

Abstract
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder in elderly people with an impairment of cognitive decline and memory loss. β-amyloid (Aβ) as a potent neurotoxic peptide has a pivotal role in the pathogenesis of AD. This disease begins with impairment in synaptic functions before developing into later neuro¬degeneration and neuronal loss. The aim of this study was to evaluate the synaptic plasticity and electrophysiological function of granule cells in hippocampal dentate gyrus (DG) after intracerebroventricular (i.c.v.) administration of aggregated Aβ (1-42) peptide in vivo.
Methods
Animals were divided to control and Aβ (1-42) groups. Long-term potentia¬tion (LTP) in perforant path-DG synapses was assessed in order to investigate the effect of aggregated Aβ (1-42) on synaptic plasticity. Field excitatory post-synaptic potential (fEPSP) slope and population spike (PS) amplitude were measured.
Results
Administration of Aβ (1-42) significantly decreased fEPSP slope and PS amplitude in Aβ (1-42) group comparing with the control group and had no effect on baseline activity of neurons.
Conclusion
The present study indicates that administration of aggregated form of Aβ (1-42) into the lateral ventricle effectively inhibits LTP in granular cells of the DG in hippocampus in vivo.
Keywords: β-Amyloid, Synaptic Plasticity, Dentate Gyrus, Long-Term Potentiation
Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder among elderly people that is accompanied with a progressive cognitive decline and memory loss. The major hallmarks of this disease include neuronal degeneration and synaptic loss with presence of extracellular β-amyloid (Aβ) deposits and intracellular neurofibrillary tangles in different areas of cerebral cortex (Armstrong 1994). Aβ is a potent neurotoxic peptide and a major constituent of the senile plaques that initiates a cascade that leads to neural dysfunction and memory impairment. This peptide is a product of β-amyloid precursor protein (APP) cleavage and is found in the brain in two main forms with 40 and 42 amino acid peptides: Aβ (1-40) and Aβ (1-42), respectively (Drachman 2006, Jolas et al 2002).
The majority of investigations suggested that impairment in memory in AD begins with changes in hippocampal synaptic functions and then gradually progresses to neurodegeneration and neuronal loss in these patients (Selkoe 2002). Long-term potentiation (LTP) in the hippocampus is the most important form of synaptic plasticity that is considered as a cellular basis of learning and memory and provides an attractive means of detecting any changes in synaptic functions (Yun et al 2006). Previous studies have shown that intracerebroventricular (i.c.v.) administration of aggregated Aβ makes changes in LTP in the hippocampus and consequently leads to the cognitive dysfunction and impairment of spatial and non-spatial forms of learning and memory in rodents (Selkoe 2008, Trubetskaya et al 2003).
Interestingly, the effects of Aβ on LTP differ in various parts of the brain and there are conflicting results from previous studies about the effect of Aβ on synaptic plasticity and neuronal excitability. For example, result of a research showed that Aβ increases the N-methyl-D-aspartate (NMDA)-receptor mediated synaptic transmission and enhances LTP in rat hippocampus in vitro (Wu et al 1995a, Wu et al 1995b), and in an extracellular single unit recording administration of Aβ increased NMDA responses in the hippocampus (Molnár et al 2004), while in other studies application of Aβ decreased neuronal excitability (Freir et al 2001, Yun et al 2006).
Previous reports also have demonstrated that the granule cell layer of dentate gyrus (DG), a sub-region of hippocampus, is involved in spatial learning and memory (Drapeau et al 2003, Kee et al 2007). Results of our previous study have shown that administration of exogenous aged Aβ (1-42) peptide in right lateral ventricle significantly impaired the spatial learning and memory in rats (unpublished data). We therefore examined the synaptic plasticity and electrophysiological function of granule cells in hippocampal dentate gyrus after i.c.v. injection of aggregated form of Aβ (1-42) peptide in vivo. For this purpose, we measured the fEPSP and PS as two components of the evoked field potential in the hippocampal DG in rats.
