

Abstract
More than half of the world’s population is infected with Helicobacter pylori, which is strongly linked to the development of chronic gastric inflammation (gastritis), peptic ulcer disease, and stomach cancer. However, for unknown reasons, the vast majority of infected individuals are asymptomatic beyond histologic inflammation. This review article will summarize current knowledge on the molecular mechanisms of H. pylori colonization of the gastric mucosa, with a particular focus on the biochemistry of MUC1 mucin in the host response to bacterial infection.
Keywords: Helicobacter pylori, stomach, epithelium, MUC1 mucin
1. Introduction
Helicobacter pylori is a Gram-negative, microaerophilic, flagellated bacterium that colonizes the human stomach [1]. H. pylori infections are acquired in childhood, particularly in developing countries and, in the absence of antibiotic therapy, persist for the lifetime of the individual [2]. Currently, greater than 50% of the world’s population is infected with H. pylori, making it the most globally common bacterial infectious agent. In spite of its high prevalence in the human population, greater than 80% of infected individuals remain asymptomatic apart from histologic gastritis. For example, from an estimated 50–100 million individuals in the U.S. currently infected with H. pylori, only about 25,000 new cases of stomach cancer are diagnosed each year [3]. It is enigmatic why only a subset of infected individuals develops serious gastric disease. In this review article, we propose a novel molecular mechanism that addresses this question, namely those defects in the anti-inflammatory MUC1 mucin glycoprotein in a limited cohort of H. pylori-infected individuals predisposes to uncontrolled inflammation that ultimately evolves to peptic ulcer disease and/or stomach cancer.
2. H. pylori infection of the gastric mucosa
The relatively high prevalence of H. pylori infection is due, in large part, to the inability of host immunity to clear bacterial colonization, in spite of an overt gastric inflammatory response [4]. Within the group of H. pylori-infected individuals who do display clinical symptoms, epidemiologic and pathologic studies have confirmed a progressive increase in disease symptomology and severity over several decades, starting with chronic inflammation, followed by the development of noncancerous and precancerous lesions, and finally gastric adenocarcinoma [5]. It has been proposed that chronic gastritis during a lifetime of bacterial infection generates a proinflammatory environment characterized by the activation of intracellular signaling pathways that promote the progression to gastric cancer [6]. Indeed, H. pylori infection is associated with increased gastric epithelial cell mutations, inhibition of apoptosis, and stimulation of angiogenesis and cell proliferation, all of which contribute to development of a cancer phenotype [7]. Therefore, better understanding of host proinflammatory pathways, as well as the counter-regulatory anti-inflammatory mechanisms, will provide important new information that is relevant for the treatment of H. pylori-infected patients who are at increased risk for developing life-threatening gastric diseases.
3. H. pylori adhesion to gastric mucosa
A central dogma in bacterial pathogenesis is that microbial adhesion to host cell surface receptors is a prerequisite for successful infection [8]. H. pylori colonization of the stomach is initiated through pathogen binding to cell surface receptors expressing the sialyl-Lewis a (sLea), Lewis b (Leb), and sialyl-Lewis x (sLex) glycoconjugates [9]. The corresponding H. pylori adhesins, BabA (blood group antigen binding adhesion) and SabA(sialic acid binding adhesion), interact with these host receptors [10]. Among the epithelial glycoproteins containing these Lewis antigens are gastric mucins. At low pH, BabA binds to the soluble MUC5AC mucin glycoprotein, whereas under neutral pH conditions BabA binds both to the MUC5AC mucin and to the cell-associated MUC1 mucin [11,12].
4. Gastric mucins
Mucus is a viscous, gel-like material containing a mixture of high molecular weight, extensively glycosylated mucin proteins that are produced by most secretory epithelia [13]. Currently, 20 mucin genes have been identified and their encoded glycoproteins have been classified as secreted mucins and cell-associated mucins [13–15]. By convention, mucin genes are designated as MUC in humans and Muc in animals, followed by an Arabic numeral indicating their order of discovery. Secreted mucins accumulate in large secretory granules of specialized epithelial cells known as mucous/goblet cells. Under the appropriate extracellular stimulus, exocytosis of the secretory granules releases the packaged mucin molecules that subsequently establish intermolecular associations to form a continuous, viscoelastic layer covering the epithelial mucosa. Transmembrane mucins are membrane-embedded glycoproteins that are localized on the apical surface of epithelial cells.
