

Abstract
Exposing rodents to brief episodes of hypoxia mimics the hypoxemia and the cardiovascular and metabolic effects observed in patients with obstructive sleep apnoea (OSA), a condition that affects between 5% and 20% of the population. Apart from daytime sleepiness, OSA is associated with a high incidence of systemic and pulmonary hypertension, peripheral vascular disease, stroke and sudden cardiac death. The development of animal models to study sleep apnoea has provided convincing evidence that recurrent exposure to intermittent hypoxia (IH) has significant vascular and haemodynamic impact that explain much of the cardiovascular morbidity and mortality observed in patients with sleep apnoea. However, the molecular and cellular mechanisms of how IH causes these changes is unclear and under investigation. This review focuses on the most recent findings addressing these mechanisms. It includes a discussion of the contribution of the nervous system, circulating and vascular factors, inflammatory mediators and transcription factors to IH-induced cardiovascular disease. It also highlights the importance of reactive oxygen species as a primary mediator of the systemic and pulmonary hypertension that develops in response to exposure to IH.
Keywords: systemic hypertension, pulmonary hypertension, ROS, inflammation, NFAT, NF-κB, HIF, nervous system, endothelin, sleep apnoea
Introduction
Exposing rodents to brief episodes of hypoxia mimics the hypoxemia and the cardiovascular and metabolic effects observed in patients with sleep-disordered breathing (SDB) [1–11]. SDB includes different respiratory disorders – the most frequent being obstructive sleep apnoea (OSA) and hypoapnoea. OSA corresponds to recurrent episodes of complete or partial pharyngeal collapse during sleep resulting in intermittent hypoxia (IH) and sleep fragmentation [1, 12–18].
OSA affects between 5% and 20% of the population and apart from daytime sleepiness, is associated with high incidence of hypertension, peripheral vascular disease, stroke and sudden cardiac death [7, 19]. However, the mechanisms by which OSA and cardiovascular disease are linked remain obscure since patient studies are complicated by the confounding effects of disease duration and the many comorbidities present in OSA patients.
The first animal study showing that IH induces hypertension was reported by Fletcher, et al. [20]. They exposed rats to IH (3–5% nadir ambient oxygen) every 30 sec., 7 hrs per day for up to 35 days and demonstrated a significant increase in mean arterial blood pressure (MAP) over baseline of 13.7 ± 1.8 mmHg [20]. Follow-up studies have confirmed these observations despite multiple differences in the exposure paradigms, pCO2 levels, species and strains of animals [1, 5, 21–29]. In addition, some of these elegant studies have demonstrated elevations of arterial blood pressure that persist after termination of the intermittent hypoxic exposure. Therefore, it is now established that systemic hypertension [20], dyslipidemia [30] and early atherosclerosis (14) as well as vascular remodelling [5, 20] occur after only a few weeks of IH exposure.
In summary, in animals that are not prone to hypertension, IH induces a rapid but moderate elevation in blood pressure. In contrast, in animals with a genotype that is prone to hypertension (spontaneous hypertensive rats, SHR), IH accelerates age-induced hypertension [22, 31].
The development of SDB animal models has provided convincing evidence that IH has significant vascular and haemodynamic impact that can cause the cardiovascular morbidity and mortality observed in patients with sleep apnoea. However, some investigators suggest that species differences make the applicability of animal studies to human disease states questionable. Interestingly, Tamisier, et al. recently described a human model of IH [32]. In their study, healthy volunteer human beings were exposed to IH to induce a 10% desaturation–resaturation difference (95% to 85%) every 120 sec., thus allowing 30 cyclic desaturation–resaturation sequences per hour during sleep for 14 and 28 days. CO2 accumulation was insignificant. The persons experienced spontaneous central hypopnoea during the re-oxygenation phase. These respiratory events induced arousals with significant sleep fragmentation showing adaptation after 4 weeks of exposure. Persons exposed to this model for 2 weeks displayed changes in chemosensitivity. More importantly, IH exposure induced a small but significant increase in arterial blood pressure from baseline. This study suggests that results obtained in SDB animal models can translate to human pathophysiology and confirm the clinical relevance of the experimental models that are indispensable for SDB studies. However, the mechanisms of IH-induced hypertension are still not clear and under investigation. This review focuses on the most recent findings addressing these mechanisms.