Materials and methods
Animals and treatments
Adult male Wistar rats weighing 300–350 g were obtained from Pasteur Institute of Iran. They were maintained at an ambient temperature of 22–24°C under a 12 hr light-dark cycle, with lights off at 7:00 pm. Food and water provided ad libitum.
Animals were randomly divided into two control and Aβ (1-42) groups (n=7 in each group). The Aβ (1-42) peptide fragment was purchased from Bachem (Switzerland). Congo red and poly l-lysine solution were obtained from Sigma (St. Louis, USA). Aβ (1-42) was dissolved in sterile double distilled water at a concentration of 2.25 mg/ml according to the manufacturer’s instructions and 2 nmol in 4 μl was administered intracerebroventricularly (i.c.v.), as previously described (Dall’Igna et al 2007). To obtain the aggregated form, the peptide solution was placed in an incubator at 37 °C for 72 h.
Surgical procedures
The rats were anesthetized intraperitoneally with chloral hydrate (350 mg/kg) and placed in a Stoelting stereotaxic instrument (Stoelting Co., Illinois, USA). The scalp was incised and retracted and small hole was drilled at an appropriate location in the skull to allow the insertion of an injection cannula. Aβ or sterile double distilled water was then injected through a stainless steel cannula into the right lateral ventricle (AP: −0.8, ML: 1.6 and DV: 3.5 mm below dura) during 5 min by a Hamilton micro syringe and the needle was left in place for 5 min before it was slowly withdrawn. Coordinates were chosen based on Paxinos and Watson rat brain atlas (Paxinos and Watson, 2007).
Electrophysiological study
Two weeks after i.c.v. injection of Aβ or vehicle, rats were anesthetized with urethane (1.8 g/kg, i.p.) and their heads were fixed in a stereotaxic head-holder. A heating pad was used to maintain body temperature at 36.5±0.5°C. The skull was exposed and two small holes were drilled at the positions of stimulating and recording electrodes. The exposed cortex was kept moist by the application of paraffin oil. A concentric bipolar stimulating electrode (stainless steel, 0.125 mm diameter, Advent, UK) was placed in the perforant pathway (AP=−8.1 mm; ML=4.3 mm; DV=3.2 mm), and a stainless steel recording electrode was lowered into DG until the maximal response was observed (AP=−3.8 mm; ML=2.3 mm; DV=2.7–3.2 mm) (Paxinos and Watson 2007). In order to minimize trauma to brain tissue, the electrodes were lowered very slowly (0.2 mm/min). Extracellular evoked responses were obtained from the dentate granule cell population following stimulation of the perforant pathway. Extracellular field potentials were amplified (100×) and filtered (1 Hz to 3 kHz band pass) using a DAM80 differential amplifier (WPI, USA). Signals were passed through an analogue-to-digital interface (Powerlab/4SP, AD Instruments, Australia) to a computer, and data were analyzed using custom software. Stimulation intensity was adjusted to evoke about 40% of the maximal response of the population spike (PS) and field excitatory post-synaptic potential (fEPSP). The PS amplitude was measured as the difference in voltage between the peak of first positive wave and the peak of first negative deflection, and the fEPSP slope was measured as the maximum slope between the initial point of fEPSP and the first positive wave in order to measure synaptic efficacy (Reisi et al 2010). Stimulus–response or input/output (I/O) functions were acquired by systematic variation of the stimulus current (100–1000 A) in order to evaluate synaptic potency before induction of LTP. PS and fEPSP were evoked in the DG region using 0.1 Hz stimulation. Baseline recordings were taken at least 30 min prior to each experiment.
After ensuring a steady state baseline response, LTP was induced using high-frequency stimuli protocols of 400 Hz (10 bursts of 20 stimuli, 0.2 ms stimulus duration, 10 s interburst interval) at a stimulus intensity that evoked a PS amplitude and fEPSP of approximately 80% of the maximum response. All potentials employed as baseline before and after high frequency stimulation (HFS) were evoked at a stimulus intensity which produced 40% of this maximum (Reisi et al 2010). fEPSP slope and PS were recorded for the periods of 5, 15, 30, 45 and 60 min after HFS in order to determine any changes in the synaptic response of DG neurons. For each time-point, 5 consecutive evoked responses were averaged at 10 s stimulus interval.
