

Abstract
Tumour Necrosis Factor (TNF) is critical for host control of M. tuberculosis, but the relative contribution of TNF from innate and adaptive immune responses during tuberculosis infection is unclear. Myeloid versus T-cell-derived TNF function in tuberculosis was investigated using cell type-specific TNF deletion. Mice deficient for TNF expression in macrophages/neutrophils displayed early, transient susceptibility to M. tuberculosis but recruited activated, TNF-producing CD4+ and CD8+ T-cells and controlled chronic infection. Strikingly, deficient TNF expression in T-cells resulted in early control but susceptibility and eventual mortality during chronic infection with increased pulmonary pathology. TNF inactivation in both myeloid and T-cells rendered mice critically susceptible to infection with a phenotype resembling complete TNF deficient mice, indicating that myeloid and T-cells are the primary TNF sources collaborating for host control of tuberculosis. Thus, while TNF from myeloid cells mediates early immune function, T-cell derived TNF is essential to sustain protection during chronic tuberculosis infection.
Initial events during pulmonary M. tuberculosis infection involve binding and uptake of M. tuberculosis by alveolar macrophages. Subsequent immune responses are therefore dependent on initial activation of infected macrophages through, amongst others, induction of appropriate activating molecules, production of cytokines and chemokines and synthesis of bactericidal molecules. In vitro studies have shown that macrophage stimulation through M. tuberculosis or mycobacterial antigens results in secretion of TNF which is a requirement for induction of cell activation1,2,3,4. However, coactivation by IFNγ is required for full bactericidal status and inhibition of mycobacterial growth2,5. In vivo challenge studies confirmed TNF as an integral component of the host immune defence mechanism against mycobacterial infection that promotes the initiation and maintenance of granulomas during infection6,7,8,9. Among other family members, such as lymphotoxin (LT) α, LTβ, FasL, CD40L, RANKL, CD27L and OX40L10, TNF mediated protection is non-redundant despite the potential of soluble LTα to also bind and signal through both TNF receptors11,12. TNFRp55 is thought to be primarily responsible for mediating most TNF mediated effects13,14. TNF signalling can induce cell death, promote cell survival and induce proinflammatory responses under defined cellular and micro-environmental conditions14. Both 26 kd membrane-anchored and 17 kD soluble TNF forms15,16 contribute to the control of M. tuberculosis infection17,18,19. TNF is produced by multiple cell types including macrophages, neutrophils, dendritic cells, lymphocytes and NK cells as well as by cells of non-hematopoietic origin20. However, the precise role of TNF derived from these cells in infection remains unclear and requires investigation. The clinical importance of TNF to control mycobacterial replication was demonstrated during anti-TNF treatment of patients with rheumatoid arthritis, Crohn's disease or severe psoriasis, where, in some cases, patients developed spontaneous tuberculosis reactivation21,22. T cells and macrophages are crucial components of protective granulomas and are potentially relevant sources of TNF in vivo, responding to antigens or to microbial components, respectively23,24. We reported earlier that TNF derived from hematopoietic cells is essential to control mycobacterial BCG infection9. Further, cell transfer experiments suggest that TNF produced by T cells may be essential but not sufficient for antimycobacterial host defense25. It is thus important to define the contribution and kinetics of the TNF responses issued from adaptive, T lymphocytes versus innate phagocytic, macrophage/neutrophils, in the control of acute and chronic M. tuberculosis infection.
In this study we examined the role of TNF produced by macrophages and neutrophils, or by T-cells, using cell-specific gene-inactivated mice26 in M. tuberculosis infection models. We demonstrate the significance of myeloid, macrophage/neutrophil derived TNF for protective immune responses during early M. tuberculosis infection. This function becomes redundant during persistent infection, while T-cell derived TNF is essential for long-term control of chronic infection in a non-redundant fashion. Therefore, two major cellular sources of TNF in protection against mycobacteria display both redundant/overlapping and non-redundant/specific functions.
Results
TNF produced by myeloid cells is required for control of pulmonary M. tuberculosis replication during acute, but not persistent, infection
To address whether TNF derived from macrophages/neutrophils is required for protective immune functions in vivo, we first investigated disease outcome in M-TNF−/− mice during infection with virulent M. tuberculosis in comparison with either immune-competent WT mice or complete TNF deficient mice. The inability to produce any TNF rendered mice highly susceptible to M. tuberculosis aerosol inhalation challenge resulting in rapid weight loss (Fig. 1A) and death within 42 days (Fig. 1B), confirming previous reports6,18. Strikingly, both WT- and M-TNF−/− mice were resistant to infection without notable loss of bodyweight (Fig. 1A) and survived the 7 month duration of the experiment (Fig. 1B). Thus quite unexpectedly, survival of M-TNF−/− mice clearly indicated that macrophages and neutrophils were not the unique, essential sources of TNF to elicit a sustained protective response to M. tuberculosis.
Figure 1. TNF from myeloid cells is required to control acute M. tuberculosis replication in vivo.

(A–D) WT (closed circle), M-TNF−/− (grey triangle) and TNF−/− (open circle) mice were infected by aerosol inhalation with 200–500 cfu/lung of M. tuberculosis H37Rv and body weight changes (A) and mortality (B) (n = 10 mice/group) were monitored. Changes in bodyweight values are expressed as the mean of 4–10 mice/group. Data represents one of three independent experiments. The number of viable bacteria present in lungs was assessed at the indicated time points (C). The results are expressed as the mean ± SD of 5 mice/group per time point and represent one of three independent experiments. * = p < 0.05 (WT versus M-TNF−/− mice), # = p < 0.05 (WT versus TNF−/− mice.), ϕ = p < 0.05 (M-TNF−/− versus TNF−/− mice). Ziehl-Neelsen staining of lungs at 35 days post infection (D). The results represent one of four similar experiments. (Magnification = 600×). Arrows indicate the presence of AFB. (E) WT (black), M-TNF−/− (grey) and TNF−/− (clear) bone marrow derived macrophages were stimulated either with LPS (100 ng/ml) or M. tuberculosis H37Rv (at a MOI of 2:1) for 48 hrs and TNF measured by ELISA. The results are expressed as the mean ± SD of quadruplicate values and are representative of one of three independent experiments. ND: Not detected. (F) Bone marrow derived macrophages were infected with M. tuberculosis expressing luciferase and bacterial replication measured as relative luminescence after 100 hrs. The results are expressed as the mean ± SD of quadruplicate values and represent one of three independent experiments.
Several studies have correlated uncontrolled mycobacterial replication with high susceptibility in TNF−/− mice6,9,27. Our working hypothesis was that M. tuberculosis recognition may lead to suboptimal macrophage activation in the absence of TNF synthesis in M-TNF−/− mice, resulting in possible susceptibility due to a reduced capability to restrict bacterial replication during early infection. The resistant phenotype of M-TNF−/− mice was unexpected since TNF is required for optimum activation of infected macrophages to restrict M. tuberculosis growth2,28,29,30. We thus investigated the effect of TNF produced by myeloid cells on bacterial replication by comparing pulmonary bacilli growth in WT-, M-TNF−/−- and TNF−/− mice over a period of 7 months after M. tuberculosis challenge. Interestingly, M-TNF−/− mice failed to control pulmonary bacilli replication within the first 3–4 weeks of infection and developed high bacilli titers similar to TNF−/− mice which were 1–2 log10 higher than WT mice (Fig. 1C). Ziehl-Neelsen staining of infected lung tissue confirmed distinctive differences in bacilli loads in the respective genotypes (Fig. 1D). However, the susceptibility of M-TNF−/− mice was transient and the high pulmonary bacilli burden was eventually reduced to levels similar to that of WT mice during persistent infection, whereas in TNF−/− mice bacilli replication remained unrestricted. Thus, the absence of TNF production by macrophages/neutrophils was clearly associated with defective control of bacilli growth during early infection, but it could be compensated for thereafter.
