

Abstract
The migration of oligodendrocyte progenitor cells (OPCs) to the white matter is an indispensable requirement for an intact brain function. The mechanism of cell migration in general is not yet completely understood. Nevertheless, evidence is accumulating that besides the coordinated rearrangement of the cytoskeleton, a finetuned interplay of ion and water fluxes across the cell membrane is essential for cell migration. One part of a general hypothesis is that a local volume increase towards the direction of movement triggers a mechano-activated calcium influx that regulates various procedures at the rear end of a migrating cell. Here, we investigated cell volume changes of migrating OPCs using scanning ion conductance microscopy. We found that during accelerated migration OPCs undergo an increase in the frontal cell body volume. These findings are supplemented with time lapse calcium imaging data that hint an increase in calcium content the frontal part of the cell soma.
Cell migration plays an essential role in a wide variety of cell types, being crucial for organogenesis, wound healing, immune surveillance and tumor metastases formation. It is regulated by a complex interplay of the actin cytoskeleton dynamics, cell-cell and cell-substrate interactions, transporters, ion channels and aquaporins1,2,3.
Migrating cells are polarised along their axis of movement4,5,6. In most cell types, the leading edge consists of the lamellipodium, a thin, wide and highly motile cell extension7. The trailing edge of the cell consists of the cell body containing the nucleus. Migrating cells undergo changes in shape as investigated e.g. by light microscopy in Chinese Hamster Ovary (CHO) and transformed Madin-Darby canine kidney (MDCK-F) cells8,9.
The conception that local volume changes accompany cell motility evolved from the investigation of the role of ion channels and aquaporins in cell migration2,3. Ion channels regulate cell volume10 and in turn, cell volume regulates the integrity of the cytoskeleton that polymerises within the lamellipodium and thus protrudes the cell towards its direction of movement11,12,13. Furthermore, migrating nasopharyngeal carcinoma cells showed increased volume regulation compared to non-migrating ones14.
Among the ion channels, transporters and aquaporins implicated in cell migration the Na+/H+ ion exchanger NHE1, the Cl−/HCO3− anion exchanger AE2 and the aquaporin AQP1 are located at the cell front in fibroblasts, endothelial and CHO cells, respectively15,16,17. The aquaporins AQP4 and AQP9 have been found to enhance lamellipodial activity in astroglial cells and neutrophil granulocytes9,18 and AQP3 has been shown to be essential for the migration of sperm cells19. The influx of Na+ and Cl− through transporters has been proposed to cause a local increase in osmotic pressure that is accompanied by water influx through aquaporins and hence local cell swelling2,3,9,17,20. This increase in local cell volume leads to traction forces in the plasma membrane that might activate mechanosensitive Ca2+-channels. This hypothesis is supported by the finding that in migrating keratinocytes and fibroblasts the Ca2+-influx is mediated by mechanosensitive channels21,22,23. Calcium channels of the transient receptor potential (TRP) superfamily that activate upon mechanical stimulation have been shown to enhance migration or migration related processes in epithelial cells, spinal neurons and hepatoblastoma cells24,25,26,27. Furthermore, it has been shown that local calcium transients mediated by the TRP family member TRPM 7 direct the migration of human lung fibroblasts28.
Various reports suggest that the distribution of calcium concentration and its alterations play a role in cell migration. In granule cells, which migrate in a saltatory manner, the rate of calcium transients correlates with the migration velocity and an impairment of the frequency or amplitude of the transients impairs migration29,30. Furthermore during the migration of neutrophils and fish keratocytes cyclic changes in intracellular calcium concentration have been observed31,32. In eosinophils, fibroblasts and MDCK-F cells the intracellular calcium is not distributed uniformly but as a gradient with higher Ca2+ concentration in the cell body33,34,35,36. Neutrophils show a higher calcium concentration at sides of stronger adhesion37 and different regions of the cell show different decay kinetics of the calcium transients38.
At the trailing edge of the cell, the calcium regulated potassium channel KCa3.1 plays a pivotal role in cell migration. Its blockade slows down migration in epithelial cells, melanoma cells, fibroblasts and microglia5,39,40,41,42. If internal calcium signaling is switched off the entire cell volume increases as shown by cell volume measurements of fixed MDCK-F cells24. The potassium efflux, putatively accompanied by an efflux of chloride, leads to a local cell shrinkage at the trailing edge of the cell as detected by atomic force measurements of living MDCK-F cells43. Furthermore, inhibiting volume activated chloride currents also inhibits migration in glioma cells44,45.
Additionally, theoretical considerations contribute to the concept that volume changes occur during cell migration. Migration is facilitated by an osmotic gradient46 and even a simple model omitting intracellular regulation and signaling pathways but including hydrodynamic pressure and swelling stresses exhibits the prominent dynamics of the lamellipodium47.
The migration of OPCs has already been investigated in detail by video time lapse microscopy. OPCs move in a saltatory manner alternating between a resting and a moving state with a mean velocity of 10 μm/h ± 7 μm/h and a maximum velocity of 120 μm/h to 140 μm/h (both on poly-L-lysine)48. It has been reported that both increases and decreases of the basal internal calcium level impair the migration of OPCs49 and that the migration correlates with internal calcium transients50. Furthermore, the golli proteins which regulate the migration of OPCs also regulate the expression of the TRP family member TRPC 151 which is proposed to be a component of store operated calcium channels52.
To test the hypothesis that an additional mechano-sensitive component plays a role during migration, we first investigated the local changes in cellular volume during random migration of OPCs.
Cellular volume changes were determined using scanning ion conductance microscopy (SICM), a tool to investigate the topography of non-conducting surfaces submerged with electrolyte in a contact free manner53,54. This method uses the change in access resistance that occurs if an electrolyte filled glass microelectrode is approached towards a non conducting surface to determine the topography of the scanned object. The application of SICM to image living cells has been introduced by imaging melanocytes and human colon cancer cells55,56. Since then, SICM has been applied to investigate living cells in manifold studies57,58. Volume determinations by SICM have been validated by confocal light microscopy59. Enhanced by a backstep mode it enables one to monitor local volume changes of entire cell bodies60,61,62 as well as to track migrating cells63 and is thus a promising tool to investigate local volume changes in moving cells.
Using this method we now detected local swellings in the range of 15% at the frontal part of the soma preceding fast cell body propulsion.
Furthermore, we monitored whether subcellularly segregated Ca2+ changes can be detected during the random migration of OPCs. Although calcium sensitive dyes proved to be toxic during long term recordings of these cells or showed a tendency to impede OPC migration, we succeded in detecting subcellular intracellular calcium increases which were localised towards the direction of movement using very small dye concentrations.
Our findings demonstrate for the first time that local water fluxes leading to cell swelling and consequent propulsion of the nucleus occur during saltatory cell migration of OPCs. The observation of local calcium increases further hints at a mechano-sensitive component in the regulation of the internal calcium concentration during OPC migration.