Histology
On completion of electrophysiological study, rats were perfused transcardially with a cold 10% formalin solution and the brain was removed and post-fixed with the same formalin solution for 24 hr at room temperature and embedded in paraffin blocks. Subsequently, coronal sections with the thickness of 5 µm through the brain were cut using a microtome and mounted on poly-L-lysine–coated glass slides. The sections were used for evaluating the injection site and Congo red staining for detection of Aβ deposits in the hippocampus.
Aβ deposits
Modified high pH Congo red staining protocol for Amyloid was used for the detection of Aβ deposit in the brain slices. Briefly, Coronal sections were deparaffinized and hydrated in water (5 min), stained in Congo red solution (0.3% in 80% ethanol) for 10 min, rinsed in distilled water, quickly differentiated in alkaline alcohol solution, rinsed in tap water, counterstained with Gill’s hematoxylin for 30 seconds, rinsed in tap water, dipped in ammonia water for 30 seconds or until sections turned blue, rinsed in tap water, dehydrated through 95% and 100% alcohol, cleared in xylene and covered with balsam and a covers lip. In this staining method the amyloid deposits and the nuclei were stained red and blue, respectively.
Statistical analysis
Two-tailed Student’s t-test and two-way ANOVA with repeated measures were used for statistical comparison. A p-value of less than 0.05 was considered to be statistically significant. Data are expressed, as means±S.E.M for each group.
Results
Statistical analysis of data from time points before HFS did not show any difference in baseline fEPSP slope and PS amplitude between control and Aβ (1-42) groups. Therefore, i.c.v. administration of aggregated Aβ (1-42) had no effect on baseline activity of neurons. A two-way repeated measures ANOVA indicated that the fEPSP slope after HFS was significantly decreased in the Aβ (1-42) group with respect to the control group (F(1,12)=18.34 ; p<0.01) and this difference between two groups was significant in all time points (p<0.01; n=7) (Fig.1. A).
Fig. 1.

The effect of aggregated Aβ on the synaptic response of DG granule cells following a 400 Hz HFS applied to the perforant pathway. (A) The Aβ did not affect the baseline fEPSP slope, but reduced the amplification of the fEPSP slope after 400 Hz HFS in comparison with the control group (p<0.01). (B) Aβ did not affect the baseline PS amplitude. However, after the 400 Hz HFS, Aβ reduced PS amplitude comparing with the control group (p< 0.0001). The data are shown as the mean±S.E.M.
Statistical analysis carried out with a two-way repeated measures ANOVA between the control and Aβ (1-42) groups revealed that Aβ (1-42) causes a significant decrease in the PS amplitude after HFS (F (1,12)=32.99; p<0.0001), and this difference was significant between two groups during 60 min after HFS (p=0.001 for 5th, and 15th min and p<0.0001 for 30th, 45th and 60thmin) (Fig.1. B). Sample traces for each group are illustrated in Fig. 2.
Fig. 2.
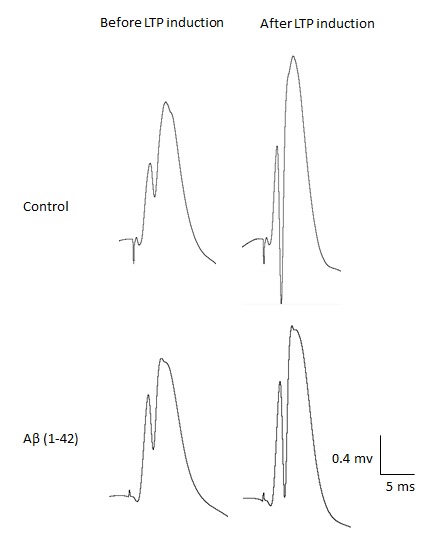
Single traces recorded before and after induction of LTP in dentate gyrus granule cells of hippocampus.
Congo red staining of slices showed Aβ deposits between DG granule cells following i.c.v. injection of 2 nmol/4µl of aggregated Aβ (1-42)that were absent in control group (Fig. 3).