Mucins expressed by the gastric mucosa include secreted mucins, e.g. MUC5AC, and membrane-tethered mucins, e.g. MUC1. The deduced amino acid sequence of the MUC1 gene indicates a multidomain structure of the encoded protein, consisting of a large molecular weight extracellular (EC) domain, a transmembrane (TM) domain, and an intracellular cytoplasmic tail (CT) domain (Fig. 1) [16,17]. Autocatalytic proteolysis of the MUC1 protein generates a heterodimeric structure that resides on the cell surface with the EC domain noncovalently associated with a small subunit containing the TM and CT domains [18,19]. The separate EC and TM/CT subunits can be detected on SDS-PAGE gels using antibodies specific for each region (Fig. 2). The EC domain contains a variable number of 20-amino acid tandem repeats (VNTRs). The CT domain contains 72 amino acids, 7 of which are evolutionarily conserved tyrosines that are potential sites of phosphorylation (Fig. 3). Similar to cytokine and growth factor receptors, MUC1 CT tyrosine phosphorylation occurs in consensus amino acid sequence motifs for signaling kinases and adaptor proteins including phosphoinositide 3-kinase (PI3K), Shc, c-Src, and Grb-2 [20,21].
Figure 1.

Schematic structure of the MUC1 mucin glycoprotein. The extracellular region consists of a variable number of tandem repeats (VNTR) and the sea urchin sperm protein, enterokinase and agrin (SEA) domain, both of which contain numerous glycan side chains. Autocatalytic proteolysis within the SEA domain (arrow) creates the noncovalently-associated, heterodimeric structure. Distal to the SEA domain is a single-pass transmembrane (TM) region, and an intracellular cytoplasmic tail (CT) region.
Figure 2.
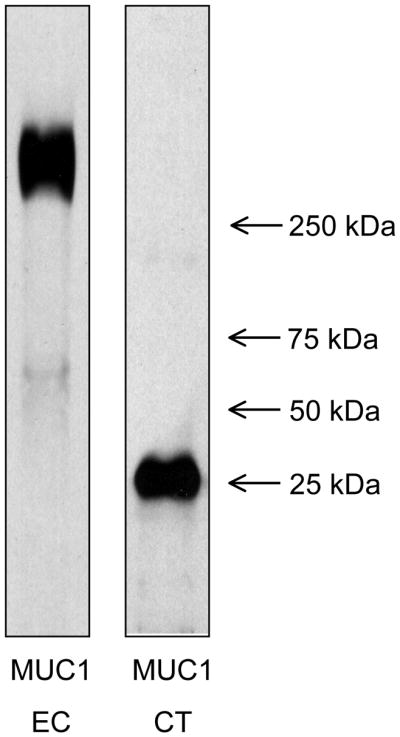
Heterodimeric structure of MUC1 mucin glycoprotein. A detergent lysate of AGS human gastric epithelial cells was resolved on denaturing SDS-PAGE gels to separate the large molecular weight MUC1 extracellular (EC) region and the smaller cytoplasmic tail (CT) region. The separated proteins were probed on immunoblots with antibodies specific for the MUC1 EC and CT regions.
Figure 3.

Schematic structure of the MUC1 CT. The 72-amino acid (aa) CT contains binding sites for PI3K, Shc, c-Src, β-catenin, and Grb-2.
5. Phosphorylation of the MUC1 CT
Given the receptor-like structure of MUC1, it is not surprising that numerous studies have shown that its CT region is phosphorylated at multiple tyrosine residues [20–32]. Chinese hamster ovary (CHO) cells over-expressing hamster Muc1 and treated with whole bacteria, or with bacterial flagellin protein, had increased phosphorylation of the Muc1 CT [28]. In another cell culture system, we utilized the human MUC1 CT that was linked to the EC domain of CD8 and the resulting CD8/MUC1 chimeric protein was expressed into COS7 cells. Treatment of CD8/MUC1-COS7 cells with anti-CD8 antibody induced tyrosine phosphorylation of the MUC1 CT at 4 tyrosine residues, Y20, Y29, Y46, and Y60 [21]. These 4 tyrosines are located within consensus amino acid sequence motifs for binding to the PI3K, Shc, c-Src, and Grb-2 signaling proteins (Fig. 3), suggesting that MUC1 CT phosphorylation activates downstream, intracellular signaling transduction.