Considering earlier reports on TNF dependent restriction of M. tuberculosis replication we wanted to confirm that early susceptibility of M-TNF−/− was indeed a function of deficient TNF synthesis and bacterial control by macrophages from these mice31,32,33. TNF release by macrophages occurs upon infection or exposure to various stimuli29,34,35. WT macrophages released TNF after infection with M. tuberculosis H37Rv (Fig. 1E), and the growth of the bacilli was restrained, as seen with the luminescent, luciferase expressing reporter M. tuberculosis H37Rv strain (Fig. 1F). In contrast, in macrophages from TNF−/− mice, the absence of TNF release, was accompanied by failure to restrain the bacilli growth. We demonstrate here that infected macrophages derived from M-TNF−/− mice that fail to produce TNF as shown earlier36 are defective in controlling bacteria growth, similar to macrophages from TNF−/− mice. Therefore, our data clearly supports the importance of TNF, and in particular macrophage-derived TNF, in controlling mycobacterial replication.
TNF produced by myeloid cells regulates lung inflammation but is not required for the initiation and maintenance of granuloma structural integrity
It is well established that containment and control of bacilli growth is dependent on the establishment of functional, bactericidal granulomas. In view of uncontrolled M. tuberculosis replication during early infection in M-TNF−/− mice, we expected that the establishment of granulomas would be defective in the absence of TNF synthesis by macrophages and neutrophils. TNF is required not only for the initiation, but also for maintenance of granulomas and we and others have previously reported the lack of granuloma formation in the absence of TNF after mycobacterial challenge6,7,8,9. However, granulomas comprise different cell types, many capable of synthesising TNF that could potentially contribute to their structural integrity. To gain further insight into the transient nature of the immune response in M-TNF−/− mice, we investigated whether macrophages and neutrophils were an essential cellular source of TNF during the establishment of granulomas. As a correlate for infection-induced inflammation we measured lung weights (Fig. 2A) and found a transient increase of lung weights in M-TNF−/− mice which peaked at day 28 post infection, with partial resolution after day 35 post infection. In contrast, sustained, uncontrolled inflammation was observed in TNF−/− mice, at significantly higher levels than in either M-TNF−/− or WT mice on day 35 post infection, when TNF−/− mice succumbed to infection. Controlled inflammation in M-TNF−/− mice was substantiated by quantitative assessment of pulmonary infiltrating leukocytes (Fig. 2B) which was significantly higher compared to WT mice, while TNF−/− mice experienced a delay in cell recruitment at day 21 followed by excessive cell infiltration in the lung 35 days post-infection.
Figure 2. TNF from myeloid cells regulates cellular recruitment for granuloma formation but is dispensable for initiation and maintenance of granuloma structure.

(A; B) WT (black), M-TNF−/− (grey), and TNF−/− (clear) mice were infected with M. tuberculosis H37Rv as described under “Materials and Methods” and lung weights (A) and cell numbers (B) assessed at the indicated time points. Data represent the mean ± SD of 5 mice per time point and one of three independent experiments. * = p < 0.05 (WT versus M-TNF−/− mice), # = p < 0.05 (WT versus TNF−/− mice.), ϕ = p < 0.05 (M-TNF−/− versus TNF−/− mice). (C–D). Macroscopic lung pathology (C) in WT, M-TNF−/− and TNF−/− mice at 28 days post infection (one of three independent experiments; n = 5 mice/strain). Low magnification (20×) of pulmonary granulomas at (i) 28 days and (ii) 166 days postinfection after haemotoxylin and eosin staining (D). Arrows indicate granulomatous structures and X indicates necrosis. The results represent one of 5 similar experiments.
We further assessed the role of macrophage and neutrophil derived TNF for structural granuloma integrity during M. tuberculosis challenge. Macroscopic lung pathology showed no distinction amongst the strains after 21 days of infection (Suppl. Fig. 1), but differences were clearly evident at 28 days post-infection (Fig. 2C). Small, defined lesions were present on the pleura of WT mice while TNF−/− mice had large and in part confluent nodules. Interestingly, M-TNF−/− mice presented with an intermediate lesion phenotype not as severe as observed in TNF−/− mice but not as small and compact as in WT mice (Fig. 2C). Histological assessment showed that, although enlarged, pulmonary granulomas in M-TNF−/− mice were well structured with no necrotic areas, in contrast to TNF−/− mice which displayed effaced structures with necrotic centers on day 28 post infection (Fig. 2D). Importantly, the kinetics of granuloma formation was normal in M-TNF−/− mice with no evidence of a delay in the onset of granuloma initiation (data not shown) as previously reported for TNF−/− mice6. Examination of tissues at higher magnification clearly showed occlusion of bronchi and blood vessels in TNF−/− mice but not in M-TNF−/− - or WT mice. Moreover, the differences noted in pulmonary pathology between WT and M-TNF−/− mice during early stages of infection were transient and clearly absent during chronic infection (Fig. 2Dii). At six months post-infection lung sections were pathologically indistinguishable, with defined structural granulomas present and no necrosis was evident in either M-TNF−/−- or WT mice (Fig. 2Dii). Therefore, TNF produced by myeloid cells regulates inflammation and cellular recruitment to the lung but is not required for initiating and maintaining the structural integrity of granulomas.
TNF produced by myeloid cells regulates pulmonary lymphocytic recruitment during early M. tuberculosis infection
Protective immunity in response to M. tuberculosis is cell-mediated37,38 and the recruitment of T cells, especially CD4+- and CD8+ T cells, is critical to control the growth of bacilli39,40. TNF is an important mediator of cellular traffic through the regulation of adhesion molecule expression, dendritic cell maturation and chemokine induction27,41. To understand the mechanism responsible for the transient nature of susceptibility in M-TNF−/− mice, we asked whether lymphocytic recruitment and activation were affected by the lack of macrophage/neutrophil derived TNF. Here, we first measured migration of lymphocytes to local sites of infection and found that M-TNF−/− mice had a small but significantly higher recruitment of both CD4+ (Fig. 3A) and CD8+ (Fig. 3B) T cells to the lung at 21 and 28 days post infection as compared to WT mice. Enhanced lymphocytic recruitment in M-TNF−/− mice was transitory and levels similar to those in WT mice were recorded at 35 days post infection. Conversely, TNF−/− mice seemed to control cell migration during the early stages of infection, but 2–4-fold increased CD4+ and CD8+ T cell levels were measured on day 35, correlating with the overwhelming bacilli burden at this latter stage of infection. The transient increase in pulmonary CD4+ and CD8+ T cells seen during early infection in M-TNF−/− mice was effectively controlled thereafter, suggesting that the control of excessive lymphocytic response was independent of macrophage/neutrophil derived TNF but likely dependent upon another source of TNF like T-cell derived TNF. We further assessed the activation status of the lung infiltrating CD4+- and CD8+ T cell subsets by flow cytometry for expression of the leukocyte homing and activation marker CD44 (Fig. 3C and 3D) and the early activation marker CD69 (Fig. 3E and 3F). CD4+- and CD8+ T cells recruited to the lung after M. tuberculosis infection expressed CD44 and CD69 in all the respective strains. Thus, the absence of TNF production by myeloid cells favoured early recruitment of CD4+ and CD8+ activated T cells to the lung in response to M. tuberculosis infection.
Figure 3. Transiently enhanced pulmonary infiltration of CD4+ and CD8+ activated T cells is independent of TNF produced by myeloid cells during M. tuberculosis infection.
