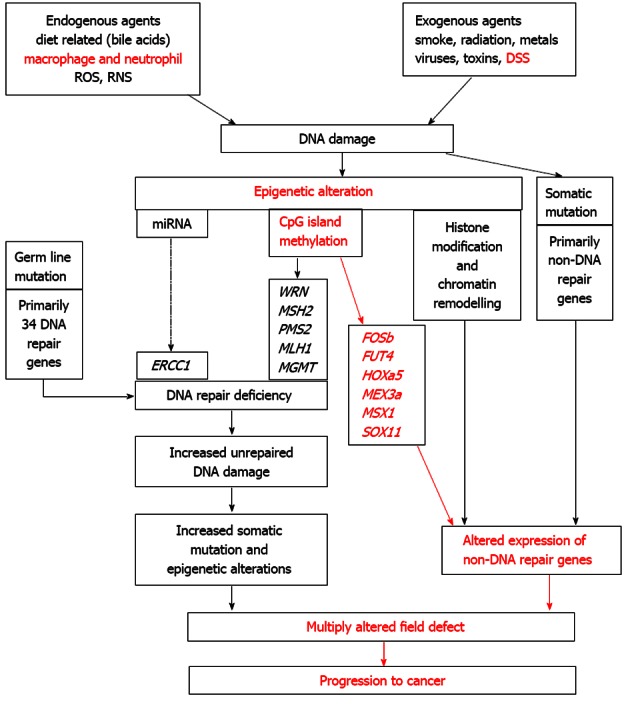
Roles of DNA damage, reduced DNA repair and epigenetic alterations in a field defect in progression to cancer.
DNA damage as a primary cause of cancer
Exogenous and endogenous agents that induce DNA damage have been identified as major causes of many common cancers (Figure 1). These include cancers of the lung (tobacco smoke[12]), colorectum (exposure to bile acids that cause increased reactive oxygen species (ROS) and reactive nitrogen species, and are produced in response to a high fat diet[13]), esophagus (exposure to stomach acids plus bile acids due to gastroesophageal reflux[14]), stomach (reactive oxygen species caused by Helicobacter pylori infection[15]), liver (Aspergillus metabolite aflatoxin B1[16]), cervix/uterus (human papillomavirus plus increased nitric oxide from tobacco smoke or other infection[17]) and melanoma (UV light from solar radiation[18]). Inherited germ line mutations in DNA repair genes similarly cause an increase in DNA damages due to a deficiency in repair capability, and these also cause increases in cancer risk. At least 34 inherited human DNA repair gene mutations increase cancer risk, including, for example, germ line mutations in the BRCA1, XPC and MLH1 genes[19]. From a study of 44 788 pairs of twins, it is estimated that overall, about 30% of cancers are familial (largely due to inherited germ line mutations or genetic polymorphisms) and 70% are sporadic[20].
DNA damages cause epigenetic changes and mutations
ROS, produced during inflammation and other types of cellular stress, cause a variety of types of DNA damage[21], some of which lead to double strand breaks[22]. During repair of double strand breaks and other types of oxidative DNA damages, methylation of promoter CpG islands in DNA and/or modification of histones can occur, causing gene silencing (Figure 1)[23,24]. These epigenetic alterations are sometimes not reversed after repair is completed[23,24]. While it has long been known that oxidative damage can cause mutation[21], it has only recently become clear that oxidative damage can also give rise to epigenetic changes (epimutation)[23,24].
Other types of DNA damage can also give rise to epimutation during DNA repair. The DNA repair enzyme Parp1 [poly(ADP)-ribose polymerase-1] acts at sites of DNA damage, especially single strand breaks, where it adds poly(ADP)-ribose to specific proteins as part of the overall DNA repair process[25]. This, in turn, directs recruitment and activation of the chromatin remodeling protein ALC1 to cause nucleosome remodeling[26]. Nucleosome remodeling has been found to cause, for instance, epigenetic silencing of DNA repair gene MLH1[27]. In addition, certain chemicals previously identified as DNA damaging agents, including benzene, hydroquinone, styrene, carbon tetrachloride and trichloroethylene, cause considerable hypomethylation of DNA, leading to epigenetic modifications, and some of this hypomethylation occurs through the activation of oxidative stress pathways[28].