Mechanisms of IH-induced systemic hypertension
Contribution of the nervous system
It has been well established that IH-induced elevation in blood pressure involves enhanced sympathetic nerve activity [33–38], increased adrenal catecholamine secretion [39–42], decreased baroreflex [28, 43] and enhanced chemoreflex sensitivity [5, 19, 21, 36, 44, 45]. These reflexes are under the control of the nucleus of the solitary tract in the brainstem. IH has been shown to induce activation of catecholaminergic structures and neuronal cell death – including hypoglossal motoneurons, pyramidal, Purkinje, catecholaminergic wake neurons and upper airway dilator motoneurons [9, 46–49]. The role each of these plays in the blood pressure response and in the decreased baroreflex sensitivity, however, is not clear.
Endoplasmic reticulum (ER) plays a central role in both adaptive and pro-apoptotic response in neurons (reviewed in [50]). The unfolded protein response (UPR) in the ER represents an adaptive response to minimize accumulation of misfolded proteins that would be toxic to the cell. This response includes reduced overall protein translation, increased production of chaperones, up-regulated clearance of improperly folded proteins and increased antioxidant capacity. Several components of this protective response are mediated by phosphorylation of eIF-2a. However, when this stress is insurmountable, the ER may lead to apoptosis by activating CAAT/enhancer-binding protein homologous protein (CHOP)/growth arrest and DNA damage inducible protein (GADD153) and caspase-7. Zhu, et al. have recently demonstrated that IH exposure induces ER injury contributing to death of a selective population of brainstem motoneurons [51]. In adult mice, IH selectively activates the PKR-like ER kinase (PERK ER) sensor of cell fate. Hypoglossal motoneurons, which innervate the upper airway, have high basal levels of CHOP/GADD153 and dephosphorylated p-eIF-2a and are not protected from apoptosis when they are exposed to IH. However, motor trigeminal neurons and occulomotor neurons do not have significant levels of CHOP/GADD153 at baseline, and can mount a protective eIF-2a response. More importantly, protection of eIF-2 dephosphorylation with systemically administered salubrinal (protein phosphatase inhibitor) throughout IH exposure prevented apoptosis in susceptible motoneurons [51]. Other areas, including phrenic and other spinal motoneurons, should now be examined to determine the contribution of neural apoptosis to the development of hypertension.
As mentioned above, substantial evidence demonstrates that hypoxia affects the synthesis of various neurotransmitters in the central and peripheral nervous systems. Synthesis of bioamines is affected in the brainstem. IH exposure through increased reactive oxygen species (ROS) produces a robust increase in tyrosine hydroxylase activity, dopamine level and tyrosine hydroxylase phosphorylation. Tyrosine hydroxylase is the rate-limiting enzyme in the biosynthesis of catecholamines [52]. In addition to biomines, neuropeptides play critical roles in neurotransmission, acting as transmitters or modulators in the brainstem; a region that has been implicated in the central nervous system control of cardiovascular responses. IH activates O2-sensitive peptidylglycine α-hydroxylating monooxygenase in rat brainstem, which is the rate-limiting enzyme for the generation of carboxy-terminal α-amidated neuropeptides. ROS increases post-translational proteolytic processing leading to elevated levels of α-amidated forms of substance P and neuropeptide Y, which contribute to IH-mediated peptidergic neurotransmission in rat brainstem [53].
Studies in rodents exposed to IH suggest enhanced carotid body (CB) responsiveness to hypoxia is involved in the autonomic alterations induced by OSA and in the development of the hypertension [5, 19, 21, 36, 44, 45]. This is supported by recordings of CB chemosensory neural discharges in situ and in vitro showing that exposure to IH increases basal CB chemosensory discharges and enhances the chemosensory response to hypoxia [44, 54, 55]. Studies with anti-oxidants suggest the involvement of ROS-mediated signalling in eliciting long-term facilitation of sensory nerve discharge (sLTF) of the CB in rodents exposed to IH [56]. Peng and colleagues have recently demonstrated increases in NADPH oxidase (NOX) activity in IH but not in control carotid bodies. This effect was associated with up-regulation of NOX2 mRNA and protein in glomus cells of the CB. sLTF was prevented by NOX inhibitors and was absent in mice deficient in NOX2. H2O2 generated from O2._ contributes to sensory plasticity of the CB [57]. These ROS-dependent functional changes may contribute to persistent reflex activation of sympathetic nerve activity in IH and sleep apnoea. In addition, recent evidence support a role for endothelin-1 (ET-1) [58–60] and increased N-methyl-D-aspartate receptors [61] as contributors to the increased CB chemoreflex sensitivity.