Results
Volume changes of mature oligodendrocytes
Successive scans (127 in total) were obtained from 21 mature oligodendrocytes to investigate volume fluctuations in stationary cells. As an example, three-dimensional plots of four successive scans of an oligodendrocyte are shown in Fig. 1A. The oligodendrocyte remained stationary (migration distances δ were 0.4 μm, 0.5 μm and 0.9 μm, respectively, indicated by the plots in Fig. 1B) during the observation time of 39 minutes and only slight changes in cell shape occured. The corresponding volumes V and relative volumes Vr are depicted in Fig. 1C. A slight swelling is visible between scans 1, 2 and 3 (V1 = 1.79 pL, V2 = 1.83 pL and V3 = 1.93 pL) whereas the cell had shrunk back to 1.76 pL in the last scan shown. Accordingly, the decadic logarithm (lg) of the relative volumes (depicted as the black bars in Fig. 1C) is positive between the first three scans and negative between the last two scans (lg Vr = 0.01; 0.02 and 0.04, respectively, corresponding to +2%, +6% and −9% at linear scale). Fig. 1D shows the histogram of the relative volumes between all successive scans performed (logarithmic scale, n = 106, bin size 0.011). Mean value is 0.00 ± 0.04 thus indicating a constant mean volume. The logarithmic representation of the relative volume was chosen because it matches a gaussian distribution (dotted line in Fig. 1D, R2 = 0.86; in contrast R2 = 0.79 when the data is plotted at a linear scale). Furthermore, applying a linear scale would assume the same probability for the cell to swell by 100% and to shrink by 100%, thus to disappear completely. In contrast, handling the data using a logarithmic scale confers an equal weight to an increase and a decrease of the cell volume by the same factor which we assume to be more realistic.
Figure 1. Determination of volume changes in mature oligodendrocytes by scanning ion conductance microscopy.

(A,B) Three dimensional (A) and top-view (B) plots of four successive scans of a mature oligodendrocyte. Scans were obtained immediately after the previous scan had finished. Lateral step size was 1 μm, vertical step size was 0.1 μm, scan size was 30 × 30 μm2, data shown interpolated by cubic splines. Grey scale indicates height. Note that the oligodendrocyte remained stationary (dislocation amounted to 0.4 μm, 0.5 μm and 0.9 μm, respectively). (C) Calculated absolute (white bars) and relative volume (black bars, logarithmically scaled axis at the right, for easier recognition changes in percent are indicated by the small grey axis) of the cell shown in (A, B). The cell slightly swelled between scans 1, 2 and 3, yielding positive relative volumes and shrank approximately to the volume of scan 1 in scan 4 yielding a corresponding negative relative volume. (D) Histogram of relative volumes of mature oligodendrocytes. Bars represent counts of relative volumes per bin, bin size is 0.011; n = 106 calculations of relative volume between two successive scans obtained from 21 different cells. Note that bins represent relative volume at a logarithmic scale. Mean is 0.00 ± 0.04 (SD). Dotted line indicates a gaussian fit to the data, R2 = 0.86.
The mean migration distance between two successive scans amounted to 0.56 μm ± 1.18 μm. This is below the lateral resolution of the scans64 indicating that mature oligodendrocytes maintain stationary.
Volume changes of oligodendrocyte progenitor cells
161 pairs of successive scans were obtained from 33 different OPCs. A summary of the analysis of velocity and volume changes is shown in Fig. 2. The histogram of the velocity distribution is shown in Fig. 2A, bin size is 1 μm/h; n = 161. Mean velocity amounted to 5.6 μm/h ± 5.1 μm/h, maximum and minimum velocities determined amounted to 30.03 μm/h and and 0.17 μm/h, respectively.
Figure 2. Average velocities and relative volume changes of oligodendrocyte progenitor cells.
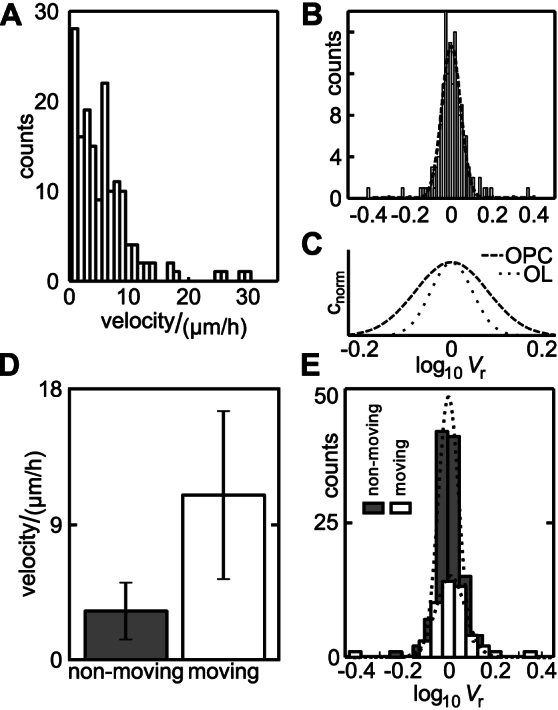
(A) Histogram of the velocities determined from SICM scans (see Material and Methods section, bin size 1 μm/h; n = 161 determinations from 33 different cells; mean velocity: 5.6 μm/h ± 5.1 μm/h (SD), minimum and maximum velocities: 0.17 μm/h and 30.03 μm/h, respectively). (B) Histogram of the relative volume measurements of OPCs. Bars represent counts of relative volume per bin, bin size is 0.011 (as in Fig. 1D), bins represent relative volumes at a logarithmic scale. Mean value: 0.00 ± 0.07 (SD). Dashed line indicates a gaussian fit to the data, R2 = 0.93. (C) Normalised fits to the relative volumes of adult oligodendrocytes (OL, dotted line, see Fig. 1D) and OPCs (dashed line, see B) on the same x-scale. (D) Mean velocities of the cells classified as moving and non-moving. Moving: v > 6.5 μm/h, yielding 30% moving cells. Error bars indicate ± SD. (E) Histograms of relative volumes of non-moving (grey bars, n = 112) and moving (white bars, n = 49) cells. Bin size 0.05, relative volume displayed at logarithmic scale. Mean values: 0.02 ± 0.10 and 0.00 ± 0.05 for moving and non-moving cells, respectively. Dotted lines indicate a gaussian fit to the respective data. R2 = 0.98 for both fits.
The histogram of the relative volumes (plotted at a logarithmic scale) is depicted in Fig. 2B with a bin size of 0.011 as in Fig. 1D. Mean value was 0.00 ± 0.07. The distributions of the relative volumes of OPCs (dashed line) and adult oligodendroctyes (OLs, dotted line) are plotted in Fig. 2C, clearly showing the larger fluctuations observed in OPCs. However, on the average the volume remained constant. To investigate specific volume changes occuring potentially in migrating OPCs we had to distinguish between moving and non-moving OPCs. A previous investigation of OPC migration had reported a mean velocity of 10 μm/h ± 7 μm/h and a fraction of about 50% of moving cells48. Hence, we classified cells as moving if their velocity exceeded 6.5 μm/h. This limit was defined as the mean minus half the SD of the velocity of moving OPCs according to the previous report.
The classification of cells into moving and non-moving cells yielded a fraction of 30% moving cells with a mean velocity of 11.1 μm/h ± 5.7 μm/h. The remaining cells displayed a mean velocity of 3.2 μm/h ± 1.9 μm/h, see Fig. 2D. The mean migration distances between two successive scans amounted to 1.82 μm ± 1.68 μm and 0.43 μm ± 0.66 μm for moving and non-moving cells, respectively. The histograms of lg Vr of moving (white bars) and non-moving (grey bars) cells are depicted in Fig. 2E. Mean values amount to 0.00 ± 0.05 for non-moving and 0.02 ± 0.10 for moving cells (0% ± 12%; n = 112 and 6% ± 27%; n = 49, respectively), indicating a slight tendency of the moving OPCs to swell. The larger variance of the moving cells (±0.10) compared with the stationary cells (±0.04 for differentiated oligodendrocytes as well as ±0.05 for progenitor cells) indicates a larger volume fluctuation of migrating cells.