Because previous studies demonstrated that the extracellular signal-regulated kinase (ERK) was constitutively activated by MUC1 in mammary epithelial cells [33], ERK1/2 phosphorylation was investigated in response to treatment of CHO-Muc1 cells with bacterial flagellin protein. Flagellin activated ERK1/2 in CHO-Muc1 cells, but not in Muc1 negative CHO cells [28]. Further, anti-CD8 antibody treatment of CD8/MUC1-COS7 cells stimulated a Ras → MEK1/2 → ERK1/2 cascade [34]. Collectively, these results demonstrated that extracellular stimulation of the full-length Muc1 molecule with flagellin, or the CD8/MUC1 chimera with anti-CD8 antibody, stimulated phosphorylation of the intracellular MUC1/Muc1 CT, leading to activation of an ERK1/2 signaling pathway.
6. MUC1 CT interaction with β-catenin
The MUC1 CT region binds to β-catenin at a -S-A-G-N-G-G-S-S-L- amino acid sequence (Fig. 3). This sequence fits a canonical β-catenin binding motif (-S-X-X-X-X-X-S-S-L-) that is conserved with other β-catenin binding partners, including the epithelial E-cadherin protein [25,32,35,36]. β-Catenin was originally identified in the Wnt signaling pathway, a major oncogenic pathway associated with gastric tumorigenesis [37]. Subsequently, β-catenin was demonstrated to form a multiprotein complex that anchors the cytoplasmic domain of E-cadherin to the actin cytoskeleton, thereby regulating epithelial barrier formation, the paracellular pathway, and polarity in normal cells [38]. However, the role of MUC1/β-catenin interaction in regulating the host gastric epithelial cell response H. pylori colonization is unknown.
7. Counter-regulatory role of gastric MUC1 mucin in H. pylori infection
Mucosal epithelial cells, including those of the gastric epithelium, express multiple surface receptors, in addition to MUC1, that signal the presence of invading pathogens and activate host inflammatory responses [39]. While much is known concerning the inflammatory pathways that are activated following microbial colonization, relatively few studies have characterized the counter-balancing, anti-inflammatory responses. We recently reported that expression of MUC1/Muc1 counter-regulated H. pylori-induced gastric inflammation [40]. Using a mouse model of H. pylori infection, Muc1 knockout (Muc1−/−) mice displayed increased bacterial load and greater levels of gene transcripts encoding the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and keratinocyte chemoattractant (KC) in the stomach, compared with Muc1+/+ mice. Knockdown of MUC1 expression using in vitro cultures of AGS human gastric epithelial cells was correlated with greater activation and nuclear translocation of nuclear factor-κB (NF-κB), decreased activity of the NF-κB inhibitor, IκBα, and augmented IL-8 production, compared with MUC1-expressing cells. By contrast, transfection of AGS cells with a MUC1 expression plasmid was associated with decreased NF-κB activation and nuclear translocation, and reduced IL-8 production, compared with cells expressing MUC1 endogenously. Cotransfection of AGS cells with MUC1 plus the IκB kinase (IKK), which phosphorylates the inhibitory IκBα protein resulting in dissociation from NF-κB and activation of NF-κB, reversed the MUC1 inhibitory effect. Finally, the MUC1 CT formed a protein complex with IKK, suggesting that MUC1 binds to IKK, thereby inhibiting formation of the catalytically active IKK complex and blocking the ability of H. pylori to stimulate IκBα phosphorylation, NF-κB activation, and downstream inflammatory responses.