Epigenetic changes in DNA repair gene expression are a likely source of genomic instability
While germ line (familial) mutations in DNA repair genes cause a high risk of cancer, in sporadic (non-familial) cancers, by contrast, somatic mutations in DNA repair genes are rarely found[5]. However, deficient expression of DNA repair genes is frequently observed within sporadic cancers, and this is almost always due to epigenetic alteration (Figure 1). Epimutation leading to silencing of a gene necessary for DNA repair will allow unrepaired damages to increase. Such additional DNA damages, in turn, will cause increased mutations and epimutations, including carcinogenic driver mutations and epimutations.
Truninger et al[29] compared the frequencies of germ line mutations, CpG island methylations and other unidentified alterations in the down-regulation of expression of DNA mismatch repair (MMR) gene MLH1 in colon cancer. They evaluated 1 048 unselected consecutive colon cancers. They found that 103 of these cancers were deficient in protein expression of MLH1. Among the MLH1 deficient cancers, 68 were sporadic and the remaining 35 were due to germ line mutations. Among the 68 sporadic MLH1 protein-deficient colon cancers, 65 (96%) were deficient due to epigenetic methylation of the CpG island of the MLH1 gene. Reduced protein expression of MLH1 in the remaining 3 sporadic MLH1 protein-deficient cancers may have been caused by over expression of the microRNA miR-155. This explanation is suggested by the finding that transfection of miR-155 into cells caused reduced expression of MLH1[30]. Furthermore, high expression of miR-155 was found in colon cancers in which protein expression of MLH1 was reduced and the MLH1 gene was neither mutated nor hypermethylated[30].
Some of the epigenetic alterations in DNA repair genes found in colon cancers, as well as in their associated field defects, are summarized in Table 1[31-35].
Table 1.
Examples of epigenetic alterations (epimutations) of DNA repair genes in colon cancers and in their field defects, with CpG island methylation indicated where known
| Reference | Epimutations in genes found in colon cancer (mechanism) | Percentage of the sporadic cancers with that epimutation | Epimutations in genes in field defect (mechanism) | Percentage of the field defect with that epimutation |
| Agrelo et al[31] | WRN (CGI) | 38% | ||
| Shen et al[32] | MGMT (CGI) | 46% | MGMT (CGI) | 23% |
| Psofaki et al[33] | MGMT (CGI) | 90% | ||
| Psofaki et al[33] | MLH1 (CGI) | 65% | ||
| Truninger et al[29] | MLH1 (CGI) | 96% | ||
| Lee et al[34] | MLH1 (CGI) | 2% | ||
| Lee et al[34] | MSH2 (CGI) | 13% | MSH2 (CGI) | 5% |
| Lee et al[34] | MGMT (CGI) | 47% | MGMT (CGI) | 11% |
| Facista et al[35] | ERCC1 | 100% | ERCC1 | 60% |
| Facista et al[35] | PMS2 | 88% | PMS2 | 50% |
| Facista et al[35] | XPF | 55% | XPF | 40% |
CGI: CpG island methylation.
Deficiencies in DNA repair genes cause increased mutation rates in MMR defective cells[36,37] and in homologous recombinational repair (HRR) defective cells[38]. Chromosomal rearrangements and aneuploidy also increase in HRR defective cells[39]. Thus, deficiency in DNA repair causes genomic instability (a mutator phenotype), the likely main underlying cause of DNA sequence alterations leading to tumorigenesis. Genomic instability permits the acquisition of a sufficient number of mutations and epimutations in tumor suppressor genes and oncogenes to fuel carcinogenesis. Deficiencies in DNA repair appear to be central to the genomic and epigenomic instability characteristic of cancer.
Figure 1 illustrates the chain of consequences of exposure of cells to endogenous and exogenous DNA damaging agents that lead to cancer. The role of germ line defects in DNA repair genes in familial cancers are also indicated. The large role of DNA damage and consequent epigenetic DNA repair deficiencies leading to sporadic cancer are emphasized. The role of directly induced somatic mutation in sporadic cancer is indicated as well. The items shown in red lettering were demonstrated in the recent article of Katsurano et al[1].
Sequence of epimutation, mutation and natural selection leading to carcinogenesis
A field defect arises when an epimutation or mutation occurs in a stem cell that provides a reproductive advantage allowing clonal descendents of that stem cell to out-compete neighboring stem cells. These cells form a patch of somewhat more rapidly growing cells (an initial field defect). As the patch enlarges at the expense of neighboring cells, an additional epimutation or mutation may arise in one of the field defect stem cells so that this new stem cell with two advantageous epimutations and/or mutations generates daughter stem cells that can out-compete the surrounding field defect of cells that have just one advantageous epimutation or mutation. As illustrated in Figure 2, this process of expanding sub-patches within earlier patches can occur multiple times until a particular constellation of epimutations and mutations results in a cancer (represented by the small dark patch in Figure 2). The cancer, once formed, continues to evolve and to produce sub clones. A renal cancer, for example, sampled in 9 areas, had 40 ubiquitous mutations, 59 mutations shared by some, but not all regions, and 29 “private” mutations only present in one region[40].
