

Abstract
We examined the microRNA signature that distinguishes the most common melanoma histological subtypes, superficial spreading melanoma (SSM) and nodular melanoma (NM). We also investigated the mechanisms underlying the differential expression of histology-specific microRNAs. MicroRNA array performed on a training cohort of 82 primary melanoma tumors (26 SSM, 56 NM), and nine congenital nevi (CN) revealed 134 microRNAs differentially expressed between SSM and NM (P<0.05). Out of 134 microRNAs, 126 remained significant after controlling for thickness and 31 were expressed at a lower level in SSM compared with both NM and CN. For seven microRNAs (let-7g, miR-15a, miR-16, miR-138, miR-181a, miR-191, and miR-933), the downregulation was associated with selective genomic loss in SSM cell lines and primary tumors, but not in NM cell lines and primary tumors. The lower expression level of six out of seven microRNAs in SSM compared with NM was confirmed by real-time PCR on a subset of cases in the training cohort and validated in an independent cohort of 97 melanoma cases (38 SSM, 59 NM). Our data support a molecular classification in which SSM and NM are two molecularly distinct phenotypes. Therapeutic strategies that take into account subtype-specific alterations might improve the outcome of melanoma patients.
INTRODUCTION
The most common histological subtypes of melanoma are superficial spreading melanoma (SSM, 70% of diagnosed cases) and nodular melanoma (NM, 20% of diagnosed cases) (Duncan, 2009). SSM (also known as radial growth phase (RGP) melanoma) is characterized by a flat appearance and a slow and horizontal growth. In contrast, NM is usually thicker than SSM and is characterized by fast growth (Liu et al., 2006). NM tends to move vertically and penetrate into the dermis and is therefore referred to as vertical growth phase (VGP) melanoma.
Currently, melanoma is perceived as a stepwise process in which normal melanocytes in the basal layer of the epidermis acquire genetic mutations that allow linear progression from RGP melanoma to VGP melanoma, and eventually metastatic melanoma (Kwong et al., 2007). However, clinicopathological data reported by our group and others suggest that SSM and NM might not always be two sequential phases of the same disease (Poetsch et al., 2003; Liu et al., 2007; Segura et al., 2008; Warycha et al., 2008; Zalaudek et al., 2008a, 2008b; Baumert et al., 2009; Geller et al., 2009; Rose et al., 2011).
Here, we use microRNA array profiling and validation to define SSM- and NM-specific microRNA alterations. Micro-RNAs are short single-stranded RNA molecules that act as posttranscriptional regulators of gene expression (Djuranovic et al., 2011). MicroRNAs were chosen to identify the differences between SSM and NM because they are accurate markers of cell identity, and their profile can unambiguously distinguish between different cell types (Kosik, 2010). Changes in the expression level of even a single microRNA can shift the expression profile of the cell, cause the activation or repression of signaling pathways, and have profound pathological consequences (Lim et al., 2005; Neveu et al., 2010).
Our data reveal microRNA alterations that cannot be explained by NM being thicker than SSM or by the linear progression model of melanoma. We therefore propose a classification in which SSM and NM are molecularly distinct phenotypes.
RESULTS
134 MicroRNAs are differentially expressed between SSM and NM
Total RNA extracted from 82 melanoma specimens (26 SSM and 56 NM, training cohort, Table 1) and 9 congenital nevi (CN) was analyzed using miRCURY LNA array (Exiqon, Vedbaek, Denmark) platform. According to both T-test and SAM analyses, SSM samples formed one cluster, whereas NM samples showed higher heterogeneity, with some of them clustered together with the SSM samples and others completely separated (Figure 1). In all, 160 (T-test) and 165 (SAM) microRNAs showed differential expression between SSM and NM. A total of 134 microRNAs overlapped between the two analyses, of which 98 are canonical microRNAs and 36 are miRPlus microRNAs. Out of the 134 microRNAs, 75 showed lower expression in SSM compared with NM, whereas the remaining 59 showed higher expression in SSM than in NM (Supplementary Table S1 online). The 98 canonical microRNAs were analyzed using the Ingenuity Software (Redwood city, CA) for pathway analysis. Among the 51 microRNAs that are currently characterized enough for functional evaluation, the majority (39, 76%) are already known to have a role in human cancer (Supplementary Figure S1 online) and some of them have specifically been shown to have a role in melanoma (Felicetti et al., 2008; Gaziel-Sovran et al., 2011).
Table 1.