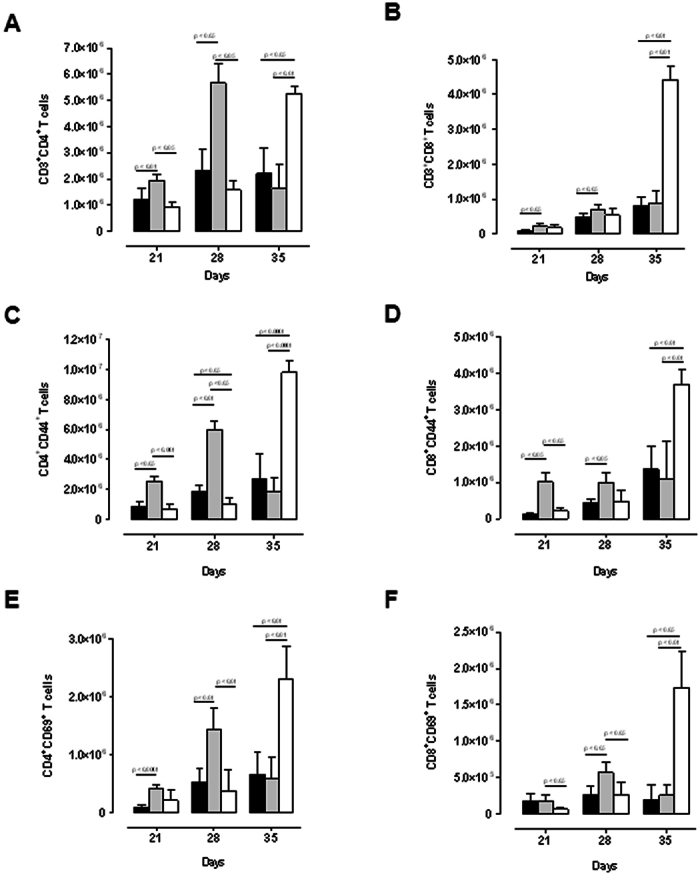
WT (black), M-TNF−/− (grey) and TNF−/− (clear) mice were infected via aerosol inhalation with 200–500 cfu/lung of Mycobacterium tuberculosis. Lung cell suspensions from infected mice were analyzed by flow cytometry to determine (A) CD3+CD4+ and (B) CD3+CD8+ T cell pulmonary populations at day 21, 28 and 35 post-infection and activation status analysed for markers CD44 (C and D) and CD69 (E and F) on CD4+ (C, E) and CD8+ (D, F) T cells by flow cytometry. The results are expressed as the mean ± SD of 5 mice per group and are from one experiment, representative of two independent experiments.
M. tuberculosis specific T cell-priming is independent of TNF produced by myeloid cells
Resistance to M. tuberculosis is highly dependent on the host's capability to induce an effective Th1 immune response40,42,43. Our studies indicated a down-regulation of CD4+- and CD8+ T cell recruitment by TNF synthesized by myeloid cells with no clear effect on lymphocyte activation. To further assess how distinct cellular sources of TNF may affect Th1 immune function we next investigated IFNγ release by primed CD4+- and CD8+ T cells in response to mycobacterial antigens in M-TNF−/− mice. Single cell suspensions from lungs harvested at 21 day post M. tuberculosis infection were restimulated ex vivo with purified protein derivative (PPD), early secreted antigen of 6 kDa (ESAT-6), or anti-CD3/anti-CD28 antibodies. Restimulation of lung infiltrating CD4+ T cells from TNF−/− mice showed compromised IFNγ expression in response to ESAT-6, PPD or anti CD3/CD28 antibodies, to only one third of that seen in WT cells, while cells from M-TNF−/− mice had a 2-fold increased IFNγ response (Fig. 4A(i and ii)). We found no differences in IFNγ synthesis of restimulated CD8 T cells amongst the mutant and control strains (data not shown). Moreover, the level of IFNγ in whole lung homogenates was increased in M-TNF−/− mice at 21 days post-infection and contracted thereafter to concentrations equivalent to WT mice, while pulmonary IFNγ in TNF−/− mice were equivalent to WT mice during the first 3 weeks and highly increased at 4 weeks (Fig. 4B) Effective control of M. tuberculosis infection is only possible when IL-12 synergizes with IFNγ to induce a Th1 immune response28,44. Pulmonary IL-12p70 levels increased strongly between 2 and 3 weeks post-infection in both WT and M-TNF−/− mice but were halved in TNF−/− mice (Fig. 4C). Therefore, the absence of TNF production by myeloid cells did not compromise M. tuberculosis specific priming and restimulation of lung infiltrating CD4+ T cells.
Figure 4. Enhanced M. tuberculosis specific priming of pulmonary CD4+ T cells in infected M-TNF−/− mice.

(A) Lung cell suspensions from M. tuberculosis H37Rv infected WT, M-TNF−/−, and TNF−/− mice harvested at 21 days post infection were restimulated with ESAT 6, PPD or antibodies to CD3 and CD28 for 72 hrs and intracellular IFNγ produced by CD4+T cells assessed by flow cytometry. Fig. 4A(i) are representative dot blots of single mice while the results in Fig. 4A(ii) are expressed as the mean ± SD of 4 mice per group and represent one of two similar experiments. (B, C) Lung concentrations of IFNγ (B) and IL-12p70 (C) were determined by ELISA in lung homogenates of M. tuberculosis H37Rv infected WT (black), M-TNF−/− (grey) and TNF−/− (open) mice at different time points. The data are expressed as the mean and ± SD of 4 mice/group for each time point and represent one of two similar experiments.
TNF release by lung infiltrating CD4+- and CD8+ T cells during M. tuberculosis infection is not compromised by the absence of TNF from myeloid origin
The overall protection against M. tuberculosis infection seen in M-TNF−/− mice indicated that TNF derived from an alternative cellular source, other than macrophages/neutrophils contributed to protective immune functions. We thus next addressed the contribution of T cells, a major cellular component of bactericidal granuloma, as a source of TNF during M. tuberculosis infection that could contribute to control bacilli replication. Adoptive transfer experiments previously illustrated that TNF secretion by lymphocytes is protective against M. tuberculosis challenge25. Moreover, it is known that T cells produce TNF upon antigen specific activation and the addition of CD4+ T cells to M. tuberculosis infected monocytes enhances TNF production by both T cells and monocytes45. We first investigated whether T cells do respond to M. tuberculosis challenge by expressing TNF in vivo and how this may be influenced by the absence of TNF from myeloid origin. TNF expression by CD4+- or CD8+ T cells isolated from M. tuberculosis infected WT mice 21 days post-infection was analyzed by flow cytometry. TNF production was absent in CD4+ T cells isolated from TNF−/− mice, as expected, while it was present or slightly increased in CD4+ or CD8+ T cells isolated from M-TNF−/− mice (Supplementary Fig. 2). Therefore, T cell derived TNF may compensate for the lack of TNF production by myeloid cells in vivo during M. tuberculosis infection.
T cell derived TNF is required for sustained control of pulmonary M. tuberculosis during chronic infection
Guided by our hypothesis that CD4+- and CD8+ T cell derived TNF may compensate for the absence of TNF produced by myeloid cells during early infection, we next addressed the specific role of T-cell derived TNF in protective immunity to M. tuberculosis and disease outcome. T cell specific TNF−/− (T-TNF−/−) mice26 were infected with M. tuberculosis and the absence of TNF release by CD4+ or CD8+ T cells verified on day 21 post-infection (Suppl. Fig. 2). Strikingly, while WT mice survived M. tuberculosis infection with no apparent loss of body mass and TNF−/− mice succumbed within 40 days with rapid bodyweight loss (Fig. 5A, B), T-TNF−/− mice survived significantly longer than TNF−/− mice, but eventually succumbed between 130–150 days post-infection accompanied by severe loss of body mass (Fig. 5A, B). Thus, ablation of TNF production by both CD4+- and CD8+ T cells led to an inability of the host to sustain immune protection during chronic M. tuberculosis infection.
Figure 5. T-TNF−/− mice succumb to chronic M. tuberculosis infection.

(A; B) WT (closed circle), T-TNF−/− (grey diamond), and TNF−/− (clear circle) mice were infected via aerosol inhalation with 200–500 cfu/lung of M. tuberculosis H37Rv are represented (n = 10 mice/strain). Change in bodyweight values (A) are expressed as the mean of 4–10 mice/group. Differences in mortality rates (B) were analysed by the student's logrank test. (C; D). The number of viable bacteria present in lungs were assessed at 33 days (C) and 150 days (D) post-infection. The results are expressed as the mean of 4 mice/group. (E; F and G). Lung weights (E), macroscopic lung pathology (F) and haemotoxylin and eosin staining of lung sections (G) from WT-, and T-TNF−/− mice infected with M. tuberculosis. Images of left lobes of WT and T-TNF−/− mice at 150 days post infection are presented (n = 4 mice/group). The data is from one experiment representative of three independent experiments.