8. Host versus bacteria initiated events in H. pylori pathogenesis
While it is well-known that pro- and anti-inflammatory responses during H. pylori infection are initiated in reaction to a variety of bacterial products, the relative roles of these host responses versus direct activity of the bacterial components in mediating pathogenesis/carcinogenesis remain controversial. A chronic inflammatory microenvironment during a lifetime of H. pylori infection has been suggested to promote host-dependent alterations in gastric epithelial cell phenotypes that ultimately culminate in neoplastic transformation. For example, augmented production of proinflammatory mediators in response to bacterial infection may increase gastric epithelial cell mutations in tumor suppressor genes, ultimately leading to cancer [7]. The perigenetic hypothesis proposes that H. pylori-induced up-regulation of inflammatory proteins, such as TNF-α, alters gastric epithelial cell adhesion and/or proliferation, thus leading to the dispersion and propagation of precancerous epithelial cells into mature tumors without the need for additional genetic mutations [41,42]. On the other hand, the H. pylori cytotoxin-associated gene A (CagA) protein has been proposed as a major factor determining the progression to gastric epithelial tumorigenesis. In the absence of bacterial exposure, Ohnishi and colleagues [43] reported that transgenic mice expressing a recombinant CagA protein in vivo developed greater gastric epithelial hyperplasia and adenocarcinomas, compared with nontransgenic mice. Further studies are required to elucidate the relative roles host versus bacterial events in H. pylori pathogenesis.
9. Role of H. pylori CagA protein in virulence
The H. pylori CagA virulence factor is a 120–145 kDa protein encoded by the 40 kb Cag pathogenicity island (PAI) [44]. Approximately 60% of H. pylori isolates in Western countries encode the Cag PAI, whereas nearly 100% of East Asian isolates are CagA positive. Individuals infected with bacterial strains carrying the Cag PAI have a stronger gastric inflammatory response, and are at a greater risk of developing gastric cancer, compared with individuals infected with strains lacking the Cag PAI [45]. The Cag PAI encodes a type IV secretion apparatus that delivers the CagA protein into gastric epithelial cells [46]. Intracellular CagA disrupts the actin cytoskeleton, modifies cell-cell adhesion, alters cell polarity, and deregulates intracellular signaling pathways [47]. Some of these effects are mediated as a result of c-Src- or Abl-dependent CagA phosphorylation at a COOH-terminal -E-P-I-Y-A- repetitive amino acid sequence, leading to activation of the SHP-2 protein tyrosine phosphatase.
CagA positive strains of H. pylori also have been shown to activate the epidermal growth factor receptor (EGFR) receptor protein tyrosine kinase [48]. Activation of EGFR by H. pylori is associated with altered signal transduction in host epithelial cells that may contribute to bacterial pathogenesis. More specifically, among the intracellular signaling cascades that are affected by CagA is the β-catenin pathway [49–51]. In this regard, binding of β-catenin to the MUC1 CT is regulated by EGFR [26]. Because the IL-8 gene promoter is transcriptionally activated by β-catenin [52], it was hypothesized that up-regulation of MUC1 expression during H. pylori infection reduces CagA-dependent β-catenin nuclear localization and concomitant IL-8 production. In support of this hypothesis, we recently reported that H. pylori-infected Muc1−/− mice had increased neutrophil infiltration of the gastric mucosa, an IL-8-dependent event, compared with Muc1+/+ mice [53]. Further, β-catenin formed protein complexes with both MUC1 and CagA in human AGS cells, and MUC1 over-expression reduced CagA/β-catenin interaction. Finally, over-expression of MUC1 decreased the levels of H. pylori-driven nuclear β-catenin in H. pylori-infected cells. These combined results suggested that H. pylori-dependent IL-8 production, neutrophil infiltration, and stomach inflammation may be reduced by future therapeutic strategies designed to increase MUC1 expression.