Local volume changes of accelerating oligodendrocyte progenitor cells
The hypothesis that cell migration is accompanied by a local volume increase at the moving front of the cell2,3,9,17,20 was tested by calculating the relative local volumes of cells prior to accelerated migration. Cell volume changes of randomly chosen pairs of migrating OPCs did not significantly differ from cell volume changes of stationary OPCs (see Fig. 2D), as expected if migrating cells oscillate between swelling and shrinkage. To investigate whether systematic volume changes occur during defined time periods of saltatory migration, we now investigated volume changes of cells during the acceleration of migration. To select accelerating cells we analysed sets of three successive scans as shown in Fig. 3A and determined the corresponding velocities. For the example shown in Fig. 3A the corresponding velocities amounted to 6.6 μm/h and 10.8 μm/h, respectively (black bars in Fig. 3B). As defined in Fig. 2 we classified cells with a velocity exceeding 6.5 μm/h as moving (indicated by the grey area in the left diagram of Fig. 3b) yielding 32 scan triples of moving and 96 scan triples of non-moving cells. Additionally, we classified a moving cell as accelerating if the second velocity was at least 1.25 fold of the velocity between the first two scans, corresponding to approximately 0.1 at a logarithmic scale (indicated by the grey area in the right diagram of Fig. 3B; for the example cell lg vr amounted to 0.22 corresponding to an increase in velocity by 65%). Nine of the 32 investigated scan triples of moving cells matched the criterion of accelerating cells.
Figure 3. Determination of velocity and volume of an accelerating progenitor cell.

(A) Velocity determination of a typical cell included in the migration analysis. Three dimensional plots of a set of three successive scans of a moving oligodendrocyte progenitor cell. Data interpolated by cubic splines, scan size 30 × 30 μm2, step size 1 μm laterally and 0.1 μm vertically. (B) Corresponding velocities between scans one, two and three (left diagram). A cell was classified as moving if the velocity between scans two and three exceeded 6.5 μm/h (according to Schmidt et al.48). Additionally, a cell was classified as accelerating if the relative velocity exceeded 0.1 at a logarithmic scale (125% in linear representation, right diagram). (C, D) Volume determination. (C) Front and rear cell body volumes of the cell shown in (A). Note that the cell swelling from about 0.8 pL to nearly 1 pL is dominated by an increase in the frontal volume. (D) Corresponding relative volumes of cell body front, rear and total indicating that the cell swelling is dominated by a swelling of the frontal part of the cell.
We now investigated whether volume changes occured between the first two scans prior to accelerated migration (see Fig. 3C). To investigate the volume of the front and rear part of the cell body separately the cell was subdivided at the level of C90, the centroid of the area exceeding 90% of the maximum cell height which approximates the location of the nucleus65. The cell shown in Fig. 3A underwent an increase in total cell soma volume from 773 fL to 983 fL (see Fig. 3C) that was dominated by an increase in the frontal soma volume (362 fL to 523 fL, black bars in Fig. 3C). The predominance of the frontal volume increase becomes more apparent in the plot of the relative volumes as depicted in Fig. 3D.
Fig. 4A shows the first scan of a set of three scans from a different cell classified as accelerating. The height profiles of the first two scans (t = 0 min and t = 12 min) along the black line that corresponds to the direction of movement of the cell are plotted in Fig. 4B (dotted and solid line, respectively). While the height of the profile nearly remained constant, a considerable extension of the cell soma front of more than 2 μm towards the direction of movement is visible.
Figure 4. Cell bodies of accelerating cells swell at their frontal part prior to body dislocation.
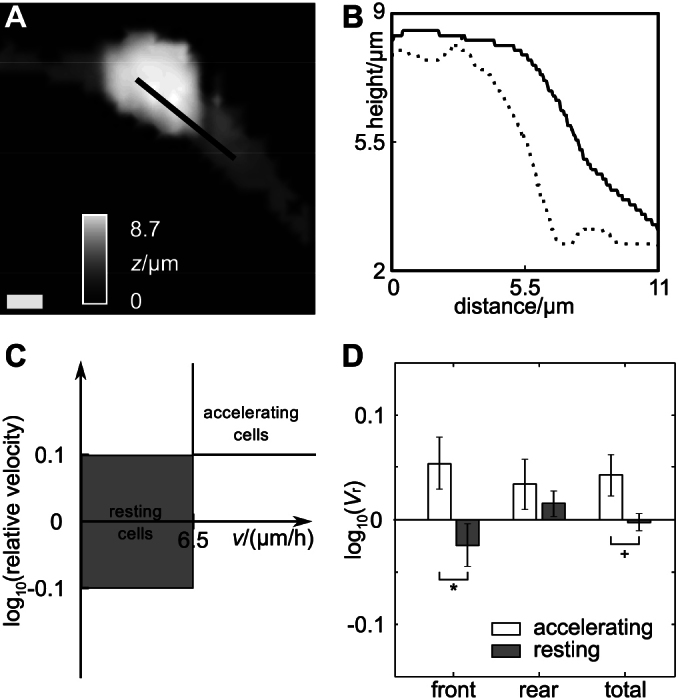
(A) Top view of an SICM scan of an oligodendrocyte progenitor cell. White lateral scale bar represents 3 μm, grey gradient indicates height. Black line indicates the position of height profiles shown in (B). Dotted line represents the profile of the scan obtained at t = 0 min, solid line represents the profile of the scan obtained at t = 12 min. Note the considerable swelling in the direction of the frontal part of the cell body. (C) Schematic illustration of the definition of accelerating and resting cells. Cells were classified as accelerating when the velocity between scans two and three of the corresponding scan triple exceeded 6.5 μm/h and the relative velocity logarithmically exceeded 1.25 (lg(vr) > 0.1). Cells were classified as resting if the velocity between scans two and three of the corresponding set of three scans was lower than 6.5 μm/h and the logarithmic representation of its relative velocity ranged between −0.1 and + 0.1. (D) Relative volume of accelerating (white bars, n = 9) and resting (grey bars, n = 12) cells between the first and second scan of a scan triple (logarithmic representation, error bars indicate SEM). Note that on the average the cell body frontal volumes increased significantly (p < 0.05, indicated by the asterisk) and the entire cell body volume with a probability of about 93.5% (p < 0.065, indicated by the plus sign) whereas the volume of the rear part of the cell body only showed insignificant changes.
To investigate whether this local cell soma swelling generally occurs prior to acceleration of migration, we compared average soma volume changes of accelerating and resting cells. Resting cells were defined as all cells displaying a velocity below 6.5 μm/h and a relative velocity in the range of 0.8 to 1.25 (−0.1 ≤ lg vr < 0.1 approximately, see Fig. 4C) from our total set of three scans of cells. Twelve out of 96 investigated sets of three scans matched the criteria for resting cells. Scan triples that were neither classified as resting nor accelerating consisted mostly of cells with no detectable increases in velocity and decelerating cells. These cells were excluded from the analysis.
As shown in Fig. 4D accelerating cells (white bars) show a significant frontal cell soma swelling compared with resting cells (grey bars; p < 0.05, indicated by the asterisk). On average, the swelling of the frontal part of the soma amounted to 15% ± 7% (0.05 ± 0.03 at a logarithmic scale). Note that here errors indicate ± SEM since we want to illustrate the significance rather than the variation. In contrast, resting cells showed a local front volume decrease of 4% ± 5% (SEM; lg Vr = 0.03 ± 0.02).