Fig. 3.
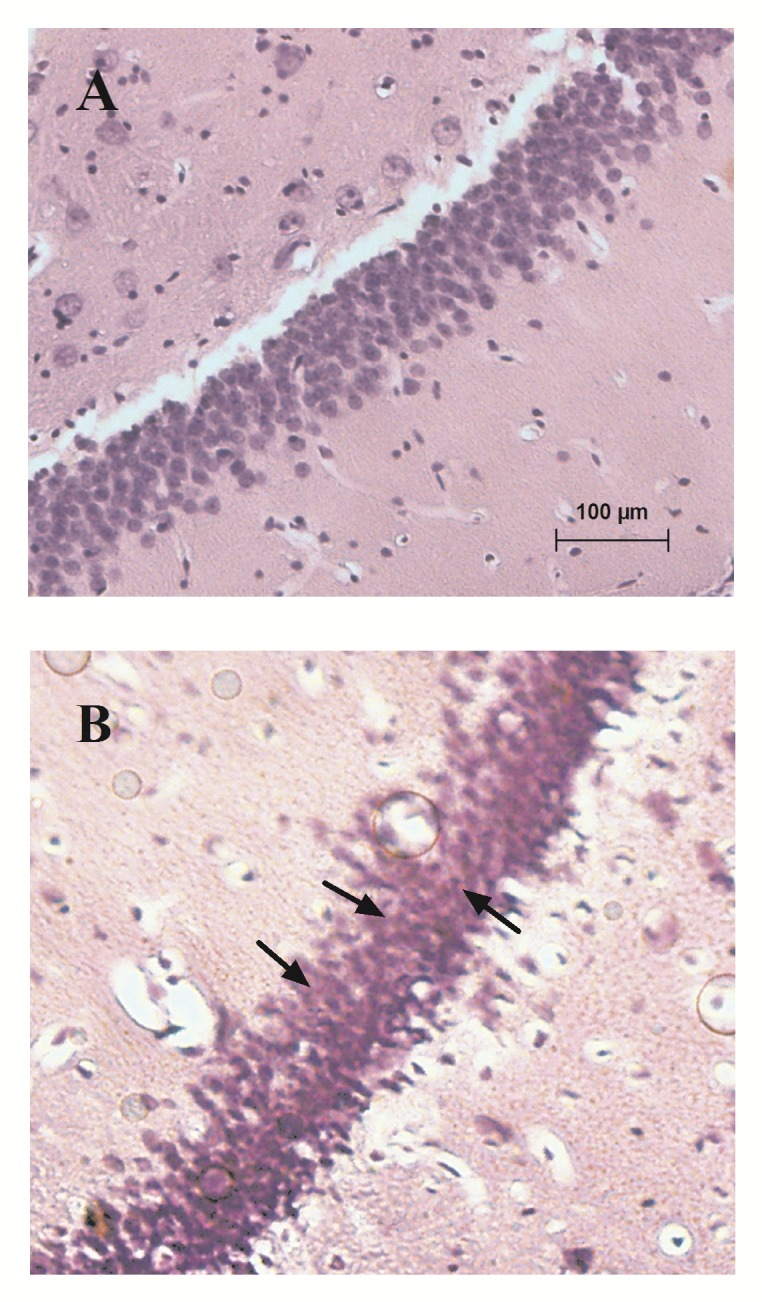
Representative Aβ deposits in the hippocampal DG granule cells: (A) control and (B) Aβ (1-42). Congo red staining of slices showed Aβ deposits (red spots indicated by arrows) between DG granule cells in Aβ (1-42) group.
Discussion
The main finding of this study was that a single unilateral microinjection of nanomolar dose of aggregated form of Aβ (1-42) into the lateral ventricle significantly inhibited HFS-induced LTP in granular cells of the DG in hippocampus in vivo. Application of this peptide had no effect on baseline activity of granule cell layer’s neurons but significantly decreased evoked field potential and this conclusion was supported by decreased fEPSP-LTP and PS-LTP in these neurons.