Contribution of circulating and vascular factors
In addition to the contribution of the sympathetic and central nervous system, other factors including circulating and tissue ROS [62, 63], angiotensin II (Ang II) [64–66] and ET-1 [23, 29, 67–69] have been implicated in the development of hypertension during IH. Indeed, ROS could be the main and common contributor to hypertension in IH and sleep apnoea. Consistent with this hypothesis is evidence that ROS generation is increased by IH in rodents [45, 62, 63, 70] and in leucocytes from patients with OSA [71].
ROS are produced as intermediates in reduction–oxidation (redox) reactions of O2 to H2O and comprise two major groups: free radicals – superoxide (.O2−), hydroxyl (OH−) and nitric oxide –, which are highly reactive and unstable; and non-radical derivatives of O2, for example, H2O2, which is less reactive and with a longer half-life than free radicals (reviewed in [72]). ROS are produced in the brain, heart, kidneys and vasculature. It is now recognized that specific enzymes, the NOX family of NADPH oxidases, may have as their sole function generation of ROS in a highly regulated fashion under physiological conditions. However, the physiological production of ROS can also occur as a by-product of other biological pathways such as peroxisomes, cytochrome P450, xanthine oxidase, uncoupled NOS, cyclooxygenase, lipoxygenase, heme oxygenases and especially mitochondrial enzymes [72]. Besides their key role in bioenergetics and ATP synthesis, it appears that mitochondria are one of the main sites of ROS generation within the cell. 80% of intracellular superoxide anion is generated by leakiness of the electron transport chain [73]. In quiescent cells, individual mitochondria undergo spontaneous bursts of superoxide generation, termed ‘superoxide flashes’[74]. Mitochondrial ROS production is similar during normoxia and hypoxia but markedly increases during reoxygenation in cardiac myocytes [74, 75]. Similar results were obtained in cortical mouse neurons exposed to IH [76] suggesting the repeated hypoxia/reoxygenation of IH leads to significant increases in ROS generation.
ROS, via oxidation of reactive cysteine residues, can activate ion channels (Ca2+ and K+ channels), redox-sensitive kinases (Src, Akt, protein kinase C [PKC], MAP kinases and ρ kinase), protein tyrosine phosphatases such as SH2-domain containing phosphatase and MAP kinase (MAPK) phosphatase, and transcription factors (nuclear factor-κ-light chain enhancer of activated B cells [NF-κB], activator protein-1 [AP-1], nuclear factor of activated T cells [NFAT][77–82], p53, Ets and hypoxia-inducible factor-1 [HIF-1]) [72].
It has been shown that IH increases in ROS production scavenge nitric oxide reducing its bioavailability leading to impaired dilation in skeletal and cerebral blood vessels [24, 26]. Impaired vasodilation contributes to hypertension. However, other studies have shown that IH leads to increased vasodilatory ROS causing a reduction in myogenic tone in skeletal muscle arterioles [83]. On the contrary, we have observed that IH augments myogenic tone in mesenteric arteries that is endothelium and ROS dependent (unpublished observations).
In spite of these disparate results the literature clearly supports a role for ROS in IH-induced hypertension. Administration of a superoxide dismutase mimetic (tempol) prevents IH-induced increases in plasma ET-1 levels and blood pressure (Fig. 1) [63].
Fig 1.