Figure 2.
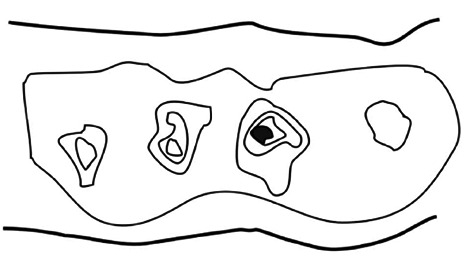
Schematic of a field defect in progression to cancer.
Figure 3 shows an opened resected segment of a human colon that has a colon cancer. There are about 100 colonic microscopic epithelial crypts per sq mm in the human colonic epithelium[41]. The colonic epithelium in the resection shown in Figure 3 has an area of about 6.5 cm by 17 cm, or 111 sq cm, or 11 100 sq mm. Thus this area has about 1.11 million crypts. There are 10-20 stem cells at the base of each colonic crypt[42,43]. Therefore there are likely 11 to 22 million stem cells in the grossly unremarkable colonic mucosal epithelium shown in Figure 3. Evidence reported by Facista et al[35], listed in Table 1 and illustrated in Figure 4, indicates that in many such resections, most of the crypts, and thus the stem cells in such an area up to 10 cm distant (in each direction) from a colon cancer (such as in the grossly unremarkable area shown in Figure 3), and the majority of their differentiated daughter cells, are epigenetically deficient for protein expression of the DNA repair genes ERCC1 and PMS2, although the epithelium is histologically normal.
Figure 3.
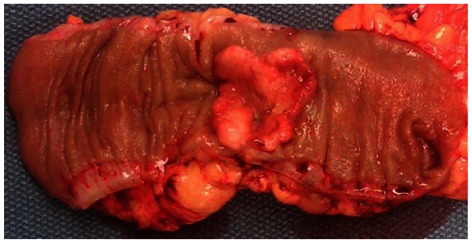
Human colon resection about 17 cm long, opened longitudinally to show inner epithelium, with cancer in center of inner surface.
Figure 4.
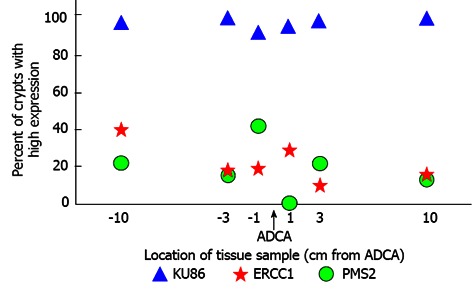
Reduced expression of DNA repair proteins ERCC1 and PMS2 and lack of reduction of DNA repair protein KU86 at distances up to 10 cm from an adenocarcinoma in a colon resection (graphed here from data reported on one of 8 similarly affected colon resections). ADCA: Adenocarcinoma.
The stem cells most distant from the cancer as well as those closer to the cancer in the resection defined by the data in Figure 4 appear to be deficient throughout the field defect for ERCC1 and PMS2. The field defect of Figure 4, containing tens of millions of stem cells, presumably arose from an initial progenitor stem cell deficient in DNA repair (due to epigenetic silencing). Because of this repair deficit, the initial stem cell was genetically unstable, giving rise to an increased frequency of epimutations and mutations in its descendents. One daughter stem cell among its descendents presumably had a mutation or epimutation that, by chance, provided a replicative advantage. This descendent then underwent clonal expansion by natural selection because of its replicative advantage. Among the further descendents of the clone, new mutations and epimutations arose frequently, since these descendents had a mutator phenotype[44], due to the repair deficiency passed down epigenetically from the original repair-defective stem cell. Among these new mutations and epimutations, some would provide further replicative advantages, giving rise to a succession of more aggressively growing sub clones (inner rings in Figure 2), and eventually to a cancer.