Our results were consistent with the previous studies which demonstrated that administration of Aβ reduces neuronal synaptic plasticity and LTP in the CA1 area of hippocampus (Freir et al 2001, Jing et al 2009). In addition, in vitro studies have shown that application of soluble form of Aβ (1-42) inhibited LTP in DG of hippocampal slices in Sprague-Dawley rats (Wang et al 2002), and in another research, a significant decrease in action potential generation was found with Aβ (1-42) in whole cell recording in hippocampal DG (Yun et al 2006).
The NMDA receptor-dependent LTP is the widely used form of synaptic plasticity that is generated by presynaptic release of glutamate and then depolarization of postsynaptic neuron that causes calcium influx by NMDA receptors. Elevation of cytoplasmic Ca 2+ concentration results in an increased number of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) type of glutamate receptor (Bredt and Nicoll 2003). Reduction of glutamate receptor is one of the main synaptotoxic effects of Aβ. Previous studies have shown that Aβ can reduce the expression of NMDA receptors and administration of this peptide reduces the NMDA and AMPA receptor-mediated synaptic transmission by increasing endocytosis of these receptors (Snyder et al 2005, Hsieh et al 2006). On the other hand, activity-dependent autophosphorylation/activation of Ca 2+ and calmodulin-dependent protein kinase ІІ (CaMKІІ) and phosphorylation of GluR1 subunits of AMPA receptors at a CaMKІІ-dependent site were demonstrated along with generation of LTP in DG of hippocampal slices (Lisman et al 2002). Meanwhile, results of previous researches show that application of Aβ (1-42) inhibits NMDA receptor-mediated synaptic transmission resulting in reduced Ca 2+ influx through the NMDA receptors and subsequently reduces the phosphorylation of CaMKІІ (Zhao et al 2004). According to these findings, in the present study, the attenuating effects of Aβ (1-42) on LTP in the granular cells of DG may be partly due to the elimination of glutamate receptors and reduced phosphorylation of CaMKІІ by this peptide.
Nevertheless, in contrast to our findings, results of some studies have demonstrated the Aβ-induced increase in synaptic currents and LTP. In a similar in vivo study, i.c.v. administration of aggregated Aβ significantly increased LTP in the CA1 area of hippocampus (Trubetskaya et al 2003), and also in another in vitro research, Aβ at a low concentration enhanced LTP in hippocampal slices (Wu et al 1995b). These studies suggested that this increasing effect of Aβ on LTP might be related to augmentation of NMDA receptor-mediated synaptic currents and increase in intracellular Ca 2+ concentration due to Aβ-induced oxidative stress.
Nicotinic acetylcholine receptors (nAChRs) in the brain are involved in learning and memory processes. Strong evidence has shown that Aβ peptides could produce cholinergic dysfunction and subsequently cognitive deficits in patients with AD (Tran et al 2002, Nordberg 2001). The α-7 nicotinic acetylcholine receptors (α7nAChRs) and α4,β2 nicotinic acetylcholine receptors (α4β2nAChRs) are two most abundant forms of nAChRs in the brain (Wu et al 2008). It is indicated that Aβ binds to α7nAChRs with high affinity and a close relationship is found between Aβ and α7nAChRs (Wang et al 2000). And it is shown that both of Aβ and α7nAChRs are present in amyloid plaques conformation in patients with AD (Dziewczapolski et al 2009). Previous studies have suggested that attachment of Aβ to α7nAChRs can initiate endocytosis of the complex of Aβ/α7nAChRs and consequently result in accumulation of intracellular Aβ and impairment of synaptic function (DAndrea and Nagele 2006). It is also proposed that α7nAChRs have a high permeability to Ca 2+ and activation of these receptors by Aβ results in Ca 2+ influx and overload of intracellular Ca 2+ that is involved in Aβ-induced synaptic dysfunction (Brzyska and Elbaum 2003).
Moreover, presynaptic α7nAChRs, which are essential for learning and memory, are also decreased in AD. Aβ binds to these receptors and decreases the release of acetylcholine (ACh) as an excitatory neurotransmitter and impairs the long-term potentiation (Parri et al 2011, Walsh et al 2002).