We next investigated whether T cell derived TNF was implicated in the control of M. tuberculosis replication during the generation of protective immune responses. Pulmonary bacilli burden measured in T-TNF−/− mice on day 33 post-infection was similar to that seen in WT mice, ca 5 log10 cfu per lung, in contrast to the 102–103 fold higher bacterial loads of TNF−/− mice (Fig. 5C). This indicated an efficient early control of bacilli replication, independent of T cell derived TNF during acute infection. Indeed, there was no significant alteration of overall pulmonary TNF mRNA and protein expression in cell-specific M-TNF−/−- or T-TNF−/− mice, as compared to WT mice, 4 weeks post M. tuberculosis infection (Suppl. Figure 3). Considering mortality of T-TNF−/− mice at 150 days post-infection we evaluated bacilli burden during chronic infection (Fig. 5D). Pulmonary bacterial burden was 10-fold higher in T-TNF−/− mice compared to WT control animals suggesting an importance of T cell derived TNF for inhibiting pulmonary bacterial replication during persistent/chronic infection (Fig. 5D). Thus, although TNF is produced by many cell types, T cell derived TNF has a non-redundant function to sustain protection during chronic mycobacterial infection that cannot be compensated for by other sources of TNF, such as myeloid cells.
Lung weights were significantly higher in T-TNF−/− mice compared to WT mice at day 150 post-infection (Fig. 5E), indicative of enhanced inflammation. During chronic infection WT mice displayed discrete lesions under the pleura; while lesions appeared coalescent and severe in T-TNF−/− mice covering most of the lung surface (Fig. 5F). Indeed, WT mice showed discrete granulomatous lesions with aggregates of lymphocytes in lung tissue (Fig. 5G), while T-TNF−/− mice demonstrated severe pulmonary histopathology with large necrotic areas, coalescent with no clear demarcated areas of lymphocytic aggregates (Fig. 5G). At higher magnification the free alveolar and airway spaces seen in WT mice were largely occluded in T-TNF−/− mice. Cellular degeneration was readily noticeable within pulmonary tissue in the T-TNF−/− mice indicating extensive necrosis. Therefore, T cell derived TNF appears critical for the structural maintenance of the granuloma during chronic M. tuberculosis infection and overall resistance to the pathogen.
Differential regulation of inflammatory genes by myeloid versus T-cell derived TNF during acute M. tuberculosis infection
The molecular mechanisms providing a non-redundant function of TNF produced by T-cells and/or macrophages remain unclear. Gene expression signatures in M. tuberculosis infected mice may provide an insight in this respect. We assessed TNF-dependent gene expression during M. tuberculosis infection and identified approximately 200 genes that were up- or down-regulated in TNF−/− mice as compared to WT mice at 2 and 3 weeks post-infection. Amongst these, we selected genes expressed preferentially in either macrophages or T-cells, and assessed the expression by quantitative PCR in M. tuberculosis infected M-TNF−/−, T-TNF−/− or TNF−/− mice (Fig. 6). Cytotoxic T cells signature consisting of CD8a, IFNγ and granzyme A as well as IFNγ-induced GTPase Irgm1/LRG47 transcription factors IRF7 or ATF3 was abnormally high in complete TNF−/− mice 4 weeks post M. tuberculosis infection, and less so in M-TNF−/− mice, while T-TNF−/− mice showed an intermediate phenotype (Fig. 6A), suggesting a limited influence of T-cell derived TNF on the regulation of these genes. Interestingly, pro-inflammatory cytokines and chemokines such as IL-1b, IL-6, IL-17a, CXCL3 or CXCL5 or s100a9 which were highly overexpressed in complete TNF−/− mice 4 weeks post-infection, were only modestly up-regulated in T-TNF−/− mice, while M-TNF−/− behaved essentially like WT mice (Fig. 6B), indicating a strong compensation of either source of TNF in this inflammatory regulation. Acute phase protein Serum Amyloid A3 (SAA3) was overexpressed in TNF−/− mice and rather strongly expressed in either T-TNF−/−- or M-TNF−/− mice 4 weeks post-infection. The expression of iNOS, a key anti-mycobacterial enzyme, was strongly increased in M-TNF−/−- as well as in complete TNF−/− mice, while T-TNF−/− mice had a low expression compared to infected WT mice 4 weeks post-infection. Therefore we showed that TNF derived from either myeloid or T cells have differential effects on genes known to contribute to the control of acute M. tuberculosis infection.
Figure 6. Inflammatory gene expression during acute M. tuberculosis infection in cell-specific TNF−/− mice.

(A; B) Analysis of selected inflammatory gene expression was performed by quantitative RT-PCR on day 28 after M. tuberculosis infection and normalized to b-actin. (n = 3–4 mice per point). Genes include CD8a, IFNγ, granzyme A,Irgm1/LRG47, IRF7 and ATF3 (Fig. 6A), the proinflammatory cytokines and chemokines IL-1b, IL-6, IL-17a, CXCL3 or CXCL5 and s100a9, the acute phase reactant, SAA and iNOS (Fig. 6B).
Deficiency in both myeloid- and T-cell derived TNF confers high susceptibility to M. tuberculosis infection and reconstitutes the phenotype of TNF−/− mice
Our data in M. tuberculosis infected M-TNF−/− mice indicated a transient dependence on TNF derived from myeloid cells to induce protection which could eventually be compensated for by other cellular sources of TNF. T cells could provide a compensatory cellular source of TNF and data from T-TNF−/− mice demonstrated the dependence on lymphocyte derived TNF for sustained protection against chronic M. tuberculosis infection. To investigate whether T cells were indeed the “compensatory” cellular source of TNF essential for early control of M. tuberculosis infection in the absence of macrophages and neutrophils TNF, we challenged mice deficient for both myeloid- and T-cell derived TNF (MT-TNF−/− mice). Strikingly, MT-TNF−/− mice were highly susceptible to M. tuberculosis infection with rapid bodyweight loss and death occurred within 40 days post-infection, similar to complete TNF−/− mice, while mice deficient for T-cell derived TNF succumbed by day 150 and M-TNF−/− mice survived the duration of the experiment (Fig. 7A,B). Lung weights, an indication of pulmonary inflammation, were markedly increased in MT-TNF−/− and TNF−/− mice, and less so in T-TNF−/− mice, compared to M-TNF−/− and WT mice at day 39 post-infection (Fig. 7C). MT-TNF−/− mice were unable to control bacterial replication, similar to TNF−/− mice, with a 100–1000 fold higher pulmonary bacterial burden compared to WT control mice (Fig. 7D). We confirmed that M-TNF−/− and T-TNF−/− mice had pulmonary bacterial burdens similar to that of WT mice at 39 days post-infection (Fig. 7D). Differences in pulmonary bacilli burden were also visible microscopically since lung sections of MT-TNF−/− and TNF−/− mice displayed widely dispersed aggregates of acid fast bacteria that appeared to be predominantly extracellular, while WT, M-TNF−/− or T-TNF−/− mice presented primarily with single acid fast bacilli sparsely distributed in alveolar macrophages (Fig. 7E). Enlarged, necrotic nodules characteristic of TNF−/− mice were also present in the lungs of MT-TNF−/− on day 39 post infection (Fig. 7E). Histopathologically, well defined granulomas were observed in WT mice, in contrast to both MT-TNF−/− and TNF−/− mice which presented with severe and necrotic lesion structures (Fig. 7F).
Figure 7. MT-TNF−/− mice are highly susceptible and succumb to acute M. tuberculosis infection.

(A; B) WT (closed circle), M-TNF−/− (grey triangle), T-TNF−/− (grey diamond), MT-TNF−/− mice (clear square) and TNF−/− (clear circle) mice infected as described were monitored for change in bodyweight (A; expressed as the mean of 4–10 mice/group) and survival (B; n = 10 mice/group) Data is from one experiment representative of three independent experiments. (C; D) The lung weights (C) and number of viable bacteria present in lungs (D) were assessed at 39 days post-infection. The results are expressed as the mean ± SD of 4–5 mice/group and are from one experiment, representative of two independent experiments. (E; F) Pulmonary bacilli burdens on day 39 were confirmed by Ziehl-Neelsen staining (E; magnification 600×). Arrows indicate the presence of single AFB within macrophages. Images of left lobes illustrating macroscopic pulmonary pathology in WT-, M-TNF−/−, T-TNF−/−, MT-TNF−/− and TNF−/− mice. (n = 5 mice/group). Images are representative of one of three independent experiments. Haemotoxylin and eosin staining show enlarged, necrotic nodules in the lungs of MT-TNF−/− and TNF−/− mice at day 39 post infection (F). Images are representative of one of three independent experiments.
