

Abstract
A high-throughput screen (HTS) of the MLPCN library using a homogenous fluorescence polarization assay identified a small molecule as a first-in-class direct inhibitor of Keap1-Nrf2 protein-protein interaction. The HTS hit has three chiral centers; a combination of flash and chiral chromatographic separation demonstrated that Keap1-binding activity resides predominantly in one stereoisomer (SRS)-5 designated as ML334 (LH601A), which is at least 100× more potent than the other stereoisomers. The stereochemistry of the four cis isomers was assigned using X-ray crystallography and confirmed using stereospecific synthesis. (SRS)-5 is functionally active in both an ARE gene reporter assay and an Nrf2 nuclear translocation assay. The stereospecific nature of binding between (SRS)-5 and Keap1 as well as the preliminary but tractable structure-activity relationships support its use as a lead for our ongoing optimization.
Keywords: Keap1, Nrf2, Inhibitors of protein-protein interaction, Nrf2 activation
Three major cellular components are involved in the regulation of cellular defense mechanisms that protect cells from oxidative stress; they are Kelch-like ECH-associated protein 1 (Keap1), nuclear factor erythroid 2-related factor 2 (Nrf2), and antioxidant response elements (ARE). The Keap1-Nrf2-ARE system is the main signaling pathway that regulates transcription of a series of cytoprotective proteins including glutathione S-transferases (GSTs), NAD(P)H:quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), and Nrf2 itself (Fig. 1). This system plays a key role in oxidative stress response, inflammation, and carcinogenesis.1–4 Targeting the Keap1-Nrf2-ARE signaling pathway represents an attractive strategy to discover chemopreventive agents for cancer as well as preventive and therapeutic agents for a variety of other diseases and conditions, including diabetes, Alzheimer’s, and Parkinson’s, that involve oxidative stress and inflammation.5–13
Fig. 1. A model depicting Nrf2 activation by a direct inhibitor of Keap1-Nrf2 interaction.

Nrf2 is sequestered in the cytosol by its protein inhibitor, Keap1, and is transcriptionally inactive. A direct inhibitor binds to the Kelch domain of Keap1 and suppresses the ubiquitination of Nrf2, which decreases degradation of Nrf2 and allows more Nrf2 to translocate to the nucleus and form a transcriptionally active complex with Maf, leading to the induced expression of Nrf2 itself and oxidative stress response enzymes that deactivate reactive oxygen species and electrophiles.
Keap1-Nrf2 protein-protein interaction is considered a critical point of the pathway that can be targeted for intervention.1 Some of the known Nrf2-activating/ARE-inducing agents are already in human clinical trials as chemopreventive agents for cancer or as therapeutic agents for conditions involving inflammation. These include sulforaphane for the treatment and prevention of prostate cancer and for the treatment of chronic obstructive pulmonary disease (COPD) and bardoxolone methyl for the treatment of advanced chronic kidney disease (CKD) in patients with type 2 diabetes mellitus.14–18 However, all currently known small-molecule Nrf2 activators/ARE inducers, including many natural products (e.g., sulforaphane, curcumin, and epigallocatechin gallate from natural sources such as fruits, vegetables, and tea products) and synthetic compounds (e.g., oltipraz, anethole dithiolethione, and bardoxolone methyl), function as eletrophiles and indirectly inhibit Keap1-Nrf2 protein-protein interaction through covalent modification of the sensitive cysteine residues found in Keap1.1,19
To discover novel non-reactive small molecules as direct inhibitors of the protein-protein interaction between Keap1 and Nrf2, we developed a fluorescence polarization (FP) assay with fluorescein-labeled 9mer Nrf2 peptide amide as the fluorescent probe and Keap1 Kelch domain as the target protein20 and used it to screen the NIH MLPCN library of small molecules. Herein, we report the discovery of a small molecule hit as a first-in-class direct small-molecule inhibitor of Keap1-Nrf2 protein-protein interaction. Because of the presence of three chiral centers and the uncertain stereochemical composition of the hit sample, we synthesized the hit and a series of analogs with defined composition for activity confirmation, determination of stereochemical requirement for Keap1 binding, and preliminary structure-activity relationship analysis.