Results of a study performed on hippocampal CA1 area in rat demonstrated that the α4β2nAChRs are required for suppressing action of Aβ on LTP. They suggested that Aβ may bind to α4β2nAChRs and activate these receptors on GABAergic inhibitory interneurons of CA1 region of hippocampus and consequently increase the inhibitory afferent to pyramidal cells that finally lead to reduced LTP in these neurons (Wu et al 2008). According to these findings, the impairing effects of aggregated Aβ (1-42) on LTP in our study might be partly due to such changes in nAChRs function in the synapses of granular cells of DG.
Conclusion
The present study indicates that administration of a single unilateral microinjection of aggregated form of Aβ (1-42) into the lateral ventricle effectively inhibits HFS-induced LTP in the granular cells of DG in hippocampus in vivo, presumably due to Aβ-induced elimination of glutamate receptors and reduced phosphorylation of CaMKІІ and also alteration in nAChRs function in the synapses of granular cells of DG.
Ethical issues
All stages of the present study were performed using protocols approved by the Research and Ethics Committee of Tabriz University of Medical Sciences and were conducted under the recommended conditions of Guide for the Care and Use of Laboratory Animals of National Institute of Health.
Competing interests
The authors declare no competing interests.
Acknowledgments
This research was supported by Drug Applied Research Center at Tabriz University of Medical Sciences. Also, the authors would like to thank Mr. Ali-Akbar Salari for his thorough review and useful comments on the manuscript and Dr. Hanieh Samadi for their helpful assistance in the laboratory.
References
- Armstrong RA. 1994 [beta]-Amyloid (A [beta]) deposition in elderly non-demented patients and patients with Alzheimer’s disease. Neuroscience letters, 178(1), 59-62 [DOI] [PubMed] [Google Scholar]
- Bredt DS and Nicoll RA. 2003 AMPA receptor trafficking at excitatory synapses. Neuron, 40(2), 361-379 [DOI] [PubMed] [Google Scholar]
- Brzyska M and Elbaum D. 2003 Dysregulation of calcium in Alzheimer’s disease. Acta neurobiologiae experimentalis, 63(3), 171-184 [DOI] [PubMed] [Google Scholar]
- Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA and Lara DR. 2007 Caffeine and adenosine A2a receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Experimental neurology, 203(1), 241-245 [DOI] [PubMed] [Google Scholar]
- Dandrea MR and Nagele RG. 2006 Targeting the Alpha 7 Nicotinic Acetylcholine Receptor to Reduce Amyloid Accumulation in Alzheimers Disease Pyramidal Neurons. Current pharmaceutical design, 12(6), 677-684 [DOI] [PubMed] [Google Scholar]
- Drachman DA. 2006 Aging of the brain, entropy, and Alzheimer disease. Neurology, 67(8), 1340-1352 [DOI] [PubMed] [Google Scholar]
- Drapeau E, Mayo W, Aurousseau C, Le Moal M, Piazza PV and Abrous DN. 2003 Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proceedings of the National Academy of Sciences, 100(24), 14385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E and Heinemann SF. 2009 Deletion of the α7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer’s disease. The Journal of Neuroscience, 29(27), 8805-8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Holscher C and Herron CE. 2001 Blockade of long-term potentiation by β-amyloid peptides in the CA1 region of the rat hippocampus in vivo. Journal of neurophysiology, 85(2), 708-713 [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. 2006 AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron, 52(5), 831-843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing W, Guo F, Cheng L, Zhang JF and Qi JS. 2009 Arginine vasopressin prevents amyloid [beta] protein-induced impairment of long-term potentiation in rat hippocampus in vivo. Neuroscience letters, 450(3), 306-310 [DOI] [PubMed] [Google Scholar]