Patient characteristics
| Training cohort (N=82) | Validation cohort (N=97) | |
|---|---|---|
| Gender, N (%) | ||
| Male | 49 (59.8) | 64 (66.0) |
| Female | 33 (40.2) | 33 (34.0) |
| Age at diagnosis (years) | ||
| Mean±SD | 58.9±14.4 | 63.8±16.5 |
| Histological type, N (%) | ||
| SSM | 26 (31.7) | 38 (39.2) |
| NM | 56 (68.2) | 59 (60.8) |
| Thickness (mm), N (%) | ||
| ≤2 | 32 (39.0) | 30 (30.9) |
| >2 | 50 (61.0) | 67 (69.1) |
| Ulceration, N (%) | ||
| Present | 42 (51.2) | 51 (52.6) |
| Absent | 40 (48.8) | 46 (47.4) |
| Mitoses per mm2, N (%) | ||
| 0 | 5 (6.1) | 8 (8.2) |
| ≥1 | 75 (91.5) | 89 (91.8) |
| Unknown | 2 (2.4) | 0 (0.0) |
| Stage, N (%) | ||
| Stage I | 19 (23.2) | 18 (18.5) |
| Stage II | 44 (53.6) | 39 (40.2) |
| Stage III | 19 (23.2) | 38 (39.2) |
| Stage IV | 0 (0.0) | 2 (2.1) |
| LN status, N (%) | ||
| Positive | 16 (19.5) | 33 (34.0) |
| Negative | 66 (80.5) | 64 (66.0) |
Abbreviations: LN, lymph node; NM, nodular melanoma; SSM, superficial spreading melanoma.
Figure 1. Expression profiling reveals 134 microRNAs that are differentially expressed between superficial spreading melanoma (SSM) and nodular melanoma (NM).

Heat map representing the differentially expressed microRNAs according to T-test, P<0.05 (left), and SAM, false discovery rate (FDR) <10% (right). T-test and SAM identify 160 and 165 differentially expressed microRNAs, respectively. In all, 134 microRNAs overlap between the two analyses. For 126 of the 134 microRNAs, the differences between SSM and NM could not be accounted for by thickness.
To examine whether the differences in the microRNA expression levels between SSM and NM are due to differences in thickness, the 134 microRNAs differentially expressed between SSM and NM were reanalyzed by dividing the 82 samples into two groups according to their thickness (≤2 mm thick versus >2mm thick). The vast majority (126/134, 94%) of the microRNAs were differentially expressed between the two thickness groups (T-test, P<0.05). These data indicate that their expression level changes along with the increasing invasion of the tumor. Nevertheless, SSM and NM samples of comparable thickness tended to cluster together, suggesting that thickness cannot account for the differences between SSM and NM.
The differential expression level of 52 microRNAs cannot be explained by the linear progression model
To understand the relationship between microRNA alterations and melanoma progression, CN were used for comparison. According to the difference in expression level between SSM and NM, CN and SSM, and CN and NM, the microRNAs could be categorized into two groups. The larger group is composed of 52 microRNAs whose level is altered in SSM, but (closer to) normal in NM or even showing opposite alterations in the 2 subtypes, being downregulated in one and upregulated in the other. This differential expression pattern is in favor of a non-linear progression model. A smaller group, composed of 17 microRNAs whose levels progressively increase or decrease in CN→SSM→NM, is consistent with the linear progression model.
The 52 microRNAs that support the nonlinear progression model are listed in Supplementary Figure S2 online and Supplementary Table S2 online. We chose to further study the subset that is downregulated in SSM compared with both CN and NM (Figure 2). This category, composed of 31 microRNAs, is the largest.
Figure 2. MicroRNAs that are specifically downregulated in superficial spreading melanoma (SSM) compared with both congenital nevi (CN) and nodular melanoma (NM).

In all, 31 microRNAs belong to this category according to T-test and/or Mann–Whitney test (P<0.05), and they are all listed on the left. On the right, miR-15a is shown as an example in the upper panel, whereas a cartoon summarizing the relationship among CN, SSM, and NM is reported in the lower panel. The black arrow on the side indicates increasing expression levels. *P<0.05.
Prioritized microRNA loci show deletions in SSM cell lines but not in NM cell lines
To determine whether the downregulation of the prioritized microRNAs is due to selective genomic loss in SSM, the corresponding genomic loci were analyzed by real-time PCR in 10 melanoma cell lines. In all, 13 loci (48%, in gray in Figure 3) showed deletion in at least one RGP/SSM-like cell line and no deletion in VGP/NM-like cell lines. miR-491 locus was considered as part of this group because although it is partially deleted in WM983A VGP/NM-like cell line, it is also the only one that shows complete deletion in one RGP/SSM-like cell line (WM1552c). Among the remaining loci, six did not show any deletion across the 10 cell lines, whereas eight showed deletions in both the RGP/SSM-like and the VGP/NM-like cell lines. The 13 loci pointing toward selective genomic deletion in SSM were characterized further, as described below.