Therefore, absence of TNF from both T-cells and myeloid cells rendered mice essentially as susceptible as fully TNF-deficient mice, demonstrating that these are the main cellular sources involved in host control of M. tuberculosis infection. The fact that TNF production by T-cells or by macrophages/neutrophils can partially compensate each other to control of acute infection, points to some redundant functions which may be provided by either source of TNF.
Discussion
TNF is essential for the effective control of mycobacterial infection in several host species including humans7,8,9,22,27,46,47,48. We and others have shown that TNF deficient mice are highly susceptible to M. bovis BCG7,9 and M. tuberculosis6,8. Although both immune and non-immune cells are capable of TNF synthesis, the identity of the critical cell types contributing to immune protection remains undefined. In this study we investigated the role of TNF from distinct cellular sources in mediating immune protection, focusing on the contribution of myeloid cells, such as macrophages and neutrophils, and CD4+/CD8+ T cells in the host's immune response against M. tuberculosis.
Synthesis of TNF by various cell types has been demonstrated as integral to the host's immune response against pathogen challenge46. Early host immune responses against M. tuberculosis infection are characterized by leukocyte recruitment in which macrophages and neutrophils constitute >70% of inflammatory cells47. Particularly relevant to this study is the widely accepted paradigm that monocytes/macrophages are significant sources of TNF during bacterial infections and are therefore deemed to be critical for host resistance to pathogens2,31,48,49,50,51. When macrophages are infected with live bacilli or presented with either M. tuberculosis derived ligands or heat killed bacilli in vitro, rapid production of TNF is initiated, suggesting that they may contribute significantly to the host immune response in vivo6. Several studies described neutrophil mediated control of M. tuberculosis52,53 and evidence for a TNF dependent cooperative mechanism between neutrophils and alveolar macrophages to contain M. tuberculosis replication has been reported54. Furthermore, optimum macrophage activation and mycobacterial growth inhibition are acquired through synergistic effects of TNF and IFNγ production by T cells to induce nitric oxide bactericidal mechanisms5.
Utilizing mice with highly efficient, cell lineage restricted inactivation of TNF26, we were able to show here that TNF produced by myeloid cells has an important non-redundant function during early control of M tuberculosis, but unexpectedly becomes redundant during the latter stages of infection. Previous studies have shown that virulent M tuberculosis induces TNF production by macrophages to mediate apoptosis and escape55 whereas neutrophil-derived TNF was reported as important for activation of macrophages54. The absence of TNF from myeloid origin in our infection model could therefore result in reduced activation and bactericidal capacity of infected macrophages permitting increased bacterial replication during early stages of infection. Interestingly, the onset of adaptive immune responses coincided with a reduction in bacterial growth eventually resulting in burden equivalent to WT control mice. From previous studies it was concluded that the recruitment of effector antigen specific CD4+- and CD8+ T cells is critical to further enhance infected macrophage killing capacity at the site of infection and the subsequent killing of the intracellular bacilli56,57. Our data show that in the absence of TNF from macrophages and neutrophils M-TNF−/− mice have enhanced pulmonary effector CD4+- and CD8+ T cell recruitment during early infection. While the increased recruitment was associated with increased bacterial burden elevated numbers of T cells likely contributed with TNF production to compensate the lack of macrophage/neutrophil-derived TNF. Susceptibility in M-TNF−/− mice was therefore not due to the lack of T cells at local sites of infection, unlike the situation in complete TNF−/− mice where CD4+- and CD8+ T cell recruitment was initially delayed, but became unregulated during terminal stages of infection. Rather, this increased influx of CD4+- and CD8+ T cells early on in M-TNF−/− mice points to an important function of TNF produced by macrophages and neutrophils in the regulation of CD4+- and CD8+ T cell recruitment. Nonetheless, we postulated that although T cell recruitment was not impaired in M-TNF−/− mice, the effector function of recruited cells may have been compromised. Our data demonstrated that recruited CD4+- and CD8+ T cells were fully mature effector cells, expressing CD44 and CD69 activation markers. Moreover these lymphocytes were primed against M. tuberculosis and hence could potentially initiate macrophage bactericidal mechanisms. Previous studies have shown that Th1 immune function is mediated predominantly by M. tuberculosis antigen specific IFNγ expression by CD4+- and CD8+ T cells that can upregulate the antimycobacterial function of macrophages at the site of infection40. These primed pulmonary CD4+- and CD8+T cells of M-TNF−/− mice were also capable of synthesising TNF and IFNγ which resulted in the induction of nitric oxide synthesis in M. tuberculosis infected macrophages43,58,59. We therefore postulated that key to the eventual resistance to M. tuberculosis infection of M-TNF−/− mice was the potential substitution of TNF produced by myeloid cells with TNF from recruited activated T cells resulting in an effective and protective adaptive immune response.
Indeed, the transient nature of disease susceptibility in M-TNF−/− mice indicated that compensatory mechanisms and other cellular sources of TNF allowed for the eventual establishment of full protective immunity. Several studies highlighted T cells as sources of TNF that impacted on disease outcome during bacterial challenge60,61. T cell derived TNF in association with GM-CSF induce macrophage aggregation that may contribute to the formation of granulomas during host immune protective responses against mycobacterial infection61. Furthermore, a modest increase in resistance of M. tuberculosis infected RAG−/− and TNF−/− mice after transfer of T cells capable of producing TNF was reported25. Adoptive transfer approaches have inherent limitations and did not allow for direct interrogation of the role of T cell derived TNF. Here we used a loss of function approach through the use of T cell specific deletion of TNF gene in both CD4+ and CD8+ T cells which has distinct advantages over the adoptive transfer approach, especially for long-term studies of chronic infection studies. Previous reports documented distinct and non-redundant functions of TNF produced by T cells, macrophages and neutrophils for primary defence against Listeria monocytogenes whereas T cell derived TNF appeared subservient in function26. Our present findings support a clear and distinctive non-redundant function for TNF derived from T cells during persistent infection, which may be dispensable during initial immune responses. Interestingly, mortality in T-TNF−/− mice was associated with a relatively modest increase in bacterial burden compared to WT mice, as opposed to TNF−/− mice which succumbed with significantly higher bacilli burdens. However, moribund T-TNF−/− mice presented with an aggressive pulmonary pathology, indicating that T cell derived TNF is required primarily for down regulation of inflammation during persistent/chronic M. tuberculosis infection rather than for bactericidal mechanisms. Increased susceptibility and eventual mortality of TNF−/− mice may thus be a consequence of pulmonary pathology induced by an unregulated immune response. Increased recruitment of CD4+ and CD8+ T cells have previously been reported by Zganiacz et al, where the authors elegantly illustrated the regulatory function of TNF mediated through both TNFRp55 and TNFRp7562. Neutralisation of TNF led to disregulated pulmonary pathology accompanied by enhanced production of specific proinflammatory cytokines responsible for the control and migration of different leukocytes including macrophages and T cells. Although the presence of TNF produced by macrophages and/or other cell types within the granulomatous structures in T-TNF−/− mice were able to contain early mycobacterial infection, the absence of TNF production by T cells during chronic infection resulted in inefficient recruitment of effector T cells, leading to inefficient TNF dependent signalling within the microenvironment of the granuloma. Here, we convincingly demonstrated dependence on CD4+- and/or CD8+T cell derived TNF for sustaining structural and functional integrity of granulomas during chronic infection, in spite of the fact that it was not required for its initial establishment. Egen et al., 2008 have elegantly shown that effector T cells are rapidly recruited to existing granulomas and are retained within these structures with limited egress from lesions60. The authors further demonstrated that T cells within granulomas are highly motile, actively interacting with resident macrophages and apparently provided signals essential for the maintenance of granuloma structure. Anti-TNF treatment both inhibited recruitment of effector T cells to granulomas and restricted movement of T cells within granulomas. It is therefore interesting to speculate on the contribution of TNF produced by T cells in providing T cell-macrophage interaction for the maintenance of structural granuloma integrity. The lack of structural granuloma integrity in T-TNF−/− mice would argue strongly for an essential role of T cell derived TNF, possibly in its membrane-bound form, in driving T cell-macrophages interaction during structural granuloma formation. In fact, we and others showed earlier that not only soluble TNF but also membrane-bound TNF is important for the control of M. tuberculosis infection17,19. Thus, local, cell-associated TNF interactions are likely important for the multi-fold effects of TNF, and measuring soluble TNF in the lung might not give the full picture. This was also documented in an earlier study using TNF expressing BCG, where a limited, local production of TNF at the site of infection conferred protection against M. bovis BCG in TNF-deficient mice63. We report low levels of TNF expression in wild-type, M-TNF−/− or T-TNF−/− mice. Indeed, it is our experience that the pulmonary protein concentration of soluble TNF remains low, even after 1–3 months of M. tuberculosis infection, as compared to other pro-inflammatory cytokines/chemokines such as IL-1, MCP1 or RANTES which are more up-regulated and reach levels in the ng/lung range64,65).
