

Abstract
Introduction
Today, with the tremendous potential of genomics and other recent advances in science, the role of science to improve reliable DNA extraction methods is more relevant than ever before. The ideal process for genomic DNA extraction demands high quantities of pure, integral and intact genomic DNA (gDNA) from the sample with minimal co-extraction of inhibitors of downstream processes. Here, we report the development of a very rapid, less-hazardous, and high throughput protocol for extracting of high quality DNA from blood samples.
Methods
Dried, clotted and ethylene diamine tetra-acetic acid (EDTA) treated fresh and frozen blood samples were extracted using this method in which the quality and integrity of the extracted DNA were corroborated by agarose gel electrophoresis, PCR reaction and DNA digestion using restricted enzyme. The UV spectrophotometric and gel electrophoresis analysis resulted in high A260/A280 ratio (>1.8) with high intactness of DNA.
Results
PCR and DNA digestion experiments indicated that the final solutions of extracted DNA contained no inhibitory substances, which confirms that the isolated DNA is of good quality.
Conclusion
The high quality and quantity of current method, no enzymatic processing and accordingly its low cost, make it appropriate for DNA extraction not only from human but also from animal blood samples in any molecular biology labs.
Keywords: Genomic DNA extraction, Blood sample, Clotted blood
Introduction
Blood samples are remarkably valuable to the research; forensic genetic laboratories have to take genetic profiles from blood that can be deposited on a wide range of supports. Likewise, genetic, hematology, and biochemistry studies employed in human clinic, demand blood samples in order to recognize and prevent human serious disorders (Phillips et al. 2000; Wang et al. 2003).
Pure genomic DNA extraction from biological samples is the vital primary step to succeed in various molecular biological techniques such as Polymerase Chain Reaction (PCR), restriction enzyme analysis, mutation detection, genotyping as well as linkage analysis (Phillips et al. 2000; Wang et al. 2003). Moreover, DNA extraction from blood samples is the most important requirement for the determination of genetic abnormalities, epigenetic studies and various diagnostic and preventive tests (Angelini et al. 2000; Lewis et al. 2005; Phillips et al. 2000; Wang et al. 2003).
DNA extraction demands a rapid and economical procedure with minimal co-extraction of inhibitors of downstream processes. Furthermore, it needs to be flexible enough to apply to frozen and clotted samples. Moreover, it should recover high amounts of pure and integral gDNA. To reach such aims, several DNA extracting methods from blood samples have been developed and commercialized (Angelini et al. 2000; Elgort et al. 2004; Lahiri et al. 1992; Lahiri and Nurnberger Jr 1991; Pachot et al. 2007; Planelles et al. 1996). Unfortunately, there is no general DNA extraction protocol to meet all these criteria (Clements et al. 2008; Nasiri et al. 2005). In one hand for instance, some protocols such as Phenol–Chloroform (PC) deem to be more efficient to remove PCR inhibitors in comparison with Chelex (the salting out procedure). On the other hand, the simple and rapid Chelex procedure could extract considerable amount of DNA which cannot be recovered by PC method (Castella et al. 2006; Cattaneo et al. 1997). Most of conventionally used protocols for genomic DNA isolation necessitate an overnight incubation with proteinase K enzyme for cell lysing and protein digest that can lead to some nuclease degradation in 37 ˚C. Besides, RNA contamination can routinely be removed by using of RNAase A enzyme, which makes these procedures time and cost-consuming (Nasiri et al. 2005).
In recent years some innovative methods have been introduced using chromatography columns or absorbing DNA on silica and coated magnetic bead matrix which are easy to perform, however, they are not economical when a high throughput amount of extracted DNA is required. Moreover, physical separation of DNA from these matrixes must be done to eliminate the inhibitory effect of matrix in downstream processes such as PCR (Nasiri et al. 2005).
Due to increasing demands for rapid and efficient DNA extraction procedure in different fields of biology; molecular medicine, medical biotechnology and diagnosis of human disorders, the present study has aimed to introduce a novel, rapid and cost-effective method for DNA extraction. It provides high yields of remarkably pure DNA from human blood samples which is suitable for most of molecular biology processes.
Materials and methods
Blood samples
Blood samples were freshly collected from healthy volunteers and handled in different ways that mimicked the range of conditions; for instance, fresh and frozen samples were treated by EDTA (Merck, Darmstadt, Germany).