Figure 3. Thirteen microRNA loci are deleted in radial growth phase (RGP)/superficial spreading melanoma (SSM)–like cell lines and not in vertical growth phase (VGP)/nodular melanoma (NM)–like cell lines.

The genomic loci corresponding to the 31 microRNAs listed in Figure 2 were analyzed by real-time PCR in 10 primary melanoma cell lines. The second column indicates whether each microRNA was identified using T-test (T) or Mann–Whitney test (MW). The cluster to which each microRNA belongs and its genomic location are reported in the third and fourth column, respectively. RGP/SSM-like cell lines are in blue and VGP/NM-like cell lines are in red. The 13 loci that were subjected to further analyses are highlighted in gray.
The analysis of the genomic loci corresponding to prioritized microRNAs confirms the SSM-specific deletion in melanoma specimens
The selective genomic loss of let-7g, miR-15a~16-1, miR-17~92, miR-29b-2~29c, miR-30a, miR-30c-2, miR-138-1, miR-138-2, miR-181a-2~181b-2, miR-191~425, miR-491, miR-933, and miRPlus-F1147 loci in SSM was confirmed on genomic DNA extracted from 9 SSM and 16 NM specimens, whereas 13 CNs were used as control. In all, 2 out of the 9 SSM and 12 out of the 16 NM samples were originally present in the training cohort. The additional ones included were chosen so that the overall clinicopathological characteristics of the 25 samples used for genomic DNA extraction were not different from those used for RNA extraction and microRNA array (P>0.05, data not shown). The results of this analysis are reported in Figure 4 and Supplementary Figure S3 online. Six loci (let-7g, miR-15a~16-1, miR-138-2, miR-181a-2~181b-2, miR-191~425, and miR-933; Figure 4) showed lower copy number in SSM versus NM (P<0.05), supporting the results obtained by microRNA array and by genomic PCR on melanoma cell lines. miR-138-2, miR-191~425, and miR-933 loci showed lower copy number compared with CN as well.
Figure 4. Validation of the genomic loss of let-7g, miR-15a, miR-16, miR-138, miR-181a, miR-191, and miR-933 in melanoma specimens.

Copy number of the microRNA loci corresponding to the seven microRNAs in congenital nevi (CN) (black), superficial spreading melanoma (SSM) (blue), and nodular melanoma (NM) (red) primary tumor specimens. The bicistronic miR-15a~16-1 expresses both miR-15a and miR-16. *P<0.05; **P<0.01; ***P<0.001.
miR-15a~16-1 is a bicistronic cluster expressing miR-15a and miR-16, which both belong to the subset under study (Figure 2). The miR-181a-2~181b-2 bicistronic cluster expresses miR-181a and miR-181b. Both microRNAs belong to the subset under study (Figure 2), but the differences in expression level of miR-181b across CN, SSM, and NM do not reach the required statistical significance (Supplementary Table S2 online). The bicistronic miR-191~425 cluster expresses miR-191 and miR-425. Contrary to miR-191, miR-425 is not detectable in SSM nor in NM.
Validation of microarray data by real-time PCR
The seven microRNAs that we found to be downregulated (Figure 2) and deleted (Figures 3 and 4) in SSM compared with NM (let-7g, miR-15a, miR-16, miR-138, miR-181a, miR-191, and miR-933) were further analyzed. The expression levels of these microRNAs in CN, SSM, and NM, as obtained from the microRNA array performed on the training cohort, are reported in Supplementary Figure S4a online. The differences in expression reached statistical significance in at least one of the following tests: T-test, Mann–Whitney test, or Wilcoxon’s rank-sum test.
To confirm the data obtained with the microarray platform using a different technique, we quantified the levels of mature let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191 by real-time PCR on seven SSM and seven NM samples belonging to the training cohort. As reported in Supplementary Figure S5 online, real-time PCR confirmed that all the tested microRNAs are expressed at a higher level in NM than in SSM, validating the data obtained by microRNA array.
The analysis of an independent validation cohort confirms the lower expression of the selected microRNAs in SSM compared with NM
The expression level of the selected microRNAs was checked by miRCURY LNA array (Exiqon) on 97 additional melanoma primary tumors (38 SSM and 59 NM, validation cohort, Table 1). The validation cohort showed that all the micro-RNAs of interest, except for miR-933, are expressed at a lower level in SSM compared with NM (Figure 5 and Supplementary Figure S4b online), confirming the results obtained in the training cohort.
Figure 5. Expression level of let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191 in the melanoma specimens belonging to the validation cohort.