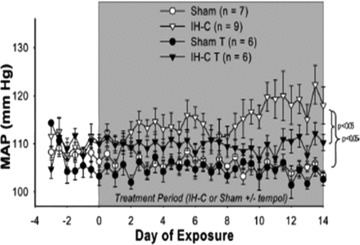
MAP was recorded daily for 3 baseline days and 14 treatment days in eucapnic intermittent hypoxia (IH-C) ± Tempol and Sham ± Tempol (T). MAP increased in IH-C-vehicle rats over the 14 days of treatment. MAP did not increase in IH-C rats treated with Tempol (1 mM). Tempol did not affect MAP in sham-operated rats. *P < 0.05, significant difference from Sham/vehicle. Reprinted with permission from Carmen M. Troncoso Brindeiro et al. [63].
Hypoxia has been shown to induce ET-1 transcription in several cell types including cardiomyocytes and endothelial cells [84–87]. In addition, ROS increases ET-1 mRNA and peptide levels in endothelial, smooth muscle and CB (glomus) cells [88, 89]. Consistent with a role of ET-1 as a contributor to IH-induced hypertension, chronic administration of an ETAR blocker (BQ123) prevents IH-induced increases in MAP in rats [68].
Our group has also demonstrated that small resistant arteries from IH-exposed rats have selective increased constrictor reactivity to ET-1 [67]. The intracellular signalling pathways that mediates ET-1 vasoconstriction are altered in IH exposed compared with air exposed rats whereas the mechanisms that mediates phenylephrine constriction are unaltered. The mechanism involves activation of PKC delta-mediated Ca2+ sensitization [90].
ROS can also increase angiotensinogen synthesis contributing to the activation of the renin-angiotensin system (RAS) [91, 92]. Interestingly, Ang II by increasing ROS production leads to up-regulation of ET-1 [93, 94]. Therefore, ROS has not only been implicated as activator of these systems but also as part of the signalling downstream of both ET-1 [95] and Ang II [93, 96–98] in the vasculature. In summary, oxidative stress and ET-1 are main contributors to the aetiology of IH-induced hypertension.
The role of Ang II in the increased blood pressure observed in animals exposed to IH and in OSA patients is still controversial. Some studies clearly demonstrate activation of RAS in animals [64] and OSA patients [99, 100]. However other studies have shown no changes in the components of RAS in patients [101–103] and that administration of an Ang II receptor inhibitor (losartan) does not prevent IH-induced hypertension in rats [37].
Although it may not be acting as a pressor agent, elevated plasma levels of Ang II in OSA patients appears to contribute to increases in plasma levels of vascular endothelial growth factor (VEGF) [100, 104]. VEGF is a potent angiogenic cytokine that also contributes to atherogenesis. Therefore, it is possible that Ang II through VEGF contributes to angiogenesis, endothelium repair and atherosclerosis more than to hypertension in OSA.
Role of transcription factors in the inflammatory and cardiovascular consequences of IH
Increases in inflammatory factors have been associated with induction of vascular damage and hypertension. Three transcription factors – NF-κB, NFAT and HIF-1– have been implicated in the initiation of the inflammatory response and the associated vascular damage induced by IH.
NF-κB
The NF-κB is a key mediator of the inflammatory response under many conditions. NF-κB is a member of the Rel family of proteins and was first described as a nuclear factor necessary for immunoglobulin k light chain transcription in B-cells [105]. NF-κB exists in the cytosol in an inactive form bound to the inhibitor, IκB. An appropriate endogenous or exogenous inflammatory stimulus triggers IκB ubiquitin-mediated degradation releasing NF-κB to translocate to the nucleus. In the nucleus, NF-κB up-regulates the transcription of several pro-inflammatory genes responsible for encoding inflammatory cytokines (tumour necrosis factor [TNF]-α, interleukin [IL]-6 and IL-8), chemokines, surface adhesion molecules (intercellular adhesion molecule-1) and enzymes (cyclo-oxygenase-2) (reviewed in [106]).
There is evidence suggesting IH increases NF-κB activity and its downstream products, particularly TNF-α in cultured cell, animal models and OSA patients. TNF-α has been shown to play a major role in inflammation-mediated vascular damage in OSA patients and animal models (reviewed in [107]).
Cultured HeLa cells (an epithelial cell line) and bovine aortic endothelial cells exposed to IH show activation of NF-κB [108]. IH activation of NF-κB is dependent on IκB kinase and p38 MAPK activity [109].