Rear soma volume changes were not significantly different from those of resting cells (accelerating cells: 9% ± 6%; lg Vr = 0.03 ± 0.02; resting cells: 4% ± 3%; lg Vr = 0.02 ± 0.01; errors indicate ± SEM) indicating that the observed volume increase prior to cell body acceleration can be mostly attributed to a swelling of the frontal part of the cell body.
Alterations in intracellular Ca2+ fluorescence during migration
To investigate whether the local volume changes might trigger increases in intracellular calcium concentration, we monitored Ca2+ transients using the Ca2+ sensitive dye Fluo8H-AM with an epifluorescent microscope. From 28 experiments, seven cells clearly identifyable as OPCs showed Ca2+ stainings clearly exceeding the background. The large number of failures is due to the low dye concentration required to observe OPC migration, the relatively large magnification used to allow spatial separation of potential Ca2+ signals as well as due to drifts of the incubation chamber and the lack of humidification during the recordings. This restricted the data analysed for Ca2+-imaging to data recorded during the first two hours of recording. We assume, that at this time the dye had less likely diffused into the cellular compartments, and that the detected signals were thus recorded from the cytosol. From the seven cells analysed, four showed migration.
Fig. 5A shows changes in the Fluo8H intensity integrated over the entire cell during the random migration of an OPC that showed both a non-migrating (red line) and a migrating phase (green line). As a control, the intensity trace of an OPC showing no migration during the entire recording is shown in Fig. 5B. The Ca2+ signal changes during migration clearly exceed the random changes during the non-migrating phase as well as those of the non-migrating OPC. The amplitude of the two largest signal changes of each of the seven OPCs, divided into migrating and non-migrating ones, is shown as box plots in Fig. 5C. Here, the median of the analysed amplitudes is shown as the horizontal line separating the light and dark grey boxes, which represent the lower (dark grey) and upper (light grey) quartile of the data. Thus 50% of the analysed data is located within the box comprising both the light and the dark grey one (interquartile range, IQR). The whiskers represent the minimal or maximal values located within 1.5 IQRs below or above the median, respectively. The median amplitudes of non-migrating (5.8%) and migrating OPCs (16.5%) clearly differ and the upper quartile of non-migrating OPCs (8.7%) does not overlap with the lower quartile of the migrating OPCs (14.1%). While in migrating OPCs all data were within 1.5 IQRs around the median (min: 8.7%, max: 22.8%), we observed an outlier (13.8%, dot in Fig. 5C) in non-migrating cells (min and max within ± 1.5 IRQ: 3.0% and 9.8%, respectively).
Figure 5. A local Ca2+ increase located at the front of the cell body can be observed in migrating OPCs.

(A) Fluorescence intensity signal indicating cytosolic free calcium load of an OPC during a phase without migration (red) and during migration (green; note that both traces were recorded from the same cell). (B) Representative trace of a cell showing no migration during the time of observation serving as control. (C) Box plots of the amplitude height of the two largest intensity changes in non-migrating (n = 6 from 3 cells) and migrating (n = 8 from 4 cells) OPCs. Boxes indicate lower quartile, median and upper quartile, respectively; whiskers indicate maximum and minimum values within 1.5 interquartile ranges (all values beyond this range indicated by dots). (D–G) Superimposition of phase contrast and differences in the fluorescences intensity. The differences are clearly located at the side of the cell pointing towards the direction of movement. Insets show the corresponding amplitude of the cytosolic free calcium load. Scale bars in all images represent 10 μm, all images are flipped such that the direction of movement points towards the lower right of the images. Scale bars in the inset in G apply to all insets. Asterisk in the colour gradient bar indicates the non-linearity due to the applied filter.
Fig. 5D–G show the location of Ca2+ signal amplitudes in migrating OPCs. The corresponding amplitudes of the integrated cytosolic free calcium load are shown in the insets (the corresponding scale is shown in Fig. 5G). Although in general Ca2+ signals were more homogeneously distributed over the entire cell (not shown), in some recordings in all four migrating OPCs, consistent local Ca2+ signal changes were located at the cell side pointing towards the direction of movement.
Discussion
A local volume increase has been postulated to link ion and water influx at the frontal part of the cell and ion and water efflux at the rear part of the cell by activating stretch-activated Ca2+-channels2,3. Here we succeded to show that in oligodendrocyte progenitor cells indeed a volume change of about 15% can be observed at the frontal part of the cell body prior to accelerated migration.
In contrast to OPCs, on average adult oligodendrocytes did neither alter their position nor volume. The observed deviation of ± 0.04 (Fig. 2) in cellular volume most likely includes both physiological volume fluctuations of the oligodendrocytes and uncertainties of the volume determination by the instrument. Furthermore, as mentioned in the Materials and Methods section, theoretical considerations estimated the error in the determination of the ratio of two volumes to amount to 8%. Thus, we conclude that volume changes exceeding 10% (that is 0.04 in logarithmic scale) on the average are not a result of the uncertainties of the applied determination method of the volume changes, but represent true cell swellings.
OPCs on average did not alter their volume systematically, but showed larger fluctuations than mature oligodendrocytes. Since it is known that only a fraction of OPCs migrate48,49,50 we devided our set of recordings into migrating and non-migrating cells by applying a threshold of 6.5 μm/h. This is in the range of the threshold for migrating cells defined previously50 and furthermore represents the mean minus half of the SD of the velocity reported by Schmidt et al48. This threshold ensures, that no stationary cell was counted as migrating. Since the determination of the position of the OPC is based on the centroid of multiple pixels, the systematic error does not exceed the size of a pixel which thus served as an upper boundary. A pixel size of 1 μm and a corresponding acquisition time of 10 min results in a maximum error of 6 μm/h. This threshold yielded a slightly smaller fraction of migrating cells than previously reported under similar conditions48. The smaller fraction of migrating OPCs is due to limitations of the SICM setup presently used. Since the cells were positioned in the center of the scanning frame, a distance of approximately 15 μm could be migrated by a cell without leaving the area of observation (resulting from half the diagonal of the scan area minus half of the cell body length (approx. 10 μm)). Additionally, we only investigated the cells that were located within the scan area for a period of at least three scans to determine changes in velocity. Hence, cells faster than 30 μm/h were discarded and do not contribute to the fraction of migrating cells analysed here. This approximation matches with the maximum velocity determined by SICM of 30.03 μm/h. During our investigations, we observed additional 19 cells that were discarded due to this limit.
While on average the volume of non-migrating OPCs was constant with a standard deviation similar to that of adult oligodendrocytes, migrating OPCs showed a slight volume increase and a larger standard deviation. This indicates that in migrating OPCs larger volume fluctuations occur.
OPCs migrate in a saltatory manner48,50, thus accelerating and decelerating. In phases of migration with small changes in velocity, potential volume increases and compensating decreases might not be detectable with the temporal resolution of the SICM presently used. Thus, only recordings of accelerating OPCs were evaluated. OPCs classified as accelerating showed a significant volume increase towards the direction of movement prior to accelerated migration if compared to resting cells. To our knowledge, our results are the first observations of a local volume increase towards the direction of movement confirming the occurence of the water fluxes postulated in a general model of cell migration2,3,17,18,20.
If, as postulated by the model, the local volume increases trigger a mechano-sensitve influx of Ca2+, a local increase in intracellular Ca2+ should be observable as well. Indeed, increases in the fluorescence intensity of the calcium binding dye Fluo-8H that exceeded the Ca2+ signal changes of non-migrating cells could be observed in randomly migrating OPCs. This is in good agreement with previously published data showing that migration velocity and intracellular Ca2+ activity correlate in OPCs50. In addition, we showed that during the random migration of OPCs, cytosolic free Ca2+ load increases in the range of 10% to 20% could be observed that were mainly located at the side of the cell body pointing towards the direction of movement.