10. Attenuation of H. pylori gastric inflammation by MUC1 and PPARγ
In addition to MUC1, interleukin-10, transforming growth factor-β, and peroxisome proliferator-associated receptor γ (PPARγ) have been identified as anti-inflammatory mediators in the gastric mucosa [54,55]. PPARs comprise a family of ligand-activated transcription factors, PPARα, β, and γ [56]. Following engagement by endogenous (free fatty acids and eicosanoids) or synthetic (thiazolidinediones) ligands, PPARs translocate to the nucleus where they heterodimerize with retinoid X receptors and transcriptionally activate gene expression. PPARs regulate multiple cellular processes, including glucose metabolism, cell differentiation, carcinogenesis, and inflammation [57]. Because activation of PPARγ is known to suppress gastric inflammatory responses to H. pylori [54], and because the MUC1 promoter contains a PPARγ response element [58], we conducted a series of experiments to investigate the potential role of MUC1 in PPARγ-dependent regulation of H. pylori-driven gastric inflammation. Our results demonstrated that treatment of AGS cells with the PPARγ agonist, troglitazone, reduced H. pylori-stimulated IL-8 levels in cell culture supernatants, compared with cells treated with H. pylori alone [59]. Moreover, following knockdown of MUC1 expression by RNA interference, no differences in IL-8 levels were seen between cells treated with troglitazone plus H. pylori versus cells treated with H. pylori alone. Finally, PPARγ was shown to bind to the MUC1 gene promoter in AGS cells, and troglitazone treatment increased MUC1 promoter activity and augmented MUC1 protein levels compared with vehicle controls. These combined results suggested that PPARγ stimulated MUC1 expression by AGS cells, thereby attenuating H. pylori-induced IL-8 production and gastric inflammatory responses.
11. Summary
Chronic inflammation of the gastric mucosa is a serious public health problem worldwide. The principal association of gastritis is with stomach infection by H. pylori. Yet, the majority of individuals colonized by H. pylori fail to progress from low-grade gastric inflammation to peptic ulcer disease and adenocarcinoma. On the basis of the studies reviewed herein, a mechanistic hypothesis to explain why a subset of H. pylori-infected patients develops the more clinically serious stomach pathologies can now be proposed. Specifically, as summarized in Fig. 4, evidence is presented that details the multiple pathways through which MUC1 mucin counter-regulates gastric inflammatory responses. We hypothesize that a defect in the anti-inflammatory activity of MUC1 mucin results in a heightened, persistent state of H. pylori-driven gastritis that creates a self-perpetuating, auto-amplifying, proinflammatory environment and ultimately promoting carcinogenesis. We hope that this review article will serve as an impetus for future studies directed at addressing this possible mechanism.
Figure 4.

Schematic illustration depicting the possible molecular mechanisms of the anti-inflammatory activities of MUC1 in gastric epithelial cells during H. pylori infection. (A) Inhibition of NF-κB signaling: H. pylori binds to cell surface receptors (e.g. Toll-like receptors, TLRs) that activate NF-κB following phosphorylation of IκBα by IKK (step 1). NF-κB enters the nucleus to activate proinflammatory (IL-8) gene expression (step 2). MUC1 blocks the ability of IKK to phosphorylate IκBα (step 3), thus inhibiting NF-κB nuclear translocation (step 4) and blocking IL-8 production (step 5). (B) Inhibition of β-catenin signaling: H. pylori delivers CagA into the cytosol (step 1). CagA binds to E-cadherin (step 2), thereby releasing β-catenin that translocates into the nucleus to activate proinflammatory (IL-8) gene expression. MUC1 binds to β-catenin (step 3) released by CagA, thus inhibiting its nuclear translocation (step 4) and blocking IL-8 production (step 5). (C) PPARγ-dependent inhibition: Endogenous PPAR-γ ligands (PPAR-L) activate PPARγ (step 1). PPARγ enters the nucleus (step 2) to activate MUC1 gene expression (step 3). MUC1 protein inhibits the H. pylori-driven NF-κB and/or β-catenin pathways (step 4), thus blocking IL-8 production (step 5).
Acknowledgments
This work was supported by U.S. Public Health Service grants AI072291 and AI083463.