Reagents and solutions
Red blood cells lysing buffer (RLB): 2M Tris-HCl (Sinagen, Iran) pH 7.6, 1M MgCl2 (Merck, Darmstadt, Germany), 3M NaCl (Merck, Darmstadt, Germany);
White blood cells lysing buffer (WLB): 2M Tris pH 7.6, 0.4 M disodium salt of ethylenediaminetetra acetic acid (Na2EDTA) (Merck, Darmstadt, Germany), pH 8, 3M NaCl, 2% cetyl trimethyl ammonium bromide (CTAB) (Merck, Hohenbrunn, Germany);
Chloroform: isoamyl alcohol (CIA) (Merck, Darmstadt, Germany) 24:1;
Isopropanol (Merck, Darmstadt, Germany);
70% and 96% ethanol (Merck, Darmstadt, Germany);
TE buffer [10 mM Tris-HCI (pH 8.0), 1mM EDTA (pH 8.0)].
DNA extraction procedure
Blood DNA extraction kit (Arman Gene Tajhiz Co., Iranian patent No. 69786) was used for isolation of chromosomal DNA from blood samples with some modifications (Atashpaz et al. 2008; Atashpaz et al. 2010; Barzegari et al. 2010).
Concisely, the extraction procedures for fresh sample were done as follow:
The peripheral blood sample was collected in EDTA vacutainer, 2 ml of which was transferred into a 2 ml tube.
Sample was centrifuged for 10 min at 6000 rpm at 4 °C.
Plasma was aspirated without touching the leukocyte layer.
One ml of RLB was added to the precipitate and mixed gently.
The sample was centrifuged for 5 min at 3000 rpm.
-
The supernatant was removed.
Note: If red blood cells still remain, resuspend the cellular pellet in 1 ml of RLB and repeat steps 4-6 for 3 or 4 times until only the white pellet appears.
One ml of WLB was added and mixed with white blood cells.
The sample was incubated at 65 °C for 30 min.
It was centrifuged at 12,000 rpm for 5 min and supernatant was transferred to a new clean tube and pellet was discarded.
An equal volume of Chloroform-Isoamilalcohol solution was added to supernatant.
The tube was centrifuged at 12000 rpm for 8 min and supernatant was transferred to a new tube.
An equal volume of chilled Isopropanol solution was added.
Sample was kept in -20 °C for 30 min.
Then tube was centrifuged at 4 °C, 12000 rpm for 10 min.
The supernatant was discarded and 300 μl of chilled 90% Ethanol was added. Tube was centrifuged at 4 °C, 12000 rpm for 5 min.
The 15 and 16 steps were repeated with chilled 70% Ethanol.
Supernatant was discarded and pellet was let to be dried at room temperature.
Pellet was dissolved in 100 μl of TE buffer or ddH2O and DNA solution was stored at -20 °C
Notification:
For frozen blood samples the procedure started from step 4.
For clotted blood samples the procedure started from step 7 by adding1 ml of WLB and incubating at 65 °C for 1 hour (shaken every 10 min). Then it was continued by step10.
Quantity and quality assessment of the extracted DNA
Quantification of extracted DNA
Quality and quantity of the extracted DNA were checked by NanoDrop 1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). By measuring the 260/230 and 260/280 nm absorbance ratios (A260/230 and A260/280) the DNA concentration, purity and protein contamination of samples were determined. We also compared our DNA extraction method with two commercial kits, Gentra Puregene Blood Kit (Gentra, Minneapolis, MN) and Dr. GenTLE® system (Takara, Osaka, Japan).
Agarose gel electrophoresis
To evaluate the probability of DNA degradation, gel electrophoresis was carried out by loading 5 µl of extracted DNA on 1% agarose gel (Invitrogen, California, USA).
Gene amplification and detection
PCR reaction was performed to check the intactness of the genomic DNA and to determine whether any inhibitory material was interfering with the reaction. For this purpose, a fragment of β-actin gene was amplified in a 25 µL volume, consisting of: 12.5 µL PCR Master Kit (2x) (Cinagen Inc., Tehran, Iran), 0.4 µM; primer sets β-actin F: 5’-TGC CCA TCT ACG AGG GGT ATG-3’ and β-actin R: 5’-CTC CTT AAT GTC ACG CAC GAT TTC-3’as from Cinagen Inc. (Tehran, Iran) and 50 ng/µl genomic DNA. The amplification program consisted of one initial denaturation at 94ºC for 4 min followed by 32 cycles of 50 s at 94 ºC for denaturation, 50 s at 58 ºC for primer annealing, 25 s at 72ºC for extension and DNA synthesis and final extension at 72 ºC for 5 min. Products were separated on 1.5% agarose gel, and photographed by /G:Box™ gel documentation system (Syngene, Cambridge, United Kingdom) after staining with ethidium bromide.