Hit Identification and Confirmation
We developed an FP assay,20 which was successfully used to screen the MLPCN library of 337,116 compounds (PubChem Assay ID: 504523, 504540). Using inhibition >3× standard deviation of DMSO wells (corresponding to 12% inhibition) for hit calling, the primary screen at 10 μM generated a list of 489 hits. After excluding fluorescent compounds, 460 of the initial hits were cherry-picked and retested in the FP assay for 8-point dose-response curves and in a thermal shift secondary assay. Among the eight confirmed hits, hit 1 (Fig. 1A) was the most promising with an IC50 of 3 μM (Fig. 2B). The binding constant (Kd) of hit 1 to Keap1 Kelch domain was confirmed to be 1.9 μM (Fig. 2C/D) using an SPR solution competition assay we recently developed.21 Hit 1 has no chemically reactive functional groups present and is not expected to modify sulfhydryl groups of cysteine residues in proteins.
Fig. 2.

(A) The structure of hit 1 identified through HTS, (B) dose-response curve of the hit 1 in the confirmative FP assay, (C) sensorgrams of the hit 1 in the SPR assay, (D) comparison of Kd of different stereoisomers of 5 (LH601) to hit 1, and (E) X-ray crystal structures of A/B as a pair and D as a single enantiomer.
Since there are three stereogenic elements in hit 1 and the two substituents on the cyclohexyl ring are known to be of cis configuration, we were expecting four stereoisomers that could be present; one of the four stereoisomers is likely to be a direct inhibitor of the Keap1-Nrf2 interaction since ligand-target interactions are often stereospecific. However, the composition of hit 1 sample in the MLPCN library was assigned with uncertainty as a pair of diastereomers. Only after our synthesis and determination of Keap1 binding of an equal mixture of all four stereoisomers did we realize that the hit 1 sample in the MLPCN library was predominantly composed of one set of enantiomers as later confirmed by chiral HPLC analysis. It became clear that we needed to determine which of the stereoisomer is responsible for binding to Keap1 Kelch domain.
Resynthesis of hit 1 for activity confirmation and stereochemistry assignment
When we synthesized hit 1 according to Scheme 1 by reacting 1-phthalimidomethyl tetrahydroisoquinoline ((±)-4) with cis-cyclohexanedicarboxylic anhydride, we obtained an expected mixture of four stereoisomers (5, LH601) based on our chiral HPLC analyses. The sample of 5 was shown to be two fold less active than the hit 1 sample obtained from the NIH MLPCN library in our SPR solution competition assay. Further analysis of the hit 1 sample from the NIH MLPCN library, we found that the hit 1 sample contained ~90% of one set of enantiomers, probably as a result of recrystallization used for purification in the commercial process. We then separated the sample of 5 into diastereomers (A/B and C/D) using flash silica gel chromatography. A/B was shown to contain the active isomer in hit 1 and was further separated into the two enantiomers A and B by preparative normal phase chiral separation on a Chiralcel OD column. The activity of isomer A, B and C/D were then compared to that of hit 1 in our FP and SPR assays. As shown in the SPR dose-response curves in Fig. 2D, we have identified the most active stereoisomer in hit 1 being the isomer A (LH601A) with a Kd of 1 μM while its enantiomer B has a Kd of only 100 μM and the other diastereomers C/D are inactive. The four trans-stereoisomers resulting from the reaction of 1-phthalimidomethyl tetrahydroisoquinoline ((±)-4) with trans-cyclohexanedicarboxylic anhydride were also inactive.
Scheme 1.