- Jolas T, Zhang XS, Zhang Q, Wong G, Vecchio RD, Gold L, et al. 2002 Long-term potentiation is increased in the CA1 area of the hippocampus of APPswe/ind CRND8 mice. Neurobiology of disease, 11(3), 394-409 [DOI] [PubMed] [Google Scholar]
- Kee N, Teixeira CM, Wang AH and Frankl . 2007 Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nature neuroscience, 10(3), 355-362 [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H and Cline H. 2002 The molecular basis of CaMKII function in synaptic and behavioural memory. Nature Reviews Neuroscience, 3(3), 175-190 [DOI] [PubMed] [Google Scholar]
- Molnár Z, Soós K, Lengyel I, Penke B, Szegedi V and Budai D. 2004 Enhancement of NMDA responses by [beta]-amyloid peptides in the hippocampus in vivo. Neuroreport, 15(10), 1649 [DOI] [PubMed] [Google Scholar]
- Nordberg A. 2001 Nicotinic receptor abnormalities of Alzheimer’s disease: therapeutic implications. Biological psychiatry, 49(3), 200-210 [DOI] [PubMed] [Google Scholar]
- Parri HR, Hernandez CM and Dineley KT. 2011. Research update: Alpha7 nicotinic acetylcholine receptor mechanisms in Alzheimer’s disease. Biochemical pharmacology. [DOI] [PubMed]
- Paxinos G and Watson C. 2007. The Rat Brain in Stereotaxic Coordinates: Hard Cover Edition. Academic press.
- Reisi P, Babri S, Alaei H, Sharifi MR, Mohaddes G, Noorbakhsh SM, et al. 2010 Treadmill running improves long-term potentiation (LTP) defects in streptozotocin-induced diabetes at dentate gyrus in rats. Pathophysiology, 17(1), 33-38 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 2002 Alzheimer’s disease is a synaptic failure. Science, 298(5594), 789-791 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. 2008 Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behavioural brain research, 192(1), 106-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. 2005 Regulation of NMDA receptor trafficking by amyloid-β. Nature neuroscience, 8(8), 1051-1058 [DOI] [PubMed] [Google Scholar]
- Tran MH, Yamada K and Nabeshima T. 2002 Amyloid β-peptide induces cholinergic dysfunction and cognitive deficits: a minireview. Peptides, 23(7), 1271-1283 [DOI] [PubMed] [Google Scholar]
- Trubetskaya V, Stepanichev MY, Onufriev M, Lazareva N, Markevich V and Gulyaeva N. 2003 Administration of aggregated beta-amyloid peptide (25–35) induces changes in long-term potentiation in the hippocampus in vivo. Neuroscience and Behavioral Physiology, 33(2), 95-98 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. 2002 Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 416(6880), 535-539 [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. 2002 Soluble oligomers of [beta] amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain research, 924(2), 133-140 [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DHS, Davis CB and Shank RP. 2000 Amyloid Peptide Aβ1‐42 Binds Selectively and with Picomolar Affinity to α7 Nicotinic Acetylcholine Receptors. Journal of neurochemistry, 75(3), 1155-1161 [DOI] [PubMed] [Google Scholar]
- Wu J, Anwyl R and Rowan MJ. 1995. a beta-Amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport, 6(17), 2409 [DOI] [PubMed] [Google Scholar]
- Wu J, Anwyl R and Rowan MJ. 1995. b [beta]-amyloid-(1-40) increases long-term potentiation in rat hippocampus in vitro. European journal of pharmacology, 284(3), R1-R3 [DOI] [PubMed] [Google Scholar]
- Wu M, HE Y, Guo F and Qi J. 2008 [alpha] 4 [beta] 2 nicotinic acetylcholine receptors are required for the amyloid [beta] protein-induced suppression of long-term potentiation in rat hippocampal CA1 region in vivo. Brain research bulletin, 77(2-3), 84-90 [DOI] [PubMed] [Google Scholar]
- Yun SH, Gamkrelidze G, Stine WB, Sullivan PM, Pasternak JF, Ladu MJ, et al. 2006 Amyloid-beta1–42 reduces neuronal excitability in mouse dentate gyrus. Neuroscience letters, 403(1), 162-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Watson JB and Xie CW. 2004 Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. Journal of neurophysiology, 92(5), 2853-2858 [DOI] [PubMed] [Google Scholar]