The study by Katsurano et al[1] identified 14 genes that were epigenetically silenced or considerably reduced in expression due to CpG island methylation within at least 4 out of 5 of the cancers arising in their DSS induced mouse model of colon cancer. These appear to be “driver” epimutations. They then evaluated the non-neoplastic epithelial cells in the scraped off distal half of mouse colons undergoing DSS-induced tumorigenesis at 2 wk, 5 wk, 8 wk and 15 wk after transitory initial exposure of the mice to DSS. These epithelial cells constitute a field defect from which a mouse colon cancer is likely to arise, since 80%-100% of the mouse colons in their repeated experiments developed tumors 15 wk after exposure to DSS. By 5 to 8 wk after DSS exposure, and before any grossly visible tumors had formed, 6 of the possible “driver” epimutations present in the cancers were not only present, but were also increasing in extent with time, in the mouse colonic field defect.
Based on their own experiments and the literature, Katsurano et al[1] proposed that macrophages and neutrophils in the mouse colonic epithelium were the source of reactive oxygen species causing the DNA damage that initiated the tumorigenesis (Figure 1). However, even though these inflammatory cells were diminishing in frequency in the epithelium by 2 wk after their initial great increase upon DSS exposure, the level of CpG island methylation of the 6 possible “driver” genes FOSb, FUT4, HOXa5, MEX3a, MSX1 and SOX11 continued to increase in the isolated epithelial cells. This increase in the percentage of CpG island methylation of these 6 genes, as tumorigenesis progressed, may have been due to clonal expansion of epithelial cells that initially had these 6 methylated genes.
The work by Katsurano et al[1] constitutes the first mouse model of carcinogenesis in which the unique finding was made that DNA methylation frequency of some genes increased even as the initial inflammation causing DNA damage was decreasing. This work adds important experimental support for the idea that epimutation, natural selection and clonal expansion are key factors driving colon carcinogenesis.
Footnotes
P- Reviewers Zocco MA, Cheng KS, Tandon RK S- Editor Jiang L L- Editor A E- Editor Lu YJ
References
- 1.Katsurano M, Niwa T, Yasui Y, Shigematsu Y, Yamashita S, Takeshima H, Lee MS, Kim YJ, Tanaka T, Ushijima T. Early-stage formation of an epigenetic field defect in a mouse colitis model, and non-essential roles of T- and B-cells in DNA methylation induction. Oncogene. 2012;31:342–351. doi: 10.1038/onc.2011.241. [DOI] [PubMed] [Google Scholar]
- 2.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963–968. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 3.Rubin H. Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture. Bioessays. 2011;33:224–231. doi: 10.1002/bies.201000067. [DOI] [PubMed] [Google Scholar]
- 4.Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, Tavaré S, Shibata D. Genetic reconstruction of individual colorectal tumor histories. Proc Natl Acad Sci USA. 2000;97:1236–1241. doi: 10.1073/pnas.97.3.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balaguer F, Link A, Lozano JJ, Cuatrecasas M, Nagasaka T, Boland CR, Goel A. Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res. 2010;70:6609–6618. doi: 10.1158/0008-5472.CAN-10-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng G, Kakar S, Kim YS. MicroRNA-124a and microRNA-34b/c are frequently methylated in all histological types of colorectal cancer and polyps, and in the adjacent normal mucosa. Oncol Lett. 2011;2:175–180. doi: 10.3892/ol.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, et al. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene. 2008;27:3880–3888. doi: 10.1038/onc.2008.10. [DOI] [PubMed] [Google Scholar]
- 10.Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303–311. doi: 10.1111/j.1399-0004.2011.01809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baldassarre G, Battista S, Belletti B, Thakur S, Pentimalli F, Trapasso F, Fedele M, Pierantoni G, Croce CM, Fusco A. Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol. 2003;23:2225–2238. doi: 10.1128/MCB.23.7.2225-2238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham FH, Fiebelkorn S, Johnson M, Meredith C. A novel application of the Margin of Exposure approach: segregation of tobacco smoke toxicants. Food Chem Toxicol. 2011;49:2921–2933. doi: 10.1016/j.fct.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol. 2011;85:863–871. doi: 10.1007/s00204-011-0648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldman A, Shahidullah M, Goldman D, Khailova L, Watts G, Delamere N, Dvorak K. A novel mechanism of acid and bile acid-induced DNA damage involving Na+/H+ exchanger: implication for Barrett’s oesophagus. Gut. 2010;59:1606–1616. doi: 10.1136/gut.2010.213686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handa O, Naito Y, Yoshikawa T. Redox biology and gastric carcinogenesis: the role of Helicobacter pylori. Redox Rep. 2011;16:1–7. doi: 10.1179/174329211X12968219310756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc Natl Acad Sci USA. 2002;99:6655–6660. doi: 10.1073/pnas.102167699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei L, Gravitt PE, Song H, Maldonado AM, Ozbun MA. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus-infected cells. Cancer Res. 2009;69:4878–4884. doi: 10.1158/0008-5472.CAN-08-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanavy HE, Gerstenblith MR. Ultraviolet radiation and melanoma. Semin Cutan Med Surg. 2011;30:222–228. doi: 10.1016/j.sder.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Bernstein H, Payne CM, Bernstein C, Garewal H and Dvorak K. Cancer and aging as consequences of un-repaired DNA damage. In: Kimura H, Suzuk A, editors. New Research on DNA Damage. New York: Nova Publishers; 2008. pp. 1–47. [Google Scholar]
- 20.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 21.Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531:37–80. doi: 10.1016/j.mrfmmm.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 2011;39:6986–6997. doi: 10.1093/nar/gkr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol. 2005;83:354–364. doi: 10.1139/o05-038. [DOI] [PubMed] [Google Scholar]
- 26.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc Natl Acad Sci USA. 2009;106:13770–13774. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, Egger G, Gal-Yam EN, Jones PA. Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island. Cancer Cell. 2007;12:432–444. doi: 10.1016/j.ccr.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabish AM, Poels K, Hoet P, Godderis L. Epigenetic factors in cancer risk: effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells. PLoS One. 2012;7:e34674. doi: 10.1371/journal.pone.0034674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128:1160–1171. doi: 10.1053/j.gastro.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 30.Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, Adair B, Vannini I, Fanini F, Bottoni A, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci USA. 2010;107:6982–6987. doi: 10.1073/pnas.1002472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agrelo R, Cheng WH, Setien F, Ropero S, Espada J, Fraga MF, Herranz M, Paz MF, Sanchez-Cespedes M, Artiga MJ, et al. Epigenetic inactivation of the premature aging Werner syndrome gene in human cancer. Proc Natl Acad Sci USA. 2006;103:8822–8827. doi: 10.1073/pnas.0600645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 33.Psofaki V, Kalogera C, Tzambouras N, Stephanou D, Tsianos E, Seferiadis K, Kolios G. Promoter methylation status of hMLH1, MGMT, and CDKN2A/p16 in colorectal adenomas. World J Gastroenterol. 2010;16:3553–3560. doi: 10.3748/wjg.v16.i28.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Lee JH. Promoter methylation status of hMLH1, hMSH2, and MGMT genes in colorectal cancer associated with adenoma-carcinoma sequence. Langenbecks Arch Surg. 2011;396:1017–1026. doi: 10.1007/s00423-011-0812-9. [DOI] [PubMed] [Google Scholar]
- 35.Facista A, Nguyen H, Lewis C, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Krouse RS, Bernstein H, Payne CM, et al. Deficient expression of DNA repair enzymes in early progression to sporadic colon cancer. Genome Integr. 2012;3:3. doi: 10.1186/2041-9414-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM. Elevated levels of mutation in multiple tissues of mice deficient in the DNA mismatch repair gene Pms2. Proc Natl Acad Sci USA. 1997;94:3122–3127. doi: 10.1073/pnas.94.7.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis. 2006;27:2402–2408. doi: 10.1093/carcin/bgl079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tutt AN, van Oostrom CT, Ross GM, van Steeg H, Ashworth A. Disruption of Brca2 increases the spontaneous mutation rate in vivo: synergism with ionizing radiation. EMBO Rep. 2002;3:255–260. doi: 10.1093/embo-reports/kvf037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.German J. Bloom’s syndrome. I. Genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196–227. [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein C, Facista A, Nguyen H, Zaitlin B, Hassounah N, Loustaunau C, Payne CM, Banerjee B, Goldschmid S, Tsikitis VL, et al. Cancer and age related colonic crypt deficiencies in cytochrome c oxidase I. World J Gastrointest Oncol. 2010;2:429–442. doi: 10.4251/wjgo.v2.i12.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolas P, Kim KM, Shibata D, Tavaré S. The stem cell population of the human colon crypt: analysis via methylation patterns. PLoS Comput Biol. 2007;3:e28. doi: 10.1371/journal.pcbi.0030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Willis ND, Przyborski SA, Hutchison CJ, Wilson RG. Colonic and colorectal cancer stem cells: progress in the search for putative biomarkers. J Anat. 2008;213:59–65. doi: 10.1111/j.1469-7580.2008.00917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011;11:450–457. doi: 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]