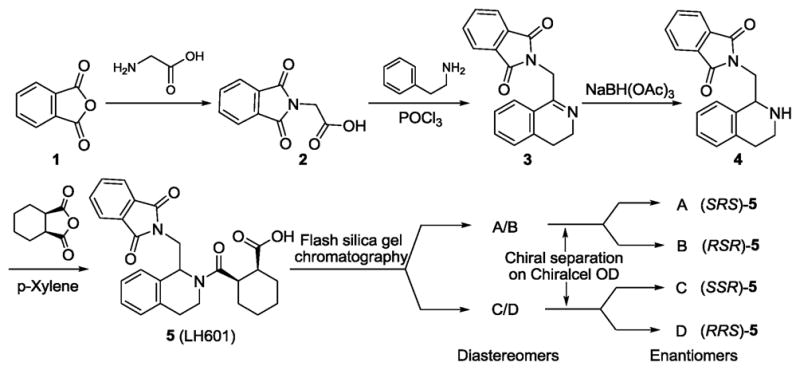
Synthesis and separation of hit 1 stereoisomers.
X-ray crystallography was used to assign the stereochemistry of the four isomers. We first attempted but failed to grow single crystals of pure isomer A. Interestingly, the A isomer could only be crystallized as a pair with its enantiomer B even when using 96% e.e. of A. Fortunately, isomer D readily crystallized as single crystals of the pure D (Fig. 2E), which was used to assign the absolute stereochemistry of all four isomers as indicated in Scheme 1.
We then undertook a stereospecific synthesis of (SRS)-5 as further confirmation of its absolute stereochemistry as shown in Scheme 2. Starting from the commercially available (S)-tetrahydroisoquinoline-1-carboxylic acid ((S)-6), we protected the secondary amine with Cbz and reduced the carboxylic acid to give Cbz-protected (S)-(tetrahydroisoquinolin-1-yl)methanol ((S)-7). Mitsunobu reaction, using phthalimide in the presence of DIAD and PPh3, followed by hydrogenolysis afforded stereospecifically the (S)-4. Alternatively, (S)-4 was prepared from the racemic amine by resolution through crystallization of their diastereomeric salts with (−)-dibenzoyl L-tartaric acid ((−)DBTA). A known alkaloid-mediated desymmetrization procedure of cis-cyclohexanedicarboxylic anhydride with benzyl alcohol in the presence of quinidine22,23 was used to control the chiral centers on the cyclohexyl ring to obtain (1R,2S)-2-(benzyloxycarbonyl)cyclohexane carboxylic acid ((R,S)-8). Simple amide bond coupling between the optically active (S)-4 and (R,S)-8 produced the benzyl protected (SRS)-9, which upon hydrogenolysis gave the desired enantiomer (SRS)-5.
Scheme 2.
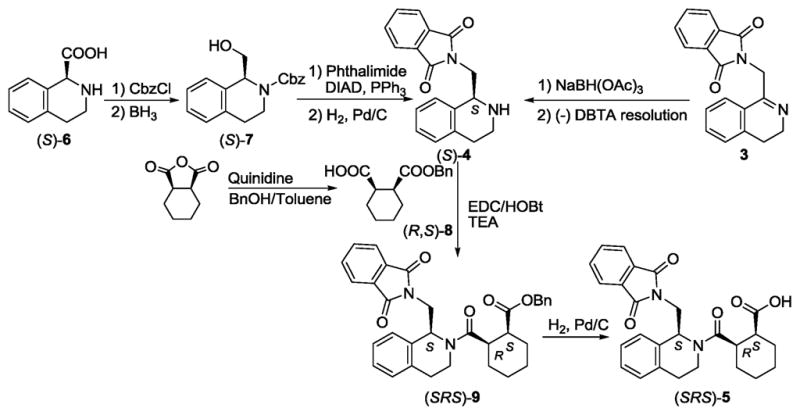
Stereospecific synthesis of (SRS)-5
After confirming the Keap1-binding activity of (SRS)-5 in our FP and SPR assays, we determined its cellular activity in two cell-based functional assays. Fig. 3A illustrates the ARE-inducing activity of (SRS)-5 (isomer A) with an EC50 of 18 μM as compared to >100 μM for its enantiomer (RSR)-5 (isomer B) and its diastereomers C/D in the CellSensor® ARE-bla HepG2 cell line, obtained from Invitrogen, where ARE controls the expression of β-lactamase.24 Fig. 3B demonstrates that (SRS)-5 promotes the nuclear translocation of Nrf2 with a similar EC50 of 12 μM in the PathHunter® U2OS Keap1-Nrf2 functional assay, obtained from DiscoveRx, which uses β-galactosidase-based enzyme fragment complementation technology and luminescence for the detection of Nrf2 nuclear translocation.25 All these data indicate that (SRS)-5 is cell permeable and is capable of inhibiting the Keap1-Nrf2 interaction, leading to the dissociation of Nrf2 from Keap1 in the cytosol, its subsequent translocation to the nucleus, and the upregulation of ARE-controlled genes.