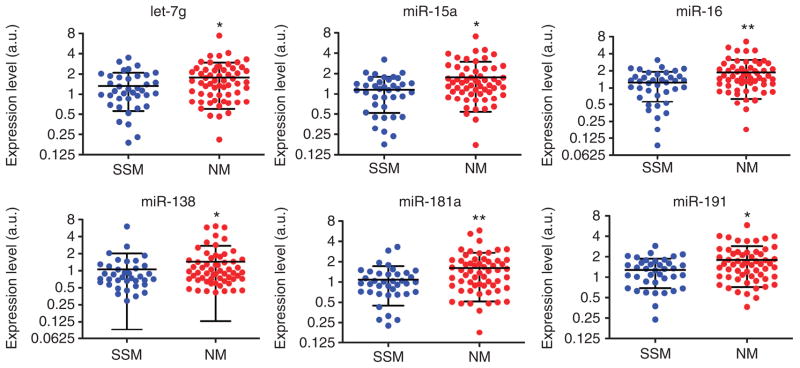
The difference in expression level of the indicated microRNAs between the 38 superficial spreading melanoma (SSM) and the 59 nodular melanoma (NM) samples belonging to the validation cohort is reported. *P<0.05; **P<0.01.
The validation cohort was also used to explore the prognostic value of let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191. The expression level of these microRNAs in the samples of patients who did or did not recur over a median follow-up period of 33 months (5–150 months range) was compared. The SSM samples show a general trend toward lower expression level in the patients who recurred (let-7g, P–value=0.067; miR-15a, P-value=0.071). The difference was statistically significant for miR-191 (P-value=0.029). Interestingly, this trend is not evident in NM samples and is actually reversed in the case of miR-138 (P-value=0.031).
The identified microRNAs that are selectively downregulated and deleted in SSM share overlapping predicted targets
The six microRNAs that we found to be downregulated in both the training and in the validation cohort (let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191) were further analyzed. TargetScan prediction algorithm (http://www.targetscan.org/) was used to obtain the predicted targets for each of them. As these microRNAs are co-downregulated and possibly co-deleted in SSM, we decided to focus on their common targets. The predicted targets that are shared by each microRNA with the other five are indicated in Supplementary Figure S6a online and listed in Supplementary Table S3 online. On average, each microRNA shares 22.4% of its predicted targets with the others. As reported in Supplementary Figure S6b online, 13 targets are shared by three of the microRNAs. Although their role in melanoma has not been studied yet, some (ADAMTS5, EN2, MSI1, TEAD1) are known to have an oncogenic role in other cancer types.
DISCUSSION
Investigations in almost every solid malignancy suggest that a thorough understanding of the molecular defects of tumor histological subtypes is critical for the development of effective targeted therapy. The identification of distinct molecular alterations characterizing acral lentiginous melanoma, mucosal melanoma, and uveal melanoma has prompted the initiation of phase II trials evaluating the efficacy of the c-KIT inhibitor Imatinib as part of a subtype-specific melanoma treatment strategy (Eton et al., 2004; Wyman et al., 2006). Acral lentiginous melanoma, mucosal melanoma, and uveal melanoma, however, comprises only a small percentage of melanomas diagnosed in the United States (<5%). On the contrary, superficial spreading and nodular melanoma histological subtypes, which represent 70 and 20% of newly diagnosed cases, respectively, are not currently taken into account in decision making, and their molecular classification has not been defined yet.
Although previous studies have mainly focused on the identification of microRNAs associated with melanoma progression (Mueller et al., 2009; Caramuta et al., 2010; Philippidou et al., 2010), in this study we have identified the 134-microRNA signature that distinguishes SSM and NM primary melanoma subtypes. The subset of microRNAs that we found selectively downregulated in SSM compared with both nevi and NM was further studied in order to formally prove that SSM and NM are two different biological entities that do not always arise one from the other. By using an initial screening in “RGP/SSM-like” and “VGP/NM-like” melanoma cell lines, and then SSM and NM primary melanoma specimens, we were in fact able to identify seven microRNAs whose genomic loci are selectively lost in SSM. We argue that, although the compensatory amplification of the remaining allele can in theory take place, it is unlikely that a genomic locus that is partially lost at a certain point of tumor progression is regained at later stages. Therefore, we consider the microRNA loci that we found selectively lost in SSM as a proof of principle that SSM and NM are not sequential phases of melanoma progression.