The same group also demonstrated increases in TNF-α and the NF-κB-dependent cytokine, IL-8 in plasma of OSA patients [108, 110]. In mice, IH increased the expression of NF-κB -dependent gene product, iNOS, in cardiovascular tissues (heart, aorta and lung) of mice [111]. Finally, OSA patients have significantly elevated monocyte and neutrophil NF-κB activity, which was normalized following elimination of apnoeas with continues positive airway pressure (CPAP) therapy [111, 112].
Interestingly, NF-κB is a redox-sensitive transcription factor [113, 114] that can initiate inflammatory and immune responses by promoting activation of endothelial cells, leucocytes and platelets. These activated cells express adhesion molecules and pro-inflammatory cytokines that may contribute to endothelial injury and dysfunction and consequently to the development of cardio- and cerebro-vascular morbidities in IH.
NFAT
Another member of the Rel family of transcription factors that has recently been implicated in the development of hypertension in IH exposed mice is the nuclear factor of activated T cells (NFAT) (Fig. 2) [29]. First identified in activated T cells, the calcium (Ca2+)-dependent transcription factor, NFAT, has since been shown to play a role in non-immune cells, including cells of the cardiovascular system (reviewed in [115]). The NFAT transcription factor family is comprised four well-characterized members, designated NFATc1 (NFAT2/c), NFATc2 (NFAT1/p), NFATc3 (NFAT4/x) and NFATc4 (NFAT3). The signature of this family is a shared NFAT homology region in the N-terminal portion of the protein. This region mediates binding of the Ca2+-activated phosphatase calcineurin and other regulatory functions necessary for nuclear translocation and contains nuclear localization and nuclear export sequences along with phosphorylation sites for a number of serine/threonine kinases. The C-terminal region of NFAT proteins contains a DNA-binding domain. NFAT activation is regulated primarily through control of its subcellular localization. In unstimulated cells, NFAT is a hyperphosphorylated cytosolic protein. An elevation in intracellular Ca2+, induced by a variety of mechanisms, increases the activity of the Ca2+-calmodulin-dependent phosphatase, calcineurin. Activated calcineurin dephosphorylates the regulatory region of NFAT, inducing a conformational change that exposes nuclear localization signals and allows import of NFAT into the nucleus (reviewed in [115, 116]). NFAT binds DNA with very low affinity in the absence of a cofactor so that formation of an NFAT-cofactor-DNA ternary complex is required for significant NFAT-mediated transcriptional activity. Egr-1 [117, 118], activator protein 1 (AP-1) [119, 120], YY1 [121], the MADS family member – MEF2 [122–124] and members of the GATA family [125, 126] (reviewed in [127]) have been shown to be NFAT cofactors.
Fig 2.
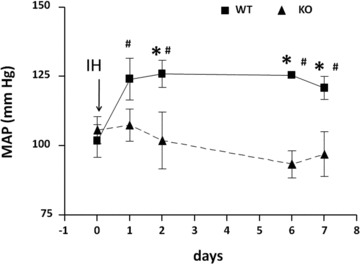
MAP values, including those for the day before IH exposure (day 0 = air exposure), for NFATc3 wild-type and knockout mice. *P < 0.05 versus knockout; #P < 0.05 versus day 0 (two-way repeated-measures anova and Holm–Sidak test). Reprinted with permission from Sergio de Frutos et al. [29].
NFAT activation is generally associated with an increase in gene expression [116, 127]. However, several reports have shown that NFAT can also function as a repressor of gene expression [128–131].
Activators of Gq-couple receptors (i.e. ET-1, Ang II, catecholamines, uridine 5′-triphosphate), elevated plasma glucose levels and increased intravascular pressure are potent activators of NFAT [115, 132–136]. NFAT activation not only plays an important role in modulating the immune response and inflammation [116], it is also important in vascular development [137, 138], maintenance of the contractile phenotype of vascular smooth muscle cells (VSMC) [126, 139, 140] and regulation of VSMC contractility through the down-regulation of voltage-dependent (KV 2.1) channel and large conductance (BK) potassium channels [128, 131, 141].