Fig. 3.
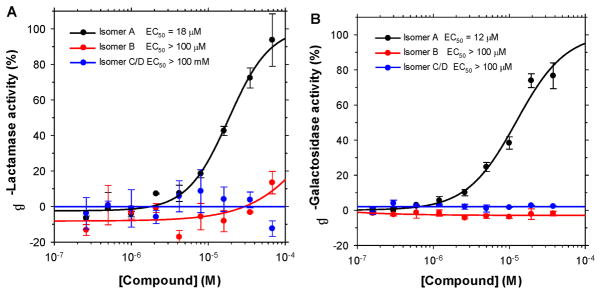
ARE-inducing activity of (SRS)-5 (isomer A) in CellSensor® β-lactamase reporter assay (A) and its effect on Nrf2 nuclear translocation in the PathHunter® U2OS Keap1-Nrf2 Functional Assay (B) in comparison to other stereoisomers.
We also synthesized several analogs of (SRS)-5 to derive preliminary structure-activity relationships suggesting that (SRS)-5 is viable lead for further structure optimization into more potent direct inhibitors of Keap1-Nrf2 interaction. The following is a summary of some of the more important SARs (Fig. 4): 1) Keap1 binding activity resides primarily in one stereoisomer. As discussed earlier, (SRS)-5 (isomer A) is at least 100 times more active than other stereoisomers (B/C/D and trans isomers). 2) Acidic functionality is preferred on the cycloalkane ring. The corresponding methyl ester was completely inactive and the corresponding amide was about 20-fold less active than (SRS)-5, suggesting that the free carboxylic acid group in (SRS)-5 is optimal for binding to Keap1 Kelch domain. 3) A one-carbon linker between tetrahydroisoquinoline (THIQ) and phthalimido group is optimal. Analogs with one-carbon extension, i.e., an ethylene linker instead of a methylene, between THIQ and phthalimido group, are inactive. 4) One of the carbonyls in phthalimido group can be removed to give a lactam with only slight decrease in Keap1 binding. Additional analogs are being synthesized in our continuing lead optimization efforts to explore the chemical spaces around the various points of the scaffold to improve binding affinity, membrane permeability, and other physico-chemical and pharmaceutical properties.
Fig. 4. Preliminary structure-activity relationships.
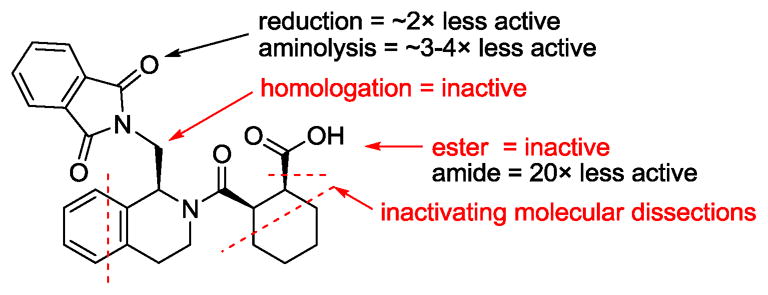
The activity noted was based on the solution competition SPR assay.
We docked (SRS)-5 to the Nrf2 peptide binding site in Keap1 Kelch domain starting from the cocrystal structure of Keap1 Kelch domain with the 16mer Nrf2 peptide (PDB code: 2FLU); Fig. 5A shows a representative binding pose of (SRS)-5 in the Nrf2 peptide binding site of Keap1 Kelch domain and Fig. 5B depicting the overlay of (SRS)-5 to Nrf2 peptide as bound to Keap1 and major interactions between Keap1 and the ligands. Based on this model, the binding of (SRS)-5 to Keap1 is strengthened by two π-cation interactions between THIQ and Arg415 and between phthalimido and Arg380 in addition to the hydrogen-bonding interactions that are also observed between Keap1 and Nrf2 peptide.