SSM is the most common histopathological subtype of melanoma and its incidence has risen in the past decades in spite of increased surveillance and earlier detection (Linos et al., 2009). Although SSM tends to be less aggressive than NM, it does have the ability to metastasize and be fatal. Furthermore, owing to the higher prevalence rate of SSM compared with NM, SSM accounts for a significant portion of melanoma-related deaths (Gimotty et al., 2007; Shaikh et al., 2011). Therefore, it is of pivotal importance to identify the alterations that can specifically drive SSM pathogenesis and that can potentially be exploited to develop tailored therapeutic approaches.
Our microarray analysis on the 82 primary melanoma samples composing the training cohort and the 97 primary melanoma samples composing the validation cohort indicates that let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191 are consistently expressed at lower levels in SSM than in NM (Figures 2 and 5), possibly because of genomic deletion (Figures 3 and 4). Several pieces of evidence suggest that the diminished expression of these six microRNAs in SSM may contribute to the onset of this melanoma subtype, as described below.
First, these microRNAs have been shown to have an oncosuppressive role in other cancers (see Supplementary Table S4 online for references). In general, they are expressed at a lower level in tumors compared with non-tumoral tissues, and oppose tumor progression by decreasing the abundance of multiple oncogenic targets. Interestingly, some of these targets, such as WNT3A, are known to have an oncogenic role in melanoma as well, although their possible differential contribution to the pathogenesis of SSM and NM has not been considered before.
The canonical WNT1/WNT3A-β-catenin pathway is crucial in the early phases of melanoma. β-Catenin causes the malignant transformation of melanocytes, because it directly represses p16/CDKN2A transcription and, consequently, provides melanocytes with an escape from senescence (O’Connell and Weeraratna, 2009). The loss of p16 is known to be a more common event in SSM than in NM, and the mechanisms reported thus far (genomic deletion, promoter methylation, and mutation) fail to explain all the cases where the loss occurs (Poetsch et al., 2003). As amplification or activating mutations in WNT3A have not been detected, we postulate that the loss of miR-15a~miR-16-1 locus might be an SSM-specific mechanism by which WNT3A is upregulated, WNT signaling increases, and p16 transcription is blunted, a mechanism that has not been previously reported. It is also noteworthy that another target of miR-15a/miR-16, the proto-oncogene c-MYB, is upregulated in SSM compared with NM according to two previously published data sets (Jaeger et al., 2007; Scatolini et al., 2010). As c-MYB has been recently shown to cooperate with the WNT signaling pathway in colorectal cancer tumorigenesis (Ciznadija et al., 2009), it is tempting to speculate that the downregulation/deletion of miR-15a/16 might result in two, cooperative oncogenic hits.
Second, the oncosuppressive activity of the identified microRNAs is reinforced by some of the predicted targets that they share. Multiple microRNAs are known to bind to the same mRNA and cooperate in its downregulation (Hobert, 2004). As we found that let-7g, miR-15a, miR-16, miR-138, miR-181a, and miR-191 were co-downregulated and codeleted, we looked for those genes that are predicted to be targeted by at least two of them and hence may be the most affected from the concomitant decrease in their levels. We found 13 predicted targets that are shared by three micro-RNAs (Supplementary Figure S6 online). Interestingly, most of these targets are expressed at a higher level in SSM than in NM according to two previously published data sets (Jaeger et al., 2007; Scatolini et al., 2010) (Supplementary Figure S7 online), a pattern that is consistent with the downregulation/deletion of the selected microRNAs in SSM compared with NM.
Two among the predicted overlapping targets, TNRC6B and CNOT6L, are known to have a role in microRNA-guided posttranscriptional repression (Baillat and Shiekhattar, 2009; Piao et al., 2010). CNOT6L is a deadenylase that removes poly(A) tails from mRNAs destabilized by microRNAs (Baillat and Shiekhattar, 2009; Piao et al., 2010). These two predicted targets suggest that the deletion/downregulation of the identified microRNAs could impact the microRNA network as a whole.
Many of the remaining predicted genes have a well-established oncogenic role in other cancer types (see Supplementary Table S5 online for references), and might be responsible for the potential oncosuppressive functions of the microRNAs that we have found deleted/downregulated in SSM. Indeed, MSI1, which is a cancer stem cell marker, has been recently shown to be directly targeted by miR-138 (Vo et al., 2011), validating the prediction obtained using TargetScan. Furthermore, MSI1 is known to sustain the cancer stem cell pool by activating the WNT pathway (Sanchez-Diaz et al., 2008; Wang et al., 2008). Therefore, it is possible that the concomitant downregulation of miR-15a/16 and miR-138 could cause the SSM-specific activation of the WNT signaling by multiple mechanisms.