Smooth muscle-specific expression of NFAT isoforms has been described. NFATc1 and NFATc2 were the first reported in thoracic aortic smooth muscle cells from rat [142]. More recently human aortic smooth muscle was shown to express NFATc1 and c3, but not NFATc2 or c4 [126]. Extensive studies of the cerebral vasculature reveal NFATc3 and c4, but not NFATc1 or c2 [132–135]. A recent study in the rat pulmonary circulation shows that NFATc4 is the only isoform expressed in PASMC, whereas we showed that NFATc3 is expressed in murine pulmonary arteries [143] and mesenteric arteries [29].
We have recently shown that systemic hypertension caused by IH in mice is associated with NFATc3 activation in aorta and mesenteric arteries [29]. In the same study, we demonstrated that genetic ablation or pharmacological inhibition of NFATc3 prevents IH-induced hypertension (Fig. 2), demonstrating the importance of NFAT in IH-induced cardiovascular pathology. We have also found that NFATc3 is required for IH-induced mesenteric arterial remodelling with increased expression of the hypertrophic/differentiation marker smooth muscle α-actin (unpublished observations). Smooth muscle α-actin has been identified as a gene regulated by NFATc3 in cooperation with serum response factor and AP-1 [140, 143].
NFAT is also an inflammatory modulator that regulates the expression of TNF-α[144], IL-6 [145, 146] and osteopontin (OPN) [147]. TNF-α, described above, and IL-6 are clearly elevated in patients with SDB [148], suggesting they may be mediators of SDB-associated cardiovascular disease. In particular, IL-6 appears to contribute to atherogenesis [149] and is associated with higher mortality in patients with coronary arterial disease and a higher risk for myocardial infarction in otherwise healthy men [150, 151].
OPN is a multifunctional protein important in proliferation, migration and remodelling in the vasculature. OPN is also linked to multiple human diseases involving systemic inflammation and autoimmunity [152]. OPN plasma levels are increased in hypertension [153, 154], coronary artery disease [155] and by hypoxia [156]. OPN expression is induced by a variety of growth factors and cytokines including IL-1, TNF-α and Ang II [157–159]. In models of systemic inflammatory response, OPN levels have been found to directly correlate with secretion of IL-6 [160]. Interestingly, ROS also enhance expression of OPN in VSMC [161, 162].
ROS also activate NFAT in many different cell types including neuronal-like cells, immune cells, osteoclasts and cardiac myocytes [77–82]. The mechanism by which ROS activates NFAT has not been completely established but, in cardiomyocytes, H2O2 induces a time- and dose-dependent activation of NFAT [81]. This activation appears to be mediated by ERK-dependent activation of AP-1, a cofactor of NFAT. This evidence further supports a role for ROS as an upstream activator of a complex signalling pathway initiated by IH that leads to changes in inflammatory and vascular-specific gene expression.
HIF-1
Hypoxia-inducible factor (HIF)-1 is a heterodimeric transcription factor formed by a constitutively expressed β-subunit and an α-subunit that contains an oxygen-dependent ubiquitin- proteasomal degradation domain [163, 164]. Therefore, HIF-1 is suppressed in normoxia.
However, in hypoxia, HIF-1 is stabilized by inhibition of its degradation allowing binding to the regulatory regions of its target genes and regulation of their expression [165]. HIF-1 is the major regulator of oxygen homeostasis within cells by mediating the shift to increased glycolysis and anaerobic metabolism at low oxygen tensions through down-regulation of mitochondrial oxygen consumption [166]. One of the first genes shown to be regulated by HIF-1 was erythropoietin (EPO) whose gene promoter contains hypoxia response elements. Up-regulation of EPO leads to increased red blood cell production and enhanced blood oxygen-carrying capacity [167]. Studies from our group have shown that IH without CO2 supplementation (hypocapnia) increases haematocrit in rats [168]. However, IH with CO2 supplementation (eucapnia) does not increase haematocrit in either rats [168] or mice [29] similar to OSA patients that have no or modest elevation in haematocrit [169]. These results suggest that HIF-1-mediated EPO up-regulation is blunted under hypoxic-eucapnic conditions.
HIF-1 also contributes to inflammation by increasing the survival of pro-inflammatory cells [170].