References
- 1.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311–5. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 2.Lindkvist P, Asrat D, Nilsson I, Tsega E, Olsson GL, Wretlind B, et al. Age at acquisition of Helicobacter pylori infection: comparison of a high and a low prevalence country. Scand J Infect Dis. 1996;28(2):181–4. doi: 10.3109/00365549609049072. [DOI] [PubMed] [Google Scholar]
- 3.Anderson WF, Camargo MC, Fraumeni JF, Jr, Correa P, Rosenberg PS, Rabkin CS. Age-specific trends in incidence of noncardia gastric cancer in US adults. JAMA. 2010;303(17):1723–8. doi: 10.1001/jama.2010.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: Insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133(1):288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988;48(13):3554–60. [PubMed] [Google Scholar]
- 6.McNamara D, El-Omar E. Helicobacter pylori infection and the pathogenesis of gastric cancer: a paradigm for host-bacterial interactions. Dig Liver Dis. 2008;40(7):504–9. doi: 10.1016/j.dld.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 7.MacFarlane AJ, Stover PJ. Convergence of genetic, nutritional and inflammatory factors in gastrointestinal cancers. Nutr Rev. 2007;65(12 Pt 2):S157–66. doi: 10.1111/j.1753-4887.2007.tb00355.x. [DOI] [PubMed] [Google Scholar]
- 8.Beachey EH. Bacterial adherence: Adhesin-receptor interactions mediating the attachment of bacteria to mucosal surface. J Infect Dis. 1981;143(3):325–45. doi: 10.1093/infdis/143.3.325. [DOI] [PubMed] [Google Scholar]
- 9.Marshall B. Helicobacter pylori: 20 years on. Clin Med. 2002;2(2):147–52. doi: 10.7861/clinmedicine.2-2-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindén SK, Wickstrom C, Lindell G, Gilshenan K, Carlstedt I. Four modes of adhesion are used during Helicobacter pylori binding to human mucins in the oral and gastric niches. Helicobacter. 2008;13(2):81–93. doi: 10.1111/j.1523-5378.2008.00587.x. [DOI] [PubMed] [Google Scholar]
- 11.Lindén S, Mahdavi J, Hedenbro J, Boren T, Carlstedt I. Effects of pH on Helicobacter pylori binding to human gastric mucins: identification of binding to non-MUC5AC mucins. Biochem J. 2004;384(Pt 2):263–70. doi: 10.1042/BJ20040402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindén SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, et al. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 2009;5(10):e1000617. doi: 10.1371/journal.ppat.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lillehoj EP, Kim KC. Airway mucus: Its components and function. Arch Pharm Res. 2002;25(6):770–80. doi: 10.1007/BF02976990. [DOI] [PubMed] [Google Scholar]
- 14.Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86(1):245–78. doi: 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- 15.Voynow JA, Gendler SJ, Rose MC. Regulation of mucin genes in chronic inflammatory airway diseases. Am J Respir Cell Mol Biol. 2006;34(6):661–5. doi: 10.1165/rcmb.2006-0035SF. [DOI] [PubMed] [Google Scholar]
- 16.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. 1990;265(25):15286–93. [PubMed] [Google Scholar]
- 17.Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA. Cloning and sequencing of a human pancreatic tumor mucin cDNA. J Biol Chem. 1990;265(25):15294–9. [PubMed] [Google Scholar]
- 18.Parry S, Silverman HS, McDermott K, Willis A, Hollingsworth MA, Harris A. Identification of MUC1 protolytic cleavage sites in vivo. Biochem Biophys Res Commun. 2001;283(3):715–20. doi: 10.1006/bbrc.2001.4775. [DOI] [PubMed] [Google Scholar]
- 19.Lillehoj EP, Han F, Kim KC. Mutagenesis of a Gly-Ser cleavage site in MUC1 inhibits ectodomain shedding. Biochem Biophys Res Comm. 2003;307(3):743–9. doi: 10.1016/s0006-291x(03)01260-9. [DOI] [PubMed] [Google Scholar]