Third, genomic loss has been previously associated with the downregulation of the identified microRNAs, supporting our results. The miR-15a~16-1 locus on 13q14.3 and the let-7g locus on 3p21.1 are located in minimal deleted regions, as reported in B-CLL and epithelial cancer, respectively (Calin et al., 2004). In addition, miR-138-2 locus on 16q13 is deleted in ovarian cancer and melanoma (Zhang et al., 2006). It is noteworthy that the analysis of a melanoma SNP array recently published by our group (Rose et al., 2011) indicates that all the loci corresponding to the identified microRNAs are not part of large deletions. Some of them are indeed located close to other important tumor suppressor genes with a role in melanoma, such as BRCA1-associated protein 1 on 3p21.1 together with let-7g, and RAS association domain family member 1 on 3p21.3 together with miR-191~425 (Harbour et al., 2010; Yi et al., 2011). Nevertheless, the “focal” nature of the deletion of the identified microRNA loci suggest that they might be “drivers” and not simply “passengers” in SSM pathogenesis (Akavia et al., 2010).
In conclusion, in this study we have identified downregulation and loss in microRNA genes that are specific for the SSM (but not NM) subtype. We propose that these alterations may constitute an SSM-specific “point of entry” for the activation of oncogenic and possibly cancer stem cell–related pathways. Overall, our data lend support to the incorporation of genetic signature into the histopathologic classification of melanoma subtypes.
MATERIALS AND METHODS
Human melanoma tissues
Patients were prospectively enrolled into the Interdisciplinary Melanoma Cooperative Group (IMCG) database (Wich et al., 2009) since August 2002. All patients provided written consent at enrollment and the Internal Review Board of NYU approved the study. Information about the characteristics of the primary tumors was collected, including histologic subtype, age at diagnosis, stage, thickness, ulceration, number of mitoses per mm2, lymph node (LN) status and recurrence. Tumors designated as NM demonstrate aggressive vertical growth with large dermal nests and sheets of cytologically atypical melanocytes. To distinguish NM from SSM that extends into the dermis, NM cannot have evidence of melanoma cells in the epidermis extending beyond 3 rete ridges lateral to the dermal component (Smoller, 2006). The study was conducted according to Declaration of Helsinki Principles.
Melanoma specimens were sectioned on Leica Membrane Slides PEN and laser-capture microdissected, using a LMD6000 Leica Laser Micro-Dissection System (NYU CI Histopathology Core; Buffalo Grove, IL). Two to ten 10-μm slides were used per specimen. Total RNA from these samples was extracted using miRNeasy FFPE Tissue Kit (Qiagen, Hilden, Germany) and quantified using a Nanodrop Spectrophotometer (Thermo Scientific, Wilmington, DE).
Human melanoma cell lines
Normal melanocytes were obtained from infant foreskin (NHM) and grown in supplemented melanocyte growth medium (PromoCell, Heidelberg, Germany).
Primary RGP/SSM-like (WM35, WM1552c, WM1575, WM3211) and VGP/NM-like (WM98.1, WM278, WM793B, WM853.2, WM902B, WM983A) melanoma cell lines were purchased from the Wistar Institute (Philadelphia, PA) and cultured in Mel 2% medium (Fang et al., 2005). They were not further tested or authenticated.
MicroRNA array
Following quality control, 300 ng of RNA was labeled with Cy3 and co-hybridized with a Cy5-labeled reference sample (equal mixture of all arrayed samples) to miRCURY LNA version 11.0 array platform (miRBase v. 14.0, Exiqon) on Tecan HS4800 Pro automated hybridization stations and scanned on Agilent G25605BA Microarray Scanners (Santa Clara, CA). Image analyses (ImaGene 8.0 software, Biodiscovery, El Segundo, CA) were performed in order to quantify the signals of the arrays. Technical quality assessment of the data was performed based on results from synthetic spike-in control RNAs. The microRNA array used in this study contains probes for 26 control small RNAs, 79 viral microRNAs, and 1264 human microRNAs. The sequence of 838 out of the 1264 human microRNAs (hsa-miR) is available at http://www.mirbase.org/, while the remaining 426 microRNAs (hsa-miRPlus) are proprietary small RNAs identified by EXIQON. MicroRNA array results were analyzed as described in Supplemental Text S1 (online).
Real time PCR on genomic DNA and on mature microRNAs
Real time PCR was performed according to standard procedures, which are described in detail in Supplemental Text S1 and in Supplementary Table S6 online.
Supplementary Material
Acknowledgments
We thank Ting Tu, Daniel Lackaye, Michelle Ma, and Holly Greenwald for the management of the clinical information of the Interdisciplinary Melanoma Cooperative Group (IMCG) patients. We thank Erica Friedman for her help with real-time PCR. PJC was partially supported by the Clinical Translational Science Center (CTSC) grant UL1-RR024996. This work was supported by the Chemotherapy Foundation, the Elsa U. Pardee Foundation, and the National Cancer Institute Cancer Center Support grant 5 P30 CA 016087-27.