Another HIF-1 gene product is VEGF, which it has been demonstrated to be elevated in OSA patients with severe nocturnal hypoxemia [171]. VEGF induces migration of mature endothelial cells towards hypoxic areas of tissue, thereby promoting angiogenesis [172]. IH increases HIF-1 in endothelial cells in culture [173, 174] to mediate cell migration and promote an angiogenic phenotype [174]. Therefore, HIF-1-mediated VEGF up-regulation could be involved in IH-induced angiogenesis.
Interestingly, HIF-1α partially deficient mice do not display the typical cardio-respiratory response to IH. Specifically, IH did not: elicit sLTF of the chemoreceptor activity, augment hypoxic ventilatory response, cause LTF of breathing, elevate blood pressure, elevate brain ROS levels, up-regulate brain HIF-1α or increase plasma noradrenaline [45]. This study suggests that IH involves complex positive interactions between HIF-1 and ROS.
The contribution of HIF-1α to IH-induced hypertension may involve up-regulation of ET-1. The pre-pro ET-1 promoter contains hypoxia response elements and it has been demonstrated that pre-pro ET-1 transcription is increased by hypoxia via recruitment of HIF-1, AP-1, GATA-2, CAAT-binding factor (NF-1) and CREB binding protein (p300/CBP) to the transcripsome [175–177]. In addition, IH-induced ET-1 up-regulation in the CB requires HIF-1 [178]. Consistent with a role for HIF-1 and ET-1 in IH, Belaidi, et al. recently demonstrated a major role for HIF-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of OSA [69]. They found that IH accelerates the development of hypertension and enhances infarct size in SHR compared with that seen in control rats. This was accompanied by an increase in myocardial pre-pro ET-1 levels, ET-1 protein and ET-A receptor expression and by enhanced coronary vascular reactivity to ET-1 in SHR only. Myocardial HIF-1 activity was also increased and in vivo chromatin immunoprecipitation assay on myocardial extracts provided evidence that HIF-1α targets the ET-1 gene in SHR after IH. Moreover, administration of bosentan, a mixed ET receptor antagonist, during IH prevented both the increase in blood pressure and in infarct size. This study demonstrates that activation of the ET system, mediated by HIF-1 activity, is responsible for the enhanced susceptibility to IH and for its associated cardiovascular consequences leading to hypertension and ischemic injury.
Yuan G, et al. have investigated the molecular mechanism of HIF-1α up-regulation during IH [179]. The mechanism includes both increased mTOR-dependent HIF-1α synthesis during re-oxygenation and decreased hydroxylase-dependent HIF-1α degradation during hypoxia. HIF-1α up-regulation also appears to require increased generation of ROS by NOX. Anti-oxidants prevented IH decreased hydroxylation of HIF-1α protein. Ca2+-dependent activation of phospholipase Cγ and PKC are also required for decreased HIF-1α degradation.
There is also an active NF-κB binding site in the proximal promoter site of the HIF-1 gene and NF-κB appears to regulate basal levels of HIF-1 gene expression [180]. Indeed, hypoxia up-regulation of HIF-1 transcription appears to be through a NF-κB-dependent mechanism and vice versa [180–182]. Interestingly these studies further established a role for ROS as a factor that contributes to the significant crosstalk between NF-κB and HIF-1, suggesting mutual dependence between the pathways. The O2 regulated α subunit of HIF-2α, also known as endothelial PAS domain protein-1, is another member of the HIF family [183]. HIF-2α shares 80% sequence homology to HIF-1α and also interacts with HIF-1β[184]. Similar to HIF-1, continuous hypoxia leads to HIF-2α accumulation and the resulting transcriptional activation of VEGF [185]. In contrast to the up-regulation of HIF-1α, it has been shown that IH down-regulates HIF-2α protein levels via activation of calpain proteases [186]. IH-evoked HIF-2α degradation led to inhibition of SOD2 transcription, resulting in greater oxidative stress. Systemic administration of calpain inhibitors restored HIF-2α levels in IH-exposed rats and prevented IH-induced oxidative stress and cardiovascular abnormalities.
Together these studies suggest that IH activates HIF-2α leading to increases in ROS, which act in concert with the low O2 levels to up-regulate HIF-1 and contribute to IH-induced hypertension.