- 20.Zrihan-Licht S, Baruch A, Elroy-Stein O, Keydar I, Wreschner DH. Tyrosine phosphorylation of the MUC1 breast cancer membrane protein. Cytokine receptor-like molecule. FEBS Lett. 1994;356(1):130–6. doi: 10.1016/0014-5793(94)01251-2. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Lillehoj EP, Kim KC. Identification of four sites of stimulated tyrosine phosphorylation in the MUC1 cytoplasmic tail. Biochem Biophys Res Comm. 2003;310(2):341–6. doi: 10.1016/j.bbrc.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Ren J, Kufe D. Interaction of human MUC1 and β-catenin is regulated by Lck and ZAP-70 in activated Jurkat T cells. Biochem Biophys Res Commun. 2004;315(2):471–6. doi: 10.1016/j.bbrc.2004.01.075. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Kufe D. The human DF3/MUC1 carcinoma-associated antigen signals nuclear localization of the catenin p120ctn. Biochem Biophys Res Commun. 2001;281(2):440–3. doi: 10.1006/bbrc.2001.4383. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction of glycogen synthase kinase 3β with the DF3/MUC1 carcinoma-associated antigen and β-catenin. Mol Cell Biol. 1998;18(12):7216–24. doi: 10.1128/mcb.18.12.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. J Biol Chem. 2001;276(9):6061–4. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Ren J, Yu WH, Li Q, Kuwahara H, Yin L, et al. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and β-catenin. J Biol Chem. 2001;276(38):35239–42. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Chen W, Ren J, Yu WH, Li Q, Yoshida K, et al. DF3/MUC1 signaling in multiple myeloma cells is regulated by interleukin-7. Cancer Biol Ther. 2003;2(2):187–93. doi: 10.4161/cbt.2.2.282. [DOI] [PubMed] [Google Scholar]
- 28.Lillehoj EP, Kim H, Chun E, Kim KC. Pseudomonas aeruginosa stimulates phosphorylation of the airway epithelial membrane glycoprotein Muc1 and activates MAP kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):809–15. doi: 10.1152/ajplung.00385.2003. [DOI] [PubMed] [Google Scholar]
- 29.Quin RJ, McGuckin MA. Phosphorylation of the cytoplasmic domain of the MUC1 mucin correlates with changes in cell-cell adhesion. Int J Cancer. 2000;87(4):499–506. doi: 10.1002/1097-0215(20000815)87:4<499::aid-ijc6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 30.Singh PK, Wen Y, Swanson BJ, Shanmugam K, Kazlauskas A, Cerny RL, et al. Platelet-derived growth factor receptor β-mediated phosphorylation of MUC1 enhances invasiveness in pancreatic adenocarcinoma cells. Cancer Res. 2007;67(11):5201–10. doi: 10.1158/0008-5472.CAN-06-4647. [DOI] [PubMed] [Google Scholar]
- 31.Wykes M, MacDonald KPA, Tran M, Quin RJ, Xing PX, Gendler SJ, et al. MUC1 epithelial mucin (CD227) is expressed by activated dendritic cells. J Leukoc Biol. 2002;72(4):692–701. [PubMed] [Google Scholar]
- 32.Yamamoto M, Bharti A, Li Y, Kufe D. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and β-catenin in cell adhesion. J Biol Chem. 1997;272(19):12492–4. doi: 10.1074/jbc.272.19.12492. [DOI] [PubMed] [Google Scholar]
- 33.Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic MUC1 interacts with EGFR and correlates with MAP kinase activation in mouse mammary gland. J Biol Chem. 2001;276(16):13057–64. doi: 10.1074/jbc.M011248200. [DOI] [PubMed] [Google Scholar]
- 34.Meerzaman D, Shapiro PS, Kim KC. Involvement of the MAP kinase ERK2 in MUC1 mucin signaling. Am J Physiol Lung Cell Mol Physiol. 2001;281(1):86–91. doi: 10.1152/ajplung.2001.281.1.L86. [DOI] [PubMed] [Google Scholar]
- 35.Schroeder JA, Adriance MC, Thompson MC, Camenisch TD, Gendler SJ. MUC1 alters β-catenin-dependent tumor formation and promotes cellular invasion. Oncogene. 2003;22(9):1324–32. doi: 10.1038/sj.onc.1206291. [DOI] [PubMed] [Google Scholar]
- 36.Molock KE, Lillehoj EP. Biochemical interactions among intercellular adhesion molecules expressed by airway epithelial cells. Biochem Biophys Res Commun. 2006;343(2):513–19. doi: 10.1016/j.bbrc.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Oshima H, Oshima M. Mouse models of gastric tumors: Wnt activation and PGE2 induction. Pathol Int. 2010;60(9):599–607. doi: 10.1111/j.1440-1827.2010.02567.x. [DOI] [PubMed] [Google Scholar]