Abbreviations
- CN
congenital nevi
- NM
nodular melanoma
- SSM
superficial spreading melanoma
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
References
- Akavia UD, Litvin O, Kim J, et al. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–17. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Shiekhattar R. Functional dissection of the human TNRC6 (GW182-related) family of proteins. Mol Cell Biol. 2009;29:4144–55. doi: 10.1128/MCB.00380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumert J, Schmidt M, Giehl KA, et al. Time trends in tumour thickness vary in subgroups: analysis of 6475 patients by age, tumour site and melanoma subtype. Melanoma Res. 2009;19:24–30. doi: 10.1097/CMR.0b013e32831c6fe7. [DOI] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramuta S, Egyhazi S, Rodolfo M, et al. MicroRNA expression profiles associated with mutational status and survival in malignant melanoma. J Invest Dermatol. 2010;130:2062–70. doi: 10.1038/jid.2010.63. [DOI] [PubMed] [Google Scholar]
- Ciznadija D, Tothill R, Waterman ML, et al. Intestinal adenoma formation and MYC activation are regulated by cooperation between MYB and Wnt signaling. Cell Death Differ. 2009;16:1530–8. doi: 10.1038/cdd.2009.94. [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R. A parsimonious model for gene regulation by miRNAs. Science. 2011;331:550–3. doi: 10.1126/science.1191138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan LM. The classification of cutaneous melanoma. Hematol Oncol Clin North Am. 2009;23:501–13. ix. doi: 10.1016/j.hoc.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Eton O, Billings LA, Kim KB, et al. Phase II trial of imatinib mesylate (STI-571) in metastatic melanoma. J Clin Oncol (2004 ASCO Annual Meeting Proceedings) 2004;22:14S. [Google Scholar]
- Fang D, Nguyen TK, Leishear K, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–37. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Felicetti F, Errico MC, Bottero L, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–54. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- Gaziel-Sovran A, Segura MF, Di Micco R, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell. 2011;20:104–18. doi: 10.1016/j.ccr.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller AC, Elwood M, Swetter SM, et al. Factors related to the presentation of thin and thick nodular melanoma from a population-based cancer registry in Queensland Australia. Cancer. 2009;115:1318–27. doi: 10.1002/cncr.24162. [DOI] [PubMed] [Google Scholar]
- Gimotty PA, Elder DE, Fraker DL, et al. Identification of high-risk patients among those diagnosed with thin cutaneous melanomas. J Clin Oncol. 2007;25:1129–34. doi: 10.1200/JCO.2006.08.1463. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. Common logic of transcription factor and microRNA action. Trends Biochem Sci. 2004;29:462–8. doi: 10.1016/j.tibs.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Jaeger J, Koczan D, Thiesen HJ, et al. Gene expression signatures for tumor progression, tumor subtype, and tumor thickness in laser-microdissected melanoma tissues. Clin Cancer Res. 2007;13:806–15. doi: 10.1158/1078-0432.CCR-06-1820. [DOI] [PubMed] [Google Scholar]
- Kosik KS. MicroRNAs and cellular phenotypy. Cell. 2010;143:21–6. doi: 10.1016/j.cell.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Kwong L, Chin L, Wagner SN. Growth factors and oncogenes as targets in melanoma: lost in translation? Adv Dermatol. 2007;23:99–129. doi: 10.1016/j.yadr.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Linos E, Swetter SM, Cockburn MG, et al. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129:1666–74. doi: 10.1038/jid.2008.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Dowling JP, Murray WK, et al. Rate of growth in melanomas: characteristics and associations of rapidly growing melanomas. Arch Dermatol. 2006;142:1551–8. doi: 10.1001/archderm.142.12.1551. [DOI] [PubMed] [Google Scholar]
- Liu W, Kelly JW, Trivett M, et al. Distinct clinical and pathological features are associated with the BRAF (T1799A(V600E)) mutation in primary melanoma. J Invest Dermatol. 2007;127:900–5. doi: 10.1038/sj.jid.5700632. [DOI] [PubMed] [Google Scholar]
- Mueller DW, Rehli M, Bosserhoff AK. miRNA expression profiling in melanocytes and melanoma cell lines reveals miRNAs associated with formation and progression of malignant melanoma. J Invest Dermatol. 2009;129:1740–51. doi: 10.1038/jid.2008.452. [DOI] [PubMed] [Google Scholar]