Digestion with restriction enzymes
Digestion of extracted DNA with restriction enzyme was carried out to check its quality, purity, and intactness. Briefly, each reaction in a total volume of 20 µl contained: 1 µg DNA, 2 µl of 10x Tango buffer (Fermentas, GmbH, Germany) and 1 µl of restriction enzyme, EcoR I (10 u/µl) (Fermentas, GmbH, Germany) incubated at 37 °C for 3h. Products were separated on 1% agarose gel, and photographed by the UV fluorescence after staining with ethidium bromide.
Result and discussion
Accurate understanding of material function in molecular biology has enhanced the capability of the scientist to develop the alternative methods for DNA extraction from a variety of samples. It is obvious that the choice of the methodology will depend on several factors, including cost, time, simplicity, and robustness.
Numerous genomic DNA isolation protocols have been optimized for blood samples (Angelini et al. 2000; Budowle and van Daal 2009; Elgort et al. 2004; Lahiri et al. 1992; Lahiri and Nurnberger Jr 1991; Nasiri et al. 2005; Pachot et al. 2007; Planelles et al. 1996) most of which were verified to be reproducible and yielded sufficiently high-quality DNA for genetic analysis. However, in most of these methods, enzymes (such as proteinase K and RNAse A) or toxic organic solvents (such as phenol or guanidine isothiocyanate) have been exploded (Ding 1992; Pachot et al. 2007; Planelles et al. 1996). There are only a few methods that are non-enzymatic and do not employ hazardous organic solvents (Lahiri and Nurnberger Jr 1991), therefore, the standard protocols for DNA extraction remain time-consuming. Since these enzymes are expensive, and most of the materials that are used routinely are toxic, it is reasonable to apply an efficient DNA extraction procedure that does not undergo these steps. In an attempt to attain these aims and simplify the procedure, in this study we described a very simple, inexpensive, rapid and less-hazardous protocol for extracting high quality DNA from blood samples.
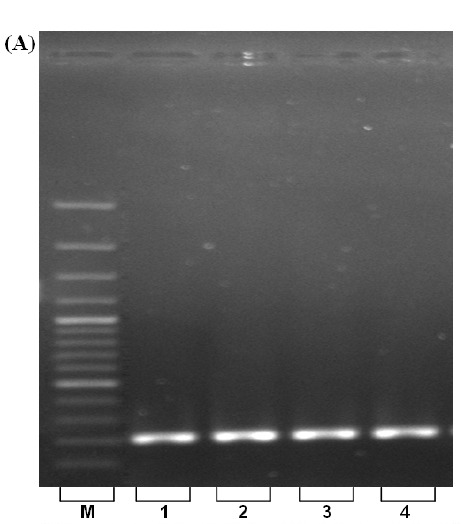
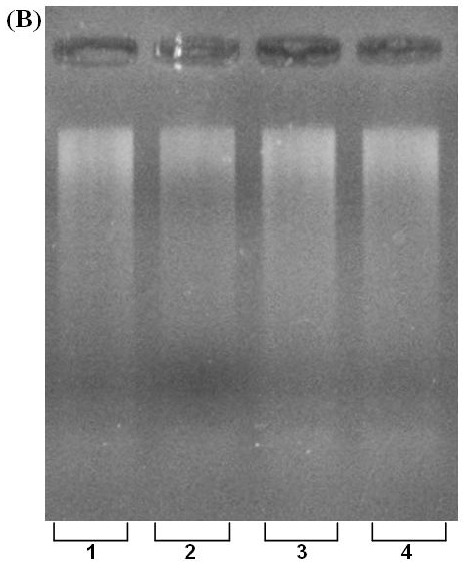
The spectrophotometric analysis for A260/280 resulted in an average of about 1.8 that indicates the extracted DNA was free from protein contamination (Table 1). Comparative spectrophotometric results obtained from two other kits; QIAGEN, TAKARA (Table 2) revealed the efficiency of our approach. Despite the fact that extracted DNA exposed to several steps, no degradation was observed that is validated by sharp and unified bands on agarose gel electrophoresis (Fig. 1, lines 1-4). Furthermore, the high quality of the isolated DNA used in PCR reaction was demonstrated with clearly recognizable bands (Fig. 2. A., lines 1-4). Enzymatic digestion of extracted DNA by EcoR I revealed that our procedure yields intact DNA with minimized inhibitory effect (Fig. 2. B lines 1-4).