The initial establishment of granuloma to facilitate myeloid-T cell interaction is significantly dependent on controlled cellular inflammation in response to infection. Th1 polarisation of the adaptive response to M. tuberculosis infection has been illustrated at the transcriptional level by persistent induction of CD8a, IFNγ and granzyme A transcripts 15–100 days post-infection66,67. Here, we show that in complete absence of TNF there was a very high induction of molecules indicative of cytotoxic T-cell signature such as CD8a, IFNγ and granzyme A, of ATF3, a positive regulator of IFNγ expression68, of IFNγ-induced GTPase irgm1/LRG47 and IFNγ signalling pathway IRF7, 4 weeks post M. tuberculosis infection, as compared with wild-type mice. The over-induction of these genes was similar or only slightly less marked in mice deficient for T-cell derived TNF, indicating that TNF from T-cell origin is dispensable for this up-regulation. However, the over-induction of cytotoxic T cell signature genes was more limited in M-TNF−/− mice, indicating that TNF from myeloid origin is essential for the up-regulation of these genes. A strong and persistent inflammatory response was documented as early as 2–3 weeks post M. tuberculosis infection, by the transcriptional up-regulation of pro-inflammatory cytokines including TNF itself, IL-1β or IL-6, of chemokines such as CXCL5, but also of acute response proteins such as SAA3 or S100a9, and of iNOS, a key anti-mycobacterial enzyme source of reactive nitrogen66,67. We show that the inflammatory signature was strongly increased in complete TNF-deficient mice 4 weeks post M. tuberculosis infection, which may result from the very high infectious burden in these mice at this time point. Interestingly, IL-1β, IL-6, IL-17a, CXCL3, CXCL5 or S100a9 were much less up-regulated in T-TNF−/− mice, and M-TNF−/− behaved essentially like WT mice. iNOS was strongly over-expressed in mice fully deficient for TNF or in M-TNF−/− mice, while T-TNF−/− mice had a low expression, as compared to infected WT mice. Thus, TNF per se is dispensable for the induction of these inflammatory mediators and might be compensated for by other up-regulated inflammatory cytokines. The data correlates with the phenotypic profiles of the respective strains and supports the relative importance and differential effects of TNF produced by macrophages and T cells in the control of acute M. tuberculosis infection.
The similarity in phenotype between WT and T-TNF−/− mice together with the disparate phenotypes between WT and M-TNF−/− mice during acute M. tuberculosis infection indicated that TNF produced by myeloid cells is more relevant for early host immune defence as compared to T cell derived TNF. Nonetheless, we hypothesised that recruited T cells capable of TNF synthesis could contribute to rescue M. tuberculosis infected M-TNF−/− mice from early defect in resistance to infection. We therefore characterized host immune function against M. tuberculosis in MT-TNF mice, in which case macrophages/neutrophils, and T cells were incapable of TNF synthesis. We argued that deletion of TNF synthesis capability from CD4+- and CD8+ T cells in M-TNF−/− mice would render the host unable to draw on the secondary TNF cellular source to induce immune protection. The extent of susceptibility of M. tuberculosis infected MT-TNF−/− mice was indistinguishable from TNF−/− mice indicating that macrophage/neutrophils and T cells are the main cellular sources of TNF required to generate complete protection against M. tuberculosis infection. Combined TNF deficiency in myeloid cells and T cells resulted in exacerbated disease progression with uncontrolled bacterial growth in organs and severe pulmonary pathology and mortality, clearly pointing out to redundant essential functions of TNF from these distinct cellular sources.
In conclusion, this study provides significant insights on TNF cellular source requirements for host immune responses against M. tuberculosis challenge. Although widely acclaimed as the major cellular source of TNF, macrophages have a surprisingly limited role in restricting bacterial replication through TNF dependent synthesis during M. tuberculosis infection. Disease outcome resides rather with T cell derived TNF whose function appears as non-redundant. Our data imply that inhibiting TNF synthesis in T cells during latent M. tuberculosis infection may lead to an imbalance in the cytokine profile within the granuloma microenvironment with subsequent potential for reactivation and immunopathology. TNF neutralizing therapies used in severe inflammatory diseases should thus spare T-cell derived TNF and this may reduce the risk of tuberculosis reactivation. We clearly demonstrated the existence of labour division between myeloid cells and T cells in provision of protective TNF which implicates both redundant and non-redundant functions of this cytokine from distinct cellular sources.
Methods
Mice
All animal strains used in this study were previously described26. TNF “floxed” (WT control) mice were crossed with MLys-Cre deleter mice to generate myeloid, macrophage/neutrophil TNF deficient mice (M-TNF−/−) and similarly crossed with CD4-Cre deleter mice to generate T-cell TNF deficient mice (T-TNF−/−). M-TNF−/− and T-TNF mice were in turn intercrossed to generate TNF ablation in both myeloid cells and T cells (MT-TNF−/−). In addition, complete TNF deficient (TNF−/−) mice were used as controls. Animals were bred under specific pathogen free conditions at the University of Cape Town or Transgenic institute (CNRS UPS44, Orleans, France) and used between 6–12 weeks. Infected animals were maintained in individually ventilated cages or isolators under biosafety level 3 conditions.
Ethics statement
All animal experimental protocols complied with South African or French regulations. Approval was obtained from the Animal Research Ethics Committee, University of Cape Town, South Africa in accordance with the South African National Standard 10386-The care and use of animals for scientific purposes or were approved by the “Ethics Committee for Animal Experimentation of CNRS Campus Orleans” (CCO), registered (N°3) by the French National Committee of Ethical Reflexion for Animal Experimentation, under the N° CLE CCO 2011-029 in compliance with the French ethical and animal experiments regulations (see Charte Nationale, Code Rural R 214–122, 124).
Mycobacteria and infection
Mycobacterium tuberculosis H37Rv was grown in Difco Middlebrook 7H9 medium containing 0.5% glycerol and enriched with 10% OADC. Cultures were incubated at 37°C and grown until log phase, aliquoted and maintained as frozen stocks at −70°C. An aliquot of M. tuberculosis H37Rv was rapidly thawed at 37°C, passed 30× through a 29.5G needle and diluted in sterile saline. Mice were infected at doses of 200–500 cfu/lung using a Glas-Col Inhalation Exposure System Model A4224. The pulmonary infection dose was confirmed by sacrificing 10 mice 1 day after infection and plating the homogenised lung tissue on Difco Middlebrook 7H10 agar plates in 10 fold serial dilutions. Plates were semi-sealed in plastic bags, incubated for 17–21 days at 37°C and the number of mycobacterial colonies was counted.
Single cell preparation, cell restimulation, intracellular cytokine staining and analysis by flow cytometry
Lungs were perfused by injecting 5 ml of cold PBS containing 20 U/ml heparin in the right ventricle of the heart. Lungs were removed, sectioned on ice and incubated in PBS containing 50 U/ml collagenase I (Worthington Biomedical Corporation, Lakewood, NJ) and 13 ug/ml DNAse I (Boehringer-Mannheim, Germany) at 37°C for 90 minutes. For single cell suspensions lung tissue was passed through a 70 μm nylon cell strainer (Beckton and Dickinson), washed 2× with PBS and the cell concentration determined by counting in the presence of trypan blue. Cells were labelled using the following antibodies: anti-mouse CD3 (Clone 17A2; Pharmingen); anti-mouse CD8a (Clone 53–6.7; Pharmingen); anti-mouse CD4 (Clone RM4-5; Pharmingen), anti-mouse CD44 mouse (Clone IM7; Pharmingen) and anti-mouse CD69 (Clone H1.2F3; Pharmingen). 1 × 106 cells were incubated for 20 minutes with anti-CD16/32 (Clone 2.4G2; Pharmingen) to block Fc receptors in 96 well round bottom plates (Sterilin). Cells were washed with PBS/0.1% BSA/0.01% NaN3 and incubated with 2 ug/ml of appropriate antibodies for 20 minutes in the dark. Excess antibodies were removed by washing cells 2× with PBS/0.1% BSA/0.01% NaN3, then pelleted cells were fixed for 18–24 hours in 10% PBS buffered formalin.