Since OPCs died within the first hour after staining with the ratiometric dye Fura-2 and showed no migration if stained with a mixture of Fluo8-H and Calcein blue, ratiometric determinations of the calcium signal could not be recorded. In contrast to ratiometric measurements, the observed signal, which was integrated over the entire cell, does not represent the intracellular calcium activity, but the cytosolic free calcium load. Nevertheless, the spatial distribution of the calcium signal most likely comprises both volume changes and thus spatial increases in sample thickness and increases in the cytosolic calcium activity. Since the subcellular increases in cytosolic free calcium load which were located at the side of the cell pointing towards the direction of movement were accompanied by an increase in the cytosolic free calcium load integrated over the entire cell, it can be excluded that they were caused solely by volume changes. A local increase of the Fluo8H signal due to a local volume increase without accompanying calcium fluxes would instead be accompanied by a constant or decreasing signal of the calcium load of the entire cell.
Assuming a volume increase of 11% of the entire cell as determined by the SICM measurements, the observed increases in cytosolic free calcium load of 10% to 20% indicate that the increased volume does not lead to a decreased calcium activity, but is compensated either by Ca2+ influx or release from internal stores. Furthermore, assuming that the total calcium content increase occurs only at the frontal part of the cell (20% to 40% increase at the frontal part of the cell), which swells by 15% as detected by SICM measurements, the resulting calcium activity increase would be in the range of 4% to 22% (further assuming equal frontal and rear volumes before swelling and a linear relationship between fluorescence intensity and calcium activity). Although this is a very rough estimation, it hints at an increased calcium activity at the frontal part of migrating OPCs that might be mediated via mechano-sensitive channels.
However, since we found that ratiometric measurements with OPCs were not realisable, detailed analysis of the spatial calcium changes during OPC migration would require the development of a more sophisticated instrument combining SICM and confocal methods as well as a migration assay with a lower failure rate.
Furthermore, since OPCs stained with Fluo8H-AM did show practically no migration at room temperature, we observed Ca2+ fluctuations during random migration at 37°C. Under these conditions and with an increased external calcium concentration as well as supplements of PDGF and bFGF, we observed migration velocities of up to 150 μm/h, which is in good agreement with previous reports48,50. Nevertheless, as explained above, only migration velocities up to 30 μm/h could be recorded with our presently available SICM, rendering simultaneous determination of both internal Ca2+ and cellular volume changes impossible. Cells which migrated faster than 30 μm/h were not included in our SICM analysis.
Although we could not directly link temporal sequences of local volume and local calcium activity increases with the instrumentation presently available, our data show that both phenomena occur during random migration of OPCs. Since OPC migration has been shown to be correlated with intracellular Ca2+ alterations mediated by voltage operated calcium channels (VOCCs)50, our data can be interpreted in two ways: Firstly, the voltage activated increase in intracellular calcium could induce the observed volume increase, secondly, the local increase in intracellular Ca2+ triggered by the volume increase through mechano-sensitive ion channels might create the initial depolarisation required to trigger a subsequent Ca2+ influx via VOCCs.
If one considers the ratio of internal Ca2+ and particularly K+ concentration, which is in the range of 10−3 to 10−4, an increase of Ca2+ by several per cent seems unlikely to cause an osmotic effect per se. However, Ca2+ might induce reactions that increase intracellular osmolarity, either by changing the electric potential which in turn induces ion influxes or via its role as a mediator in manifold biochemical signalling cascades.
The hypothesis that the volume increase triggers a calcium increase via mechano-sensitive ion channels is supported by several observations. Firstly, if VOCCs are blocked using verapamil or nifedipine, a basal movement of OPCs remains, although severely reduced, but blocking external Ca2+ by the addition of EGTA instead exceeds the effect of VOCC blockers50.
Secondly, OPCs are known to express several ion channels and transporters that are supposed to play a role in the regulation of the osmotic pressure during migration (see introduction for details), although one should consider that not all OPCs express the same set of ion channels66. OPCs are known to express both a Na+/H+ and a Cl−/HCO− exchanger67, suggested to be the mediator of a local increase in osmotic pressure2,3,17,18,20. Futhermore, a Ca2+ dependent potassium outward current is likely to mediate a compensatory volume decrease at the rear end of the cell5,39,40,41,42,68. Potassium currents have been investigated in OPCs. Both, spindle shaped OPCs as investigated in this study and OPCs in a later phase of development show similar potassium outward currents. In later OPCs, some of these currents have been shown to depend on internal Ca2+ 69.
Although a sole efflux of K+ might be sufficient to cause a compensatory volume decrease at the rear end of the cell, most likely an accompanying efflux of Cl− occurs, since the hyperpolarisation by the K+ efflux would support chloride efflux. Virtually nothing is known about chloride currents not activated by ligands in OPCs. Nevertheless, a large intracellular Cl− concentration is found in OPC descendants70. Furthermore, both astrocytes and glial cell derived tumor cells express volume regulated chloride channels45,71,72.
In our report, we showed that scanning ion conductance microscopy is an excellent tool to quantify cellular volume changes during cell migration in culture. The application of this emerging technique to observe randomly migrating OPCs revealed for the first time that local volume changes at the frontal part of the cell body, hence in the direction of movement, occur during accelerated migration of OPCs. Recent improvements of this technique73 allow the observation of flat samples such as the lamellipodium with high spatial and temporal resolution, potentially enabling the observation of volume changes in cellular structures where volume relevant proteins are located.
Furthermore, we observed local increases in the internal calcium activity that, too, were located at the frontal part of the cell body of migrating OPCs. Albeit various interpretations of these observations are conceivable, our findings agree well with the postulated model that a local volume increase triggers a succeeding increase in internal calcium concentration. Notwithstanding that an exact determination of the causal link between volume changes and Ca2+ surges is still lacking, our data contribute a yet missing aspect to unravel the mechanism of cell migration.
Methods
Cell culture
Cells were obtained and cultured similarly as described previously74. Briefly, after passing the brains of two to four days old Sprague-Dawley (SICM observations) or Wistar (Ca2+ imaging) rats through nylon meshes of 125 μm and 36 μm pore size, the cells were collected in phosphate buffered saline (PBS, containing in mM: 138 NaCl, 8.1 Na2HPO4, 2.7 KCl, 1.47 KH2PO4) and centrifuged for 10 minutes at 900 rpm and 4°C. The cells were then resuspended in glial mixed culture medium (GMM, consisting of a mixture of HAM's F12 and DMEM (1:1) supplemented with 10% fetal calf serum, 100 U/mL penicillin and 100 μg/mL streptomycin) and pre-cultured in cell culture flasks (in humidified air containing 5% CO2 at 37°C) until a dense layer of cells had grown. Cell debris and the different glial cell types then were separated by their different adherences to the flask: To remove microglia and debris flasks were shaken for 3 hours at 180 rpm. The supernatant was replaced by fresh GMM and the cell culture flasks were shaken for additional 16 to 18 hours. The supernatant now containing OPCs was centrifuged for 5 min at 1700 rpm at 4°C. The pellet was resuspended in GMM and preplated for 45 minutes in uncoated cell culture dishes to remove cells that were not OPCs but had been detached by the shaking procedure. The supernatant containing the non adherent cells was diluted to approximately 50000 cells/mL. In dependence on the further experiments, cells were then treated differently as detailed below.