Table 1. Results of absorbance ratio measured by NanoDrop 1000 Spectrophotometer.
| OD260/OD230 ratio | OD260/OD280 ratio | DNA concentration (ng/ µl) | Sample type |
| 2.3 | 1.84 | 1630 | Frozen |
| 2.27 | 1.85 | 2342 | Fresh |
| 2.58 | 1.90 | 1015 | Clotted |
| 2.33 | 1.83 | 0865 | Dried |
Table 2. Spectrophotometer results obtained from extracted DNA using different kits; Blood DNA extraction kit (current study), Gentra Puregene Blood Kit (Gentra, Minneapolis, MN) and Dr. GenTLE® system (Takara, Osaka, Japan).
| OD260/OD230 ratio | OD260/OD280 ratio | DNA concentration (ng/ µl) | Sample type | Company | Kit name |
| 2.3 | 1.84 | 1630 | Frozen | ARMAN GENE TAJHIZ | Blood DNA Extraction |
| 2.2 | 1.85 | 2342 | Fresh | ||
| 2.5 | 1.90 | 1015 | Clotted | ||
| 2.3 | 1.83 | 0865 | Dried | ||
| 2.0 | 1.90 | 1013 | Frozen | QIAGEN | Gentra Puregene Blood |
| 2.1 | 1.83 | 1506 | Fresh | ||
| 2.0 | 1.80 | 0030 | Clotted | ||
| N/A | N/A | N/A | Dried | ||
| 1.89 | 1.66 | 0300 | Frozen | TAKARA | Dr. GenTLE® System |
| 1.93 | 1.74 | 0600 | Fresh | ||
| N/A | N/A | N/A | Clotted | ||
| N/A | N/A | N/A | Dried |
Fig. 1.
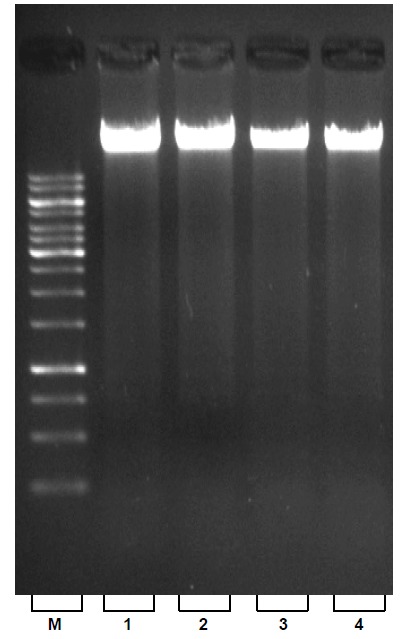
Electrophoretic analysis of total genomic DNA on 1% agarose gel; Lines 1-4: fresh, frozen, dried and clotted blood samples respectively. (M=1 Kbp molecular weight ladder).
Fig. 2. (A) PCR amplification of β-actin gene using extracted DNA. Lines 1-4: fresh, frozen, clotted and dried blood samples. M=100-bp ladder, L.6=100 bp molecular weight ladder. (B) DNA digestion with restriction enzyme, EcoR I. L. 1-4; fresh, frozen, clotted and dried blood samples, respectively.
A.
B.
Conclusion
In conclusion, this study represents a reliable approach for DNA extraction not only from fresh blood samples but also from frozen and clotted ones. Omitting enzymatic incubation (such as proteinase K and RNAase) and neutralization we reached to high throughput yield from a small amount of sample which make current method applicable in medical laboratories as well as research centers.
Ethical Issues
None to be declared.
Conflict of interests
No conflict of interest to be declared.
Acknowledgments
The authors gratefully acknowledge Dr. Yadollah Omidi, Dr. Masoud Asadi, Dr. Ahmad Khosroshahi and Mr. Sajjad Khani for their kind assistance during this work.
References
- Angelini A, Di Febbo C, Rullo A, Di FebboDi Ilio C, Cuccurullo F and Porreca E . 2000 New Method for the Extraction of DNA From White Blood Cells for the Detection of Common Genetic Variants Associated With Thrombophilia. Pathophysiol Haemost Thromb, 32(4), 180-183 [DOI] [PubMed] [Google Scholar]
- Atashpaz A, Barzegari A, Azarbaijani R. 2008. General DNA extraction kit. Iranin Patent Office, No. 48024.