- 38.Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol. 2011;192(6):907–17. doi: 10.1083/jcb.201009141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2010;90(4):417–27. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TG, Lillehoj EP. MUC1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J Biol Chem. 2010;285(27):20547–57. doi: 10.1074/jbc.M110.121319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuji S, Kawai N, Tsujii M, Kawano S, Hori M. Review article: Inflammation-related promotion of gastrointestinal carcinogenesis--a perigenetic pathway. Aliment Pharmacol Ther. 2003;18(Suppl 1):82–9. doi: 10.1046/j.1365-2036.18.s1.22.x. [DOI] [PubMed] [Google Scholar]
- 42.Suganuma M, Yamaguchi K, Ono Y, Matsumoto H, Hayashi T, Ogawa T, et al. TNF-α-inducing protein, a carcinogenic factor secreted from H. pylori, enters gastric cancer cells. Int J Cancer. 2008;123(1):117–22. doi: 10.1002/ijc.23484. [DOI] [PubMed] [Google Scholar]
- 43.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105(3):1003–8. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hatakeyama M, Higashi H. Helicobacter pylori CagA: A new paradigm for bacterial carcinogenesis. Cancer Sci. 2005;96(12):835–43. doi: 10.1111/j.1349-7006.2005.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–90. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008;10(8):1573–81. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- 47.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4(9):688–94. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 48.Keates S, Keates AC, Nath S, Peek RM, Jr, Kelly CP. Transactivation of the epidermal growth factor receptor by cag+ Helicobacter pylori induces upregulation of the early growth response gene Egr-1 in gastric epithelial cells. Gut. 2005;54(10):1363–9. doi: 10.1136/gut.2005.066977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102(30):10646–51. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bebb JR, Leach L, Zaitoun A, Hand N, Letley DP, Thomas R, et al. Effects of Helicobacter pylori on the cadherin-catenin complex. J Clin Pathol. 2006;59(12):1261–6. doi: 10.1136/jcp.2006.036772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–26. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 52.Lévy L, Neuveut C, Renard CA, Charneau P, Branchereau S, Gauthier F, et al. Transcriptional activation of interleukin-8 by β-catenin-Tcf4. J Biol Chem. 2002;277(44):42386–93. doi: 10.1074/jbc.M207418200. [DOI] [PubMed] [Google Scholar]
- 53.Guang W, Twaddell WS, Lillehoj EP. Molecular interactions between MUC1 epithelial mucin, β-catenin, and CagA proteins. Front Immunol. 2012;3:105. doi: 10.3389/fimmu.2012.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slomiany BL, Slomiany A. Suppression of gastric mucosal inflammatory responses to Helicobacter pylori lipopolysaccharide by peroxisome proliferator-activated receptor γ activation. IUBMB Life. 2002;53(6):303–8. doi: 10.1080/15216540213459. [DOI] [PubMed] [Google Scholar]
- 55.Tsujimoto H, Ono S, Ichikura T, Matsumoto Y, Yamamoto J, Hase K. Roles of inflammatory cytokines in the progression of gastric cancer: Friends or foes? Gastric Cancer. 2010;13(4):212–21. doi: 10.1007/s10120-010-0568-x. [DOI] [PubMed] [Google Scholar]
- 56.Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: Energy in the balance. Circ Res. 2011;108(8):1002–16. doi: 10.1161/CIRCRESAHA.110.226860. [DOI] [PubMed] [Google Scholar]
- 57.Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C. Peroxisome proliferator-activated receptors: Regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol. 2001;85(2–5):267–273. doi: 10.1016/s0960-0760(03)00214-0. [DOI] [PubMed] [Google Scholar]
- 58.Shalom-Barak T, Nicholas JM, Wang Y, Zhang X, Ong ES, et al. Peroxisome proliferator-activated receptor γ controls Muc1 transcription in trophoblasts. Mol Cell Biol. 2004;24(24):10661–9. doi: 10.1128/MCB.24.24.10661-10669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park YS, Guang W, Blanchard TG, Kim KC, Lillehoj EP. Suppression of IL-8 production in gastric epithelial cells by MUC1 mucin and peroxisome proliferator-associated receptor γ. Am J Physiol Gastrointest Liver Physiol. 2012 doi: 10.1152/ajpgi.00023.2012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]