- Neveu P, Kye MJ, Qi S, et al. MicroRNA profiling reveals two distinct p53-related human pluripotent stem cell states. Cell Stem Cell. 2010;7:671–81. doi: 10.1016/j.stem.2010.11.012. [DOI] [PubMed] [Google Scholar]
- O’Connell MP, Weeraratna AT. Hear the Wnt Ror: how melanoma cells adjust to changes in Wnt. Pigment Cell Melanoma Res. 2009;22:724–39. doi: 10.1111/j.1755-148X.2009.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippidou D, Schmitt M, Moser D, et al. Signatures of microRNAs and selected microRNA target genes in human melanoma. Cancer Res. 2010;70:4163–73. doi: 10.1158/0008-5472.CAN-09-4512. [DOI] [PubMed] [Google Scholar]
- Piao X, Zhang X, Wu L, et al. CCR4-NOT deadenylates mRNA associated with RNA-induced silencing complexes in human cells. Mol Cell Biol. 2010;30:1486–94. doi: 10.1128/MCB.01481-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch M, Dittberner T, Woenckhaus C. Can different genetic changes characterize histogenetic subtypes and biologic behavior in sporadic malignant melanoma of the skin? Cell Mol Life Sci. 2003;60:1923–32. doi: 10.1007/s00018-003-2324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose AE, Poliseno L, Wang J, et al. Integrative genomics identifies molecular alterations that challenge the linear model of melanoma progression. Cancer Res. 2011;71:2561–71. doi: 10.1158/0008-5472.CAN-10-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Diaz PC, Burton TL, Burns SC, et al. Musashi1 modulates cell proliferation genes in the medulloblastoma cell line Daoy. BMC Cancer. 2008;8:280. doi: 10.1186/1471-2407-8-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scatolini M, Grand MM, Grosso E, et al. Altered molecular pathways in melanocytic lesions. Int J Cancer. 2010;126:1869–81. doi: 10.1002/ijc.24899. [DOI] [PubMed] [Google Scholar]
- Segura S, Pellacani G, Puig S, et al. In vivo microscopic features of nodular melanomas: dermoscopy, confocal microscopy, and histopathologic correlates. Arch Dermatol. 2008;144:1311–20. doi: 10.1001/archderm.144.10.1311. [DOI] [PubMed] [Google Scholar]
- Shaikh WR, Xiong M, Weinstock MA. The contribution of nodular subtype to melanoma mortality in the United States, 1978 to 2007. Arch Dermatol. 2011 doi: 10.1001/archdermatol.2011.264. e-pub ahead of print 19 September 2011. [DOI] [PubMed] [Google Scholar]
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(Suppl 2):S34–40. doi: 10.1038/modpathol.3800508. [DOI] [PubMed] [Google Scholar]
- Vo DT, Qiao M, Smith AD, et al. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 2011;8:817–28. doi: 10.4161/rna.8.5.16041. [DOI] [PubMed] [Google Scholar]
- Wang XY, Yin Y, Yuan H, et al. Musashi1 modulates mammary progenitor cell expansion through proliferin-mediated activation of the Wnt and Notch pathways. Mol Cell Biol. 2008;28:3589–99. doi: 10.1128/MCB.00040-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warycha MA, Christos PJ, Mazumdar M, et al. Changes in the presentation of nodular and superficial spreading melanomas over 35 years. Cancer. 2008;113:3341–8. doi: 10.1002/cncr.23955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wich LG, Hamilton HK, Shapiro RL, et al. Developing a multidisciplinary prospective melanoma biospecimen repository to advance translational research. Am J Transl Res. 2009;1:35–43. [PMC free article] [PubMed] [Google Scholar]
- Wyman K, Atkins MB, Prieto V, et al. Multicenter phase II trial of high-dose imatinib mesylate in metastatic melanoma: significant toxicity with no clinical efficacy. Cancer. 2006;106:2005–11. doi: 10.1002/cncr.21834. [DOI] [PubMed] [Google Scholar]
- Yi M, Yang J, Chen X, et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J Cell Physiol. 2011;226:2360–9. doi: 10.1002/jcp.22568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalaudek I, Leinweber B, Hofmann-Wellenhof R, et al. The epidermal and dermal origin of melanocytic tumors: theoretical considerations based on epidemiologic, clinical, and histopathologic findings. Am J Dermatopathol. 2008a;30:403–6. doi: 10.1097/DAD.0b013e3181734e9a. [DOI] [PubMed] [Google Scholar]
- Zalaudek I, Marghoob AA, Scope A, et al. Three roots of melanoma. Arch Dermatol. 2008b;144:1375–9. doi: 10.1001/archderm.144.10.1375. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006;103:9136–41. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.