- Atashpaz S, Khani S, Barzegari A, Barar J, Vahed SZ, Azarbaijani R, et al. 2010 A Robust Universal Method for Extraction of Genomic DNA From Bacterial Species. Microbiology, 79(4), 538-542 [PubMed] [Google Scholar]
- Barzegari A, Vahed SZ, Atashpaz S, Khani S and Omidi Y . 2010 Rapid and Simple Methodology for Isolation of High Quality Genomic DNA From Coniferous Tissues (Taxus Baccata). Mol Biol Rep, 37(2), 833-837 [DOI] [PubMed] [Google Scholar]
- Budowle B and van Daal . 2009 Extracting Evidence From Forensic DNA Analyses: Future Molecular Biology Directions. Biotechniques, 46(5), 342-350 [DOI] [PubMed] [Google Scholar]
- Castella V, Dimo-Simonin N, Brandt-Casadevall C and Mangin P . 2006 Forensic Evaluation of the QIAshredder/QIAamp DNA Extraction Procedure. Forensic Sci Int, 156(1), 70-73 [DOI] [PubMed] [Google Scholar]
- Cattaneo C, Craig OE, James NT and Sokol RJ . 1997 Comparison of Three DNA Extraction Methods on Bone and Blood Stains Up to 43 Years Old and Amplification of Three Different Gene Sequences. J Forensic Sci, 42(6), 1126-1135 [PubMed] [Google Scholar]
- Clements DN, Wood S, Carter SD and Ollier WER . 2008 Assessment of the Quality and Quantity of Genomic DNA Recovered From Canine Blood Samples by Three Different Extraction Methods. Res Vet Sci, 85(1), 74-79 [DOI] [PubMed] [Google Scholar]
- Ding Y. 1992. DNA Preparation From Trace Forensic Biological Evidences. Chin Sci Technol Press, Beijing, China, 371-375.
- Elgort MG, Herrmann MG, Erali M, Durtschi JD, Voelkerding KV and Smith RE . 2004 Extraction and Amplification of Genomic DNA From Human Blood on Nanoporous Aluminum Oxide Membranes. Clin Chem, 50(10), 1817-1819 [DOI] [PubMed] [Google Scholar]
- Lahiri DK and Nurnberger Jr . 1991 A Rapid Non-Enzymatic Method for the Preparation of HMW DNA From Blood for RFLP Studies. Nucleic Acids Res, 19(19), 5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Bye S, Nurnberger JI, Hodes ME, Crisp M . 1992 A Non-Organic and Non-Enzymatic Extraction Method Gives Higher Yields of Genomic DNA From Whole-Blood Samples Than Do Nine Other Methods Tested. J Biochem Biophys Methods, 25(4), 193-205 [DOI] [PubMed] [Google Scholar]
- Lewis CM, Cler LR, Bu DW, Zochbauer-Mnller S, Milchgrub S, Naftalis EZ, et al. 2005 Promoter Hypermethylation in Benign Breast Epithelium in Relation to Predicted Breast Cancer Risk. Clin Cancer Res, 11(1), 166-172 [PubMed] [Google Scholar]
- Nasiri H, Forouzandeh M, Rasaee MJ and Rahbarizadeh F . 2005 Modified Salting Out Method: High Yield, High Quality Genomic DNA Extraction From Whole Blood Using Laundry Detergent. J Clin Lab Anal, 19(6), 229-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachot A, Barbalat V, Marotte H, Diasparra J, Gouraud A, Mougin B, et al. 2007 A Rapid Semi Automated Method for DNA Extraction From Dried-Blood Spots: Application to the HLA-DR Shared Epitope Analysis in Rheumatoid Arthritis. J Immunol Methods, 328(1-2), 220-225 [DOI] [PubMed] [Google Scholar]
- Phillips HA, Howard GCW and Miller WR . 2000 P53 Mutations As a Marker of Malignancy in Bladder Washing Samples From Patients With Bladder Cancer. Br J Cancer, 82(1), 136-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planelles D, Llopis F, Puig N and Montoro JA . 1996 A New, Fast, and Simple DNA Extraction Method for HLA and VNTR Genotyping by PCR Amplification. J Clin Lab Anal, 10(3), 125-128 [DOI] [PubMed] [Google Scholar]
- Wang SS, Thornton K, Kuhn AM, Nadeau JG and Hellyer TJ . 2003 Homogeneous Real-Time Detection of Single-Nucleotide Polymorphisms by Strand Displacement Amplification on the BD ProbeTec ET System. Clin Chem, 49(10), 1599-1607 [DOI] [PubMed] [Google Scholar]