Scanning ion conductance microscopy
For SICM measurements, 500 μl of the the cell suspension were transferred into removable glas rings (diameter 1.5 cm) which had been placed in the center of poly-L-lysine coated plastic cell culture dishes (diameter 3.5 cm). After one to three hours the rings were removed and medium was exchanged to 1 mL GMM.
SICM measurements were performed using the same instrument as previously described operating in floating backstep mode60. Scans were performed in 3.5 cm plastic petri dishes after exchanging the medium to Leibovitz-15. In order to reduce migration speed with the aim of preventing cells from escaping out of the scanning frame, neither basic fibroblast growth factor (bFGF) nor plateled derived growth factor (PDGF) was added to the culture medium.
Scanning electrodes were filled with extracellular saline (containing in mM: NaCl 110, KCl 5.4, CaCl2: 1.8, MgCl2 0.8, Glucose 10, HEPES 10). Inner diameter of the scanning probes was about 1 μm, access resistance was about 4 MOhm, scan size was 30 μm × 30 μm with a lateral step size of 1 μm and a vertical step size of 100 nm. These step sizes allow recordings with a voxel volume in the same range as the volume of the confocal spot of a confocal microscope. Acquisition time was about 10 minutes per scan. Data are shown interpolated by cubic splines.
Volume determination
Volume calculations of mature oligodendrocytes were performed as described previously60 by calculating the sum of the volumes of each single column above each pixel: V = ΔxΔyΣizi; where Δx and Δy denote the lateral scan step size and zi denotes the detected height of the pixel i. Volume calculations of OPCs were performed by applying a boundary delimitation algorithm (BDA) as detailed in65. This method circumvents the problems that occur in the separation of processes from cell somata by defining a certain height threshold74,75. In brief, after aligning the cell parallel to the x-axis polynomials of third degree were fitted along the contour of sections parallel to the x, z-plane of the cell. The cell was subdivided at the level of C90, a single data point used to approximate the location of the nucleus. It was defined by the centroid of the area covered by the pixels exceeding 90% of the maximal cell height.
Relative volumes were calculated as Vr,n = Vn+1/Vn where n denotes the scan number. The influence of the lateral step size on the volume determinations was investigated by simulating scans of increasing step sizes on the basis of scans with small step sizes. The use of a step size of 1 μm led to an underestimation of the volume by 8% ± 4%; n = 9. While the underestimation is cancelled when calculating the ratio of two volumes, the error of 4% duplicates. Therefore we estimate that the determination of the ratio of two volumes is subject to a systematic error of 8%.
Velocity determination
Cell body position was approximated by C20, the centroid of the area exceeding 20% of the maximal cell height. Its coordinates were calculated as described before65. Velocities were calculated as vn = dn/Δtn where dn is the distance between the positions of C20 of the corresponding scans n and n + 1, Δtn denotes the time interval between the corresponding scans. For the calculations of the average migration distance, the direction of movement was taken into account. We defined the longest distance that was migrated without a change in direction as positive.
Data processing
SICM data were processed using Matlab (R2008a) and ImageJ (1.42q) software. Statistical calculations were performed in R (2.7.1). Tests for significance are Welch two sample t-tests. Errors indicate standard deviations (σ) unless otherwise noted. Standard error of the mean was calculated as 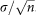 .
.
Before applying the BDA the SICM data was filtered using a threshold filter clipping every z-value below 1 μm to zero and plane corrected when necessary.
Ca2+ imaging
One milliliter of the cell suspension (see section Cell Culture) was transferred into poly-L-lysine coated plastic cell culture dishes. After adherence of the cells, medium was changed to proliferation medium (PM, containing DMEM, 1 × B27 (Gibco), 10 ng/ml PDGF, 10 ng/mL bFGF, 2 mM glutamin, 100 U/mL penicillin).
We tested ratiometric methods to investigate the calcium activity. However, OPCs stained with both Fluo-8H and Calcein blue did not show migration and OPCs stained with Fura-2 quickly died within one hour after staining. Thus, for Ca2+ recordings, cells were stained with 0.5 μM Fluo8H-AM in PM for 30 minutes at 37°C, 5% CO2 in humidified air. After rinsing with PBS, the medium was exchanged to fresh PM. Here, in contrast to SICM investigations, a medium containing growth factors was used since otherwise the Fluo-8H stained cells did not migrate. The intracellular Ca2+ activity during the random migration of OPCs was observed with an Olympus IX81 inverted microscope (FITC channel) using a 20fold objective, equipped with an incubation chamber (H.Sauer Laborbedarf, Germany), at 37°C and 5% CO2. Fluo8H intensity was recorded with a monochrome CCD camera equipped with an ICX 0285 sensor (Sony) featuring 1376 × 1032 pixels. No additional binning was applied. Exposure time was 1000 ms, images were recorded during four to six hours at 30 s intervals.
Fluo-8H intensity was analysed within a ROI comprising the entire cell during the time of observation, thus integrating over the entire cell volume and therefore limiting the volume dependence of the fluorescence signal to the activity depending binding properties of the dye. The fluorescence signal in this case represents the free calcium load of the cell.
The use of a single calcium sensitive dye as well as an epifluorescent microscope limits the interpretation of the Ca2+ recordings. Under these recording conditions, changes in the fluorescence signal can occur due to several causes: First, a change in intracellular Ca2+ activity is induced by transmembrane Ca2+ fluxes or release/uptake from intracellular stores at constant cell volume. Second, a volume change of the cells would affect the fluorescence signal in a ROI comprising the entire cell as follows: A sole redistribution of volume would lead to no changes in the signal since a volume and thus sample thickness increase at one position would be compensated by a local volume and thus sample thickness decrease at a another position. A volume increase without changes of the cytoplasmatic free Ca2+ load would lead to a dilution of both, Ca2+ and dye. Even if the activity dependence of the fluorescence intensity is neglected, this could at maximum lead to a constant signal integrated over the entire cell, but not an increase in intensity. Therefore, an increase in the fluorescence signal within a ROI comprising the entire cell can only be interpreted as a net cytosolic increase in free Ca2+ load. However, these recordings do not allow to determine the contribution of volume and calcium activity changes to the change in the fluorescence intensity signal.
Calcium recordings were corrected for background and bleaching, assuming an exponential decay, calcium signals were normalised to the mean intensity of the entire recording (F/〈F〉). Local alterations in the intracellular calcium level were calculated by subtracting the corrected fluorescence signal of two respective recordings. For easier visualisation of the localisation the differences were filtered by applying a smooth bandpass filter in the frequency domain, cutting off signals the size of which was below 3 px to 15 px and above 40 px to 100 px and subsequently normalised to the maximal difference (ΔFnorm, see Fig. 5). Note that after the application of that filter colour gradients do not represent the difference in calcium linearly. Amplitudes (ΔF/〈F〉) were determined as the maximum increase between two recordings within a time period of three minutes.
Author Contributions
PH and IDD designed the study. KM, PH and NKS recorded and analysed the data. PH and IDD wrote the manuscript. All authors reviewed and approved the final manuscript.
Acknowledgments
We thank Karen Abel, Monique Wolf, Maren D. Lange and Katrin Prost for contributing to the SICM measurements during undergraduate courses, Heidrun Breuker-Siraj for help with the cell culture, Stefan A. Mann, Jan Meijer and Bernd Kind for helpful discussions and Rolf Heumann and the RUBION-team for continuous support. Part of the work was supported by a fellowship to PH from the “Allgemeines Graduiertenkolleg der Ruhr-Universität Bochum”, some of the equipment used was initially funded by the DFG grant Schu929/51, Ca2+ imaging was supported by the MERCUR programme of the Mercator foundation (Pr-2011-0066).