Fig. 5. Docking of (SRS)-5 to the Nrf2 binding site of Keap1 Kelch domain.
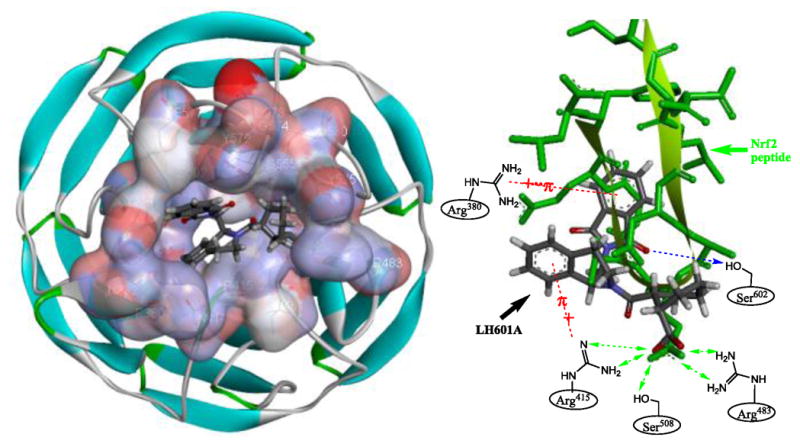
(A) Top view with (SRS)-5 (shown in stick model) bound to Keap1 Kelch domain shown in ribbon graphics and interacting Keap1 residues in VDW surface. (B) Overlay of the (SRS)-5 colored by element type with Nrf2 peptide in green as docked onto the Nrf2 binding site of Keap1 Kelch domain (rotated ~90° around the X-axis in A/B for a better view of the interactions with Keap1 residues shown at the bottom.
The solubility of (SRS)-5 is >100 μM in phosphate buffered saline and (SRS)-5 is highly stable over a period of 24 h. Even though it does not contain electrophilic groups, distinguishing it from previously reported indirect inhibitors of Keap1-Nrf2 interaction, we exposed (SRS)-5 to a high concentration (50 μM) of glutathione that we used to mimic cysteine residues. No thiol addition or decomposition could be detected over 48 h, indicating that (SRS)-5 inhibits the Keap1-Nrf2 protein-protein interaction by non-covalent binding and thus is a first-in-class direct inhibitor of Keap1-Nrf2 interaction. (SRS)-5 was designated as a probe with the NIH Molecular Libraries Program (ML334) due to these properties and its potency as an inhibitor of Keap1-Nrf2 interaction.
In summary, we have discovered, through high-throughput screening of the NIH MLPCN library of small molecules, a first-in-class direct inhibitor of the Keap1-Nrf2 protein-protein interaction. It was found that only one stereoisomer among the possible eight isomers is active. The different binding activity between the stereoisomers is expected of structurally specific ligand-target interactions. The strong binding affinity of (SRS)-5 (ML334, LH601A) over its stereoisomers and the preliminary, yet tractable, structure-activity relationships provide further evidence of its direct binding to Keap1 and support its use as a lead for structure optimization.