For intracellular staining, single cell suspensions of lungs were restimulated with plate-bound anti-mouse CD3 (5 μg/ml;clone 145-2c11) and anti-mouse CD28 (5 μg/ml; clone 37.51), ESAT-6 (10 μg/ml; Staten Serum Institute) and PPD (10 μg/ml; Staten Serum Institute) for 6 hours in the presence of Brefeldin A (5 μg/ml). Cells were labelled with either anti-mouse CD8a (Clone 53–6.7; Pharmingen) or anti-mouse CD4 (Clone RM4-5; Pharmingen) and anti-mouse IFNγ (Clone XMG1.2; Pharmingen). Analysis was done using FACScalibur (Beckton and Dickinson) incorporating Cellquest software.
For cytokine measurements samples were subjected to ELISA using antibody and protein standards obtained from RnD Systems Inc.,(Minneapolis, MN). Absorbance was measured using a Versamax Microplate Reader (Molecular Devices, LLC, CA)
Lung morphology
Lungs were fixed in phosphate buffered formalin and embedded in paraffin wax. Tissues were sectioned at 2 μm, stained with hematoxylin and eosin and Ziehl-Neelsen, and subsequently mounted with Canada balsam. Images of stained tissue sections were captured using an Olympus E330 digital camera attached to Olympus Model BX40F microscope. Macroscopic lung images were captured with Pentax Optio M40 camera.
Analysis of mRNA gene expression
Total RNA were isolated from lungs by TRIzol reagent (Sigma, St. Louis,MO) and purified by RNeasy Mini Kit (Qiagen, Valencia, CA) at the indicated times after M. tuberculosis infection. For quantitative RT-PCR 1 ug of total RNA was pretreated by DNaseI and converted to cDNA by ImProm-II™ Reverse Transcriptase (Promega, Madison, WI) using random nonamer primers. Q-PCR reactions were performed using Brillant II SYBR QPCR kits and a Stratagene Mx3005P thermocycler (Agilent, Palo Alto, CA). The following program was used: 95°C for 15 min and 40 cycles 95°C for 10 sec and 60°C for 30 sec. Primers used for quantitative PCR are provided in Supplementary Table 1.
Statistical analysis
Statistical analysis was performed by ANOVA or the Student's t test. For mortality studies, analysis was performed using the logrank test. For all tests, a p value of <0.05 was considered significant.
Author Contributions
N.A., S.G., R.K., N.H., M.B., N.C., C.F., Y.S. and V.Y. performed experiments while N.A., S.G., Y.S., S.G., S.N., V.Q. and M.J. were responsible for experimental design and data analysis. B.R., S.N. and M.J. provided materials. M.J., B.R., V.Q., S.N., S.G. and N.A. wrote the main manuscript text and all the authors reviewed the manuscript.
Supplementary Material
Supplementary Information SREP-12-04044
Acknowledgments
We thank the following institutional members: Lizette Fick, Marylin Tyler for their sterling contribution to the histopathology, Faried Abbass and Lungile Matika for technical support, the staff of the Division of Immunology and the Animal Unit at the University of Cape Town for their contribution to animal care and technical support. We are grateful to Virginie Vasseur, Sabine Charron and Rachel Vacher (CNRS, Orleans) for skilful assistance with genotyping and in vivo experiments. This study was funded by the National Research Foundation (South Africa), The Medical Research Council (South Africa), The National Health Laboratory Services (South Africa), The University of Cape Town, INTAS YSF, the European Union (TB REACT Contract no 028190), State Contract 16.512.11.2054 and RFBR grant 11-04-12159.
References
- Moreno C. et al. Lipoarabinomannan from Mycobacterium tuberculosis induces the production of tumour necrosis factor from human and murine macrophages. Clin Exp Immunol 76, 240–245 (1989). [PMC free article] [PubMed] [Google Scholar]
- Roach T. I., Barton C. H., Chatterjee D. & Blackwell J. M. Macrophage activation: lipoarabinomannan from avirulent and virulent strains of Mycobacterium tuberculosis differentially induces the early genes c-fos, KC, JE, and tumor necrosis factor-alpha. J Immunol 150, 1886–1896 (1993). [PubMed] [Google Scholar]
- Falcone V., Bassey E. B., Toniolo A., Conaldi P. G. & Collins F. M. Differential release of tumor necrosis factor-alpha from murine peritoneal macrophages stimulated with virulent and avirulent species of mycobacteria. FEMS Immunol Med Microbiol 8, 225–232 (1994). [DOI] [PubMed] [Google Scholar]
- Oswald I. P., Dozois C. M., Fournout S., Petit J. F. & Lemaire G. Tumor necrosis factor is required for the priming of peritoneal macrophages by trehalose dimycolate. Eur Cytokine Netw 10, 533–540 (1999). [PubMed] [Google Scholar]
- Flesch I. E. & Kaufmann S. H. Activation of tuberculostatic macrophage functions by gamma interferon, interleukin-4, and tumor necrosis factor. Infect Immun 58, 2675–2677 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean A. G. et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol 162, 3504–3511 (1999). [PubMed] [Google Scholar]
- Kindler V., Sappino A. P., Grau G. E., Piguet P. F. & Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56, 731–740 (1989). [DOI] [PubMed] [Google Scholar]
- Flynn J. L. et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995). [DOI] [PubMed] [Google Scholar]
- Jacobs M. et al. Correction of defective host response to Mycobacterium bovis BCG infection in TNF-deficient mice by bone marrow transplantation. Laboratory investigation; a journal of technical methods and pathology 80, 901–914 (2000). [DOI] [PubMed] [Google Scholar]
- Locksley R. M., Killeen N. & Lenardo M. J. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501 (2001). [DOI] [PubMed] [Google Scholar]
- Allie N. et al. Limited role for lymphotoxin alpha in the host immune response to Mycobacterium tuberculosis. J Immunol 185, 4292–4301 (2010). [DOI] [PubMed] [Google Scholar]
- Bopst M. et al. Differential effects of TNF and LTalpha in the host defense against M. bovis BCG. Eur J Immunol 31, 1935–1943 (2001). [DOI] [PubMed] [Google Scholar]
- Bazzoni F. & Beutler B. How do tumor necrosis factor receptors work? J Inflamm 45, 221–238 (1995). [PubMed] [Google Scholar]
- Dempsey P. W., Doyle S. E., He J. Q. & Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 14, 193–209 (2003). [DOI] [PubMed] [Google Scholar]
- Black R. A. et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733 (1997). [DOI] [PubMed] [Google Scholar]
- Moss M. L. et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385, 733–736 (1997). [DOI] [PubMed] [Google Scholar]
- Allie N. et al. Protective role of membrane tumour necrosis factor in the host's resistance to mycobacterial infection. Immunology 125, 522–534 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambuza I. et al. Efficacy of membrane TNF mediated host resistance is dependent on mycobacterial virulence. Tuberculosis (Edinb) 88, 221–234 (2008). [DOI] [PubMed] [Google Scholar]
- Fremond C. et al. Membrane TNF confers protection to acute mycobacterial infection. Respir Res 6, 136 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol 10, 411–452 (1992). [DOI] [PubMed] [Google Scholar]
- Keane J. TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatology (Oxford) 44, 714–720 (2005). [DOI] [PubMed] [Google Scholar]
- Keane J. et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 345, 1098–1104 (2001). [DOI] [PubMed] [Google Scholar]
- Kaufmann S. H. & Flesch I. E. The role of T cell--macrophage interactions in tuberculosis. Springer seminars in immunopathology 10, 337–358 (1988). [DOI] [PubMed] [Google Scholar]
- North R. J. & Jung Y. J. Immunity to tuberculosis. Annu Rev Immunol 22, 599–623 (2004). [DOI] [PubMed] [Google Scholar]
- Saunders B. M., Briscoe H. & Britton W. J. T cell-derived tumour necrosis factor is essential, but not sufficient, for protection against Mycobacterium tuberculosis infection. Clinical and experimental immunology 137, 279–287 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S. I. et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: protective and deleterious effects. Immunity 22, 93–104 (2005). [DOI] [PubMed] [Google Scholar]
- Roach D. R. et al. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 168, 4620–4627 (2002). [DOI] [PubMed] [Google Scholar]
- Flesch I. E. et al. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon gamma and tumor necrosis factor alpha. J Exp Med 181, 1615–1621 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman S. et al. Mycobacterium tuberculosis H37Ra and H37Rv differential growth and cytokine/chemokine induction in murine macrophages in vitro. J Interferon Cytokine Res 26, 27–33 (2006). [DOI] [PubMed] [Google Scholar]