References
- Lauffenburger D. A. & Horwitz A. F. Cell migration: a physically integrated molecular process. Cell 84, 359–369 (1996). [DOI] [PubMed] [Google Scholar]
- Schwab A., Nechyporuk-Zloy V., Fabian A. & Stock C. Cells move when ions and water flow. Pflugers Arch 453, 421–432 (2007). [DOI] [PubMed] [Google Scholar]
- Papadopoulos M. C., Saadoun S. & Verkman A. S. Aquaporins and cell migration. Pflugers Arch 456, 693–700 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi I. R. The polarization of the motile cell. J Cell Sci 112, 1803–1811 (1999). [DOI] [PubMed] [Google Scholar]
- Schwab A., Wojnowski L., Gabriel K. & Oberleithner H. Oscillating activity of a Ca(2+)-sensitive K+ channel. A prerequisite for migration of transformed Madin-Darby canine kidney focus cells. J Clin Invest 93, 1631–1636 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mañes S., Gómez-Moutón C., Lacalle R. A., Jiménez-Baranda S., Mira E. & Martínez-A C. Mastering time and space: immune cell polarization and chemotaxis. Semin Immunol 17, 77–86 (2005). [DOI] [PubMed] [Google Scholar]
- Abercrombie M., Heaysman J. E. & Pegrum S. M. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Exp Cell Res 67, 359–367 (1971). [DOI] [PubMed] [Google Scholar]
- Dieterich P., Klages R., Preuss R. & Schwab A. Anomalous dynamics of cell migration. Proc Natl Acad Sci U S A 105, 459–463 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadoun S., Papadopoulos M. C., Hara-Chikuma M. & Verkman A. S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 434, 786–792 (2005). [DOI] [PubMed] [Google Scholar]
- Lang F. et al. Functional significance of cell volume regulatory mechanisms. Physiol Rev 78, 247–306 (1998). [DOI] [PubMed] [Google Scholar]
- Grunnet M., MacAulay N., Jorgensen N. K., Jensen S., Olesen S.-P. & Klaerke D. A. Regulation of cloned, Ca2+-activated K+ channels by cell volume changes. Pflugers Arch 444, 167–177 (2002). [DOI] [PubMed] [Google Scholar]
- Mogilner A. & Edelstein-Keshet L. Regulation of actin dynamics in rapidly moving cells: a quantitative analysis. Biophys J 83, 1237–1258 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. L. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J Cell Biol 101, 597–602 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J. W. et al. Involvement of regulatory volume decrease in the migration of nasopharyngeal carcinoma cells. Cell Res 15, 371–378 (2005). [DOI] [PubMed] [Google Scholar]
- Grinstein S. et al. Focal localization of the NHE-1 isoform of the Na+/H+ antiport: assessment of effects on intracellular pH. EMBO J 12, 5209–5218 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M., Seeger P., Schuricht B., Alper S. L. & Schwab A. Polarization of Na(+)/H(+) and Cl(−)/HCO (3)(−) exchangers in migrating renal epithelial cells. J Gen Physiol 115, 599–608 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock C. & Schwab A. Role of the Na/H exchanger NHE1 in cell migration. Acta Physiol (Oxf) 187, 149–157 (2006). [DOI] [PubMed] [Google Scholar]
- Saadoun S., Papadopoulos M. C., Watanabe H., Yan D., Manley G. T. & Verkman A. S. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci 118, 5691–5698 (2005). [DOI] [PubMed] [Google Scholar]
- Chen Q. et al. Aquaporin3 is a sperm water channel essential for postcopulatory sperm osmoadaptation and migration. Cell Res 21, 922–933 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A. Function and spatial distribution of ion channels and transporters in cell migration. Am J Physiol Renal Physiol 280, F739–F747 (2001). [DOI] [PubMed] [Google Scholar]
- Doyle A. D. & Lee J. Cyclic changes in keratocyte speed and traction stress arise from Ca2+-dependent regulation of cell adhesiveness. J Cell Sci 118, 369–379 (2005). [DOI] [PubMed] [Google Scholar]
- Lee J., Ishihara A., Oxford G., Johnson B. & Jacobson K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 400, 382–386 (1999). [DOI] [PubMed] [Google Scholar]
- Munevar S., Wang Y.-L. & Dembo M. Regulation of mechanical interactions between fibroblasts and the substratum by stretch-activated Ca2+ entry. J Cell Sci 117, 85–92 (2004). [DOI] [PubMed] [Google Scholar]
- Fabian A. et al. TRPC1 channels regulate directionality of migrating cells. Pflugers Arch 457, 475–484 (2008). [DOI] [PubMed] [Google Scholar]
- Rao J. N. et al. TRPC1 functions as a store-operated Ca2+ channel in intestinal epithelial cells and regulates early mucosal restitution after wounding. Am J Physiol Gastrointest Liver Physiol 290, G782–G792 (2006). [DOI] [PubMed] [Google Scholar]
- Waning J. et al. A novel function of capsaicin-sensitive TRPV1 channels: involvement in cell migration. Cell Calcium 42, 17–25 (2007). [DOI] [PubMed] [Google Scholar]
- Vriens J., Janssens A., Prenen J., Nilius B. & Wondergem R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 36, 19–28 (2004). [DOI] [PubMed] [Google Scholar]
- Wei C., Wang X., Chen M., Ouyang K., Song L.-S. & Cheng H. Calcium flickers steer cell migration. Nature 457, 901–905 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H. & Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron 17, 275–285 (1996). [DOI] [PubMed] [Google Scholar]
- Komuro H. & Rakic P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J Neurobiol 37, 110–130 (1998). [PubMed] [Google Scholar]
- Mandeville J. T. & Maxfield F. R. Effects of buffering intracellular free calcium on neutrophil migration through three-dimensional matrices. J Cell Physiol 171, 168–178 (1997). [DOI] [PubMed] [Google Scholar]
- Doyle A., Marganski W. & Lee J. Calcium transients induce spatially coordinated increases in traction force during the movement of fish keratocytes. J Cell Sci 117, 2203–2214 (2004). [DOI] [PubMed] [Google Scholar]
- Hahn K., DeBiasio R. & Taylor D. L. Patterns of elevated free calcium and calmodulin activation in living cells. Nature 359, 736–738 (1992). [DOI] [PubMed] [Google Scholar]
- Brundage R. A., Fogarty K. E., Tuft R. A. & Fay F. S. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 254, 703–706 (1991). [DOI] [PubMed] [Google Scholar]
- Wei C., Wang X., Zheng M. & Cheng H. Calcium gradients underlying cell migration. Curr Opin Cell Biol 24, 254–261 (2012). [DOI] [PubMed] [Google Scholar]
- Schwab A., Finsterwalder F., Kersting U., Danker T. & Oberleithner H. Intracellular Ca2+ distribution in migrating transformed epithelial cells. Pflugers Arch 434, 70–76 (1997). [DOI] [PubMed] [Google Scholar]
- Pettit E. J. & Hallett M. B. Localised and global cytosolic Ca2+ changes in neutrophils during engagement of Cd11b/CD18 integrin visualised using confocal laser scanning reconstruction. J Cell Sci 109, 1689–1694 (1996). [DOI] [PubMed] [Google Scholar]
- Pettit E. J. & Hallett M. B. Nonuniform distribution of Ca(2+) uptake sites within human neutrophils. Biochem Biophys Res Commun 279, 337–340 (2000). [DOI] [PubMed] [Google Scholar]
- Jin M., Defoe D. M. & Wondergem R. Hepatocyte growth factor/scatter factor stimulates Ca2+-activated membrane K+ current and migration of MDCK II cells. J Membr Biol 191, 77–86 (2003). [DOI] [PubMed] [Google Scholar]