Acknowledgments
We thank Dr. Suzanne Forry-Schaudies from the National Cancer Institute for her advice and continuing efforts in overseeing the success of this project. We gratefully acknowledge the financial support of grants CA133791 and MH093197 (to L.H.) from the National Institutes of Health. This work was funded in part by the NIH-MLPCN program (1 U54 HG005032-1 awarded to S.L.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Magesh S, Chen Y, Hu L. Med Res Rev. 2012;32:687. doi: 10.1002/med.21257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nguyen T, Yang CS, Pickett CB. Free Radic Biol Med. 2004;37:433. doi: 10.1016/j.freeradbiomed.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 3.Stewart JD, Hengstler JG, Bolt HM. Arch Toxicol. 2011;85:239. doi: 10.1007/s00204-011-0694-1. [DOI] [PubMed] [Google Scholar]
- 4.Baird L, Dinkova-Kostova AT. Arch Toxicol. 2011;85:241. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 5.Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J, Johnson JA. Antioxid Redox Signal. 2009;11:497. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeong WS, Jun M, Kong ANT. Antioxid Redox Signal. 2006;8:99. doi: 10.1089/ars.2006.8.99. [DOI] [PubMed] [Google Scholar]
- 7.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Pharmacol Res. 2008;58:262. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates MS, Kensler TW. Drug News Perspect. 2007;20:109. doi: 10.1358/dnp.2007.20.2.108343. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Ichikawa T, Janicki JS, Cui T. Expert Opin Ther Targets. 2009;13:785. doi: 10.1517/14728220903025762. [DOI] [PubMed] [Google Scholar]
- 10.Yu X, Kensler T. Mutat Res. 2005;591:93. doi: 10.1016/j.mrfmmm.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Redell JB, Moore AN, Dash PK. Biochem Biophys Res Commun. 2011;407:501. doi: 10.1016/j.bbrc.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steel R, Cowan J, Payerne E, O’Connell MA, Searcey M. ACS Med Chem Lett. 2012;3:407. doi: 10.1021/ml300041g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williamson TP, Amirahmadi S, Joshi G, Kaludov NK, Martinov MN, Johnson DA, Johnson JA. Chem Biol Drug Des. 2012;80:810. doi: 10.1111/cbdd.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pergola PE, Krauth M, Huff JW, Ferguson DA, Ruiz S, Meyer CJ, Warnock DG. Am J Nephrol. 2011;33:469. doi: 10.1159/000327599. [DOI] [PubMed] [Google Scholar]
- 15.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG. N Engl J Med. 2011;365:327. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 16.Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, Sporn MB, Liby KT, Rawal B, Lee JH, Gabrilovich DI. Clin Cancer Res. 2010;16:1812. doi: 10.1158/1078-0432.CCR-09-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egner PA, Chen JG, Wang JB, Wu Y, Sun Y, Lu JH, Zhu J, Zhang YH, Chen YS, Friesen MD, Jacobson LP, Munoz A, Ng D, Qian GS, Zhu YR, Chen TY, Botting NP, Zhang Q, Fahey JW, Talalay P, Groopman JD, Kensler TW. Cancer Prev Res. 2011;4:384. doi: 10.1158/1940-6207.CAPR-10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shapiro TA, Fahey JW, Dinkova-Kostova AT, Holtzclaw WD, Stephenson KK, Wade KL, Ye L, Talalay P. Nutr Cancer. 2006;55:53. doi: 10.1207/s15327914nc5501_7. [DOI] [PubMed] [Google Scholar]
- 19.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Proc Natl Acad Sci U S A. 1993;90:2965. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoyama D, Chen Y, Huang X, Beamer LJ, Kong ANT, Hu L. J Biomol Screen. 2011;17:435. doi: 10.1177/1087057111430124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Inoyama D, Kong ANT, Beamer LJ, Hu L. Chem Biol Drug Des. 2011;78:1014. doi: 10.1111/j.1747-0285.2011.01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolm C, Schiffers I, Atodiresei I, Hackenberger CPR. Tetrahedron: Asymmetry. 2003;14:3455. [Google Scholar]
- 23.Christov C, González-Bulnes P, Malhaire F, Karabencheva T, Goudet C, Pin JP, Llebaria A, Giraldo J. ChemMedChem. 2011;6:131. doi: 10.1002/cmdc.201000378. [DOI] [PubMed] [Google Scholar]
- 24.Invitrogen. CellSensor™ ARE-bla Hep G2 Cell-based Assay Protocol. 2006. p. 8. [Google Scholar]
- 25.DiscoveRx. Product Booklet: 93-0821C3. 2011. PathHunter® Keap1-Nrf2 Functional Assay for chemiluminescent detection of activated Nrf2; p. 12. [Google Scholar]