- Eriks I. S. & Emerson C. L. Temporal effect of tumor necrosis factor alpha on murine macrophages infected with Mycobacterium avium. Infect Immun 65, 2100–2106 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch C. S., Ellner J. J., Russell D. G. & Rich E. A. Complement receptor-mediated uptake and tumor necrosis factor-alpha-mediated growth inhibition of Mycobacterium tuberculosis by human alveolar macrophages. J Immunol 152, 743–753 (1994). [PubMed] [Google Scholar]
- Denis M., Gregg E. O. & Ghandirian E. Cytokine modulation of Mycobacterium tuberculosis growth in human macrophages. International journal of immunopharmacology 12, 721–727 (1990). [DOI] [PubMed] [Google Scholar]
- Denis M. & Gregg E. O. Recombinant tumour necrosis factor-alpha decreases whereas recombinant interleukin-6 increases growth of a virulent strain of Mycobacterium avium in human macrophages. Immunology 71, 139–141 (1990). [PMC free article] [PubMed] [Google Scholar]
- Stein M. & Gordon S. Regulation of tumor necrosis factor (TNF) release by murine peritoneal macrophages: role of cell stimulation and specific phagocytic plasma membrane receptors. Eur J Immunol 21, 431–437 (1991). [DOI] [PubMed] [Google Scholar]
- Tachibana K., Chen G. J., Huang D. S., Scuderi P. & Watson R. R. Production of tumor necrosis factor alpha by resident and activated murine macrophages. J Leukoc Biol 51, 251–255 (1992). [DOI] [PubMed] [Google Scholar]
- Togbe D. et al. T cell-derived TNF down-regulates acute airway response to endotoxin. Eur J Immunol 37, 768–779 (2007). [DOI] [PubMed] [Google Scholar]
- Cooper A. M. & Flynn J. L. The protective immune response to Mycobacterium tuberculosis. Current opinion in immunology 7, 512–516 (1995). [DOI] [PubMed] [Google Scholar]
- Cooper A. M. T cells in mycobacterial infection and disease. Current opinion in immunology 21, 378–384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Z., Wang J., Croitoru K. & Wakeham J. Protection by CD4 or CD8 T cells against pulmonary Mycobacterium bovis bacillus Calmette-Guerin infection. Infection and immunity 66, 5537–5542 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C. G., Bean A. G., Hooi H., Briscoe H. & Britton W. J. Increase in gamma interferon-secreting CD8(+), as well as CD4(+), T cells in lungs following aerosol infection with Mycobacterium tuberculosis. Infection and immunity 67, 3242–3247 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan M. S., Vaporciyan A. A., Miyasaka M., Tamatani T. & Ward P. A. Tumor necrosis factor alpha regulates in vivo intrapulmonary expression of ICAM-1. Am J Pathol 142, 1739–1749 (1993). [PMC free article] [PubMed] [Google Scholar]
- Cooper A. M. et al. Disseminated tuberculosis in interferon gamma gene-disrupted mice. The Journal of experimental medicine 178, 2243–2247 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. L. et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. The Journal of experimental medicine 178, 2249–2254 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das G., Sheridan S. & Janeway C. A. Jr The source of early IFN-gamma that plays a role in Th1 priming. J Immunol 167, 2004–2010 (2001). [DOI] [PubMed] [Google Scholar]
- Tsukaguchi K., de Lange B. & Boom W. H. Differential regulation of IFN-gamma, TNF-alpha, and IL-10 production by CD4(+) alphabetaTCR+ T cells and vdelta2(+) gammadelta T cells in response to monocytes infected with Mycobacterium tuberculosis-H37Ra. Cellular immunology 194, 12–20 (1999). [DOI] [PubMed] [Google Scholar]
- Tracey K. J. & Cerami A. Tumor necrosis factor, other cytokines and disease. Annual review of cell biology 9, 317–343 (1993). [DOI] [PubMed] [Google Scholar]
- Tsai M. C. et al. Characterization of the tuberculous granuloma in murine and human lungs: cellular composition and relative tissue oxygen tension. Cellular microbiology 8, 218–232 (2006). [DOI] [PubMed] [Google Scholar]
- Ladel C. H., Szalay G., Riedel D. & Kaufmann S. H. Interleukin-12 secretion by Mycobacterium tuberculosis-infected macrophages. Infection and immunity 65, 1936–1938 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton M. J. et al. Induction of gamma interferon production in human alveolar macrophages by Mycobacterium tuberculosis. Infection and immunity 65, 5149–5156 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury M. G. & Moreno C. Effect of lipoarabinomannan and mycobacteria on tumour necrosis factor production by different populations of murine macrophages. Clinical and experimental immunology 94, 57–63 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton W. J., Meadows N., Rathjen D. A., Roach D. R. & Briscoe H. A tumor necrosis factor mimetic peptide activates a murine macrophage cell line to inhibit mycobacterial growth in a nitric oxide-dependent fashion. Infection and immunity 66, 2122–2127 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara I., Udagawa T. & Yamada H. Rat neutrophils prevent the development of tuberculosis. Infect Immun 72, 1804–1806 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau A. R. et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest 117, 1988–1994 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant K. V. & McMurray D. N. Guinea pig neutrophils infected with Mycobacterium tuberculosis produce cytokines which activate alveolar macrophages in noncontact cultures. Infection and immunity 75, 1870–1877 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engele M. et al. Induction of TNF in human alveolar macrophages as a potential evasion mechanism of virulent Mycobacterium tuberculosis. J Immunol 168, 1328–1337 (2002). [DOI] [PubMed] [Google Scholar]
- Orme I. M., Roberts A. D., Griffin J. P. & Abrams J. S. Cytokine secretion by CD4 T lymphocytes acquired in response to Mycobacterium tuberculosis infection. J Immunol 151, 518–525 (1993). [PubMed] [Google Scholar]
- Andersen P. & Smedegaard B. CD4(+) T-cell subsets that mediate immunological memory to Mycobacterium tuberculosis infection in mice. Infection and immunity 68, 621–629 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch I. & Kaufmann S. H. Mycobacterial growth inhibition by interferon-gamma-activated bone marrow macrophages and differential susceptibility among strains of Mycobacterium tuberculosis. J Immunol 138, 4408–4413 (1987). [PubMed] [Google Scholar]
- Bonecini-Almeida M. G. et al. Induction of in vitro human macrophage anti-Mycobacterium tuberculosis activity: requirement for IFN-gamma and primed lymphocytes. J Immunol 160, 4490–4499 (1998). [PubMed] [Google Scholar]
- Egen J. G. et al. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28, 271–284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P. F. et al. Patterns of cytokine production by mycobacterium-reactive human T-cell clones. Infection and immunity 61, 197–203 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zganiacz A. et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J Clin Invest 113, 401–413 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker L. G. et al. Immunopathologic effects of tumor necrosis factor alpha in murine mycobacterial infection are dose dependent. Infect Immun 68, 6954–6961 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremond C. M. et al. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest 114, 1790–1799 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremond C. M. et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 179, 1178–1189 (2007). [DOI] [PubMed] [Google Scholar]
- Gonzalez-Juarrero M. et al. Immune response to Mycobacterium tuberculosis and identification of molecular markers of disease. American journal of respiratory cell and molecular biology 40, 398–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D. D., Lin Y., Moreno J. R., Randall T. D. & Khader S. A. Profiling early lung immune responses in the mouse model of tuberculosis. PloS one 6, e16161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filen S. et al. Activating transcription factor 3 is a positive regulator of human IFNG gene expression. J Immunol 184, 4990–4999 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information SREP-12-04044