- Schilling T., Stock C., Schwab A. & Eder C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur J Neurosci 19, 1469–1474 (2004). [DOI] [PubMed] [Google Scholar]
- Schwab A., Reinhardt J., Schneider S. W., Gassner B. & Schuricht B. K(+) channel-dependent migration of fibroblasts and human melanoma cells. Cell Physiol Biochem 9, 126–132 (1999). [DOI] [PubMed] [Google Scholar]
- Schwab A., Schuricht B., Seeger P., Reinhardt J. & Dartsch P. C. Migration of transformed renal epithelial cells is regulated by K+ channel modulation of actin cytoskeleton and cell volume. Pflugers Arch 438, 330–337 (1999). [DOI] [PubMed] [Google Scholar]
- Schneider S. W. et al. Volume dynamics in migrating epithelial cells measured with atomic force microscopy. Pflugers Arch 439, 297–303 (2000). [DOI] [PubMed] [Google Scholar]
- McFerrin M. B. & Sontheimer H. A role for ion channels in glioma cell invasion. Neuron Glia Biol 2, 39–49 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom C. B., O'Neal J. T. & Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci 21, 7674–7683 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger M., Carin M., Medale M. & Tryggvason G. The osmotic migration of cells in a solute gradient. Biophys J 77, 1257–1267 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusela E. & Alt W. Continuum model of cell adhesion and migration. J Math Biol 58, 135–161 (2009). [DOI] [PubMed] [Google Scholar]
- Schmidt C., Ohlemeyer C., Labrakakis C., Walter T., Kettenmann H. & Schnitzer J. Analysis of motile oligodendrocyte precursor cells in vitro and in brain slices. Glia 20, 284–298 (1997). [PubMed] [Google Scholar]
- Gudz T. I., Komuro H. & Macklin W. B. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J Neurosci 26, 2458–2466 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez P. M. et al. Golli myelin basic proteins regulate oligodendroglial progenitor cell migration through voltage-gated Ca2+ influx. J Neurosci 29, 6663–6676 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez P. M., Fulton D., Spreuer V., Handley V. & Campagnoni A. T. Modulation of canonical transient receptor potential channel 1 in the proliferation of oligodendrocyte precursor cells by the golli products of the myelin basic protein gene. J Neurosci 31, 3625–3637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth J. T. et al. Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochim Biophys Acta 1763, 1147–1160 (2006). [DOI] [PubMed] [Google Scholar]
- Hansma P. K., Drake B., Marti O., Gould S. A. & Prater C. B. The scanning ion-conductance microscope. Science 243, 641–643 (1989). [DOI] [PubMed] [Google Scholar]
- Rheinlaender J., Geisse N. A., Proksch R. & Schäffer T. E. Comparison of Scanning Ion Conductance Microscopy with Atomic Force Microscopy for Cell Imaging. Langmuir 27, 697–704 (2011). [DOI] [PubMed] [Google Scholar]
- Korchev Y. E., Bashford C. L., Milovanovic M., Vodyanoy I. & Lab M. J. Scanning ion conductance microscopy of living cells. Biophys J 73, 653–658 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korchev Y. E. et al. Specialized scanning ion-conductance microscope for imaging of living cells. J Microsc 188, 17–23 (1997). [DOI] [PubMed] [Google Scholar]
- Happel P., Thatenhorst D. & Dietzel I. D. Scanning Ion Conductance Microscopy for Studying Biological Samples. Sensors 12, 14983–15008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lab M. J., Bhargava A., Wright P. T. & Gorelik J. The Scanning Ion Conductance Microscope (SICM) for Cellular Physiology. Am J Physiol Heart Circ Physiol (2012). [DOI] [PubMed] [Google Scholar]
- Korchev Y. E. et al. Cell volume measurement using scanning ion conductance microscopy. Biophys J 78, 451–457 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happel P., Hoffmann G., Mann S. A. & Dietzel I. D. Monitoring cell movements and volume changes with pulse-mode scanning ion conductance microscopy. J Microsc 212, 144–151 (2003). [DOI] [PubMed] [Google Scholar]
- Happel P., Wehner F. & Dietzel I. D. Modern Research and Educational Topics in Microscopy, chapter Scanning ion conductance microscopy–a tool to investigate electrolyte-nonconductor interfaces, 968–975. FORMATEX (2007).
- Mann S. A., Hoffmann G., Hengstenberg A., Schuhmann W. & Dietzel I. D. Pulse-mode scanning ion conductance microscopy–a method to investigate cultured hippocampal cells. J Neurosci Methods 116, 113–117 (2002). [DOI] [PubMed] [Google Scholar]
- Happel P. & Dietzel I. D. Backstep scanning ion conductance microscopy as a tool for long term investigation of single living cells. J Nanobiotechnology 7, 7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinlaender J. & Schäffer T. E. Image formation, resolution, and height measurement in scanning ion conductance microscopy. Journal of Applied Physics 105, 094905 (2009). [Google Scholar]
- Happel P., Möller K., Kunz R. & Dietzel I. D. A boundary delimitation algorithm to approximate cell soma volumes of bipolar cells from topographical data obtained by scanning probe microscopy. BMC Bioinformatics 11, 323 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R., Hamilton N. B., Bakiri Y. & Attwell D. Spiking and nonspiking classes of oligodendrocyte precursor glia in CNS white matter. Nat Neurosci 11, 450–456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussouf A. & Gaillard S. Intracellular pH changes during oligodendrocyte differentiation in primary culture. J Neurosci Res 59, 731–739 (2000). [DOI] [PubMed] [Google Scholar]
- Schwab A. et al. Subcellular distribution of calcium-sensitive potassium channels (IK1) in migrating cells. J Cell Physiol 206, 86–94 (2006). [DOI] [PubMed] [Google Scholar]
- Sontheimer H., Trotter J., Schachner M. & Kettenmann H. Channel expression correlates with differentiation stage during the development of oligodendrocytes from their precursor cells in culture. Neuron 2, 1135–1145 (1989). [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. & Steinhäuser C. Ion channels in glial cells. Brain Res Brain Res Rev 32, 380–412 (2000). [DOI] [PubMed] [Google Scholar]
- Mongin A. A., Reddi J. M., Charniga C. & Kimelberg H. K. [3H]taurine and D-[3H]aspartate release from astrocyte cultures are differently regulated by tyrosine kinases. Am J Physiol 276, C1226–C1230 (1999). [DOI] [PubMed] [Google Scholar]
- Buettner R., Papoutsoglou G., Scemes E., Spray D. C. & Dermietzel R. Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc Natl Acad Sci U S A 97, 3201–3206 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukov A., Richards O., Ostanin V., Korchev Y. & Klenerman D. A hybrid scanning mode for fast scanning ion conductance microscopy (SICM) imaging. Ultramicroscopy 121C, 1–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. A. et al. Corticosteroids reverse cytokine-induced block of survival and differentiation of oligodendrocyte progenitor cells from rats. J Neuroinflammation 5, 39 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S. A., Meyer J. W. & Dietzel I. D. Integration of a scanning ion conductance microscope into phase contrast optics and its application to the quantification of morphological parameters of selected cells. J Microsc 224, 152–157 (2006). [DOI] [PubMed] [Google Scholar]