Intermittent hypoxia induced pulmonary hypertension
Pulmonary hypertension (PH) is an additional cardiovascular complication of OSA. Patients with OSA demonstrate cyclical increases in pulmonary arterial pressure coinciding with apnoeic episodes, which has been largely attributed to acute hypoxic pulmonary vasoconstriction (HPV) [187, 188]. OSA additionally exacerbates pulmonary hypertension in patients with COPD (a condition termed ‘overlap syndrome’), and CPAP is effective in reducing the severity of pulmonary hypertension in these individuals [189, 190]. However, whether OSA directly causes sustained daytime pulmonary hypertension in the absence of co-existing chronic lung disease or obesity-related hypoventilation is debated. Interestingly, recent studies suggest that OSA is an independent risk factor in PH [187, 191–193], with an incidence of approximately 20–40% in patients without clinically identifiable cardiopulmonary disease [187, 190–197]. Supporting this finding is evidence that CPAP reduces daytime pulmonary arterial pressure in pulmonary hypertensive OSA patients without significant heart disease, lung disease [191–193] or daytime hypoxemia [191, 192]. This relationship between OSA and PH has been reviewed extensively elsewhere [187, 189, 190, 198]. Despite the growing recognition of the prevalence of this disorder, the cardiovascular sequelae leading to the progression of OSA-induced PH in human beings remain poorly characterized. However, animal models of OSA that involve chronic exposure to IH have begun to address these mechanisms of PH.
Many of the rodent models of OSA reproduce not only the systemic hypertension characteristic of OSA [23, 63, 67, 68] as detailed earlier in this review, but provide evidence for PH as well [27, 168, 199–202]. Mechanisms of IH-induced PH are generally considered to be parallel to those associated with chronic sustained hypoxia (CH), and include pulmonary vasoconstriction, vascular remodelling (characterized by smooth muscle hypertrophy and hyperplasia in small pulmonary arteries) and polycythemia (Fig. 3). The degree of pulmonary hypertension closely correlates with right ventricular hypertrophy [203], an adaptive response to the increase in afterload. Although clinical manifestations of right heart failure and peripheral oedema resulting from severe PH can develop in patients with overlap syndrome, such complications are uncommon in individuals with OSA uncomplicated by concomitant lung disease [187, 190–197].
Fig 3.
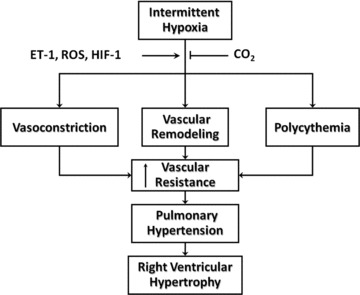
Proposed mechanisms of IH-induced pulmonary hypertension. ET-1, ROS and HIF-1 may play important roles in the vasoconstrictor, remodelling and polycythemic components of pulmonary hypertension. Supplemental CO2 blunts IH-induced right ventricular hypertrophy, arterial remodelling and polycythemia.
Whether CH and IH produce PH through similar mechanisms is not clear, but studies comparing the two models suggest that the PH resulting from IH is less severe. For example, Fagan [200] found that mice exposed to CH (barometric pressure = 410 mmHg, 4 weeks) demonstrate greater increases in right ventricular systolic pressure compared to IH mice (2 min. cycles of 10%/21% O2, 8 hrs/day, 4 weeks). Consistent with these findings, right ventricular hypertrophy was greater in CH versus IH animals, although no significant differences were observed between groups with respect to the degree of polycythemia or neomuscularization of small pulmonary arteries. A recent study from our group demonstrates a similar effect of IH (3 min. cycles of 5%/21% O2, 7 hrs/day, 2 weeks) to increase pulmonary arterial wall thickness, haematocrit, and right ventricular weight in rats [168]. However, the severity of these responses again tended to be reduced compared to effects of sustained CH (barometric pressure = 380 mmHg, 2 weeks).
Vasoconstrictor responses associated with IH have not been well studied, although HPV likely contributes to increases in pulmonary arterial pressure during hypoxic episodes, and possibly to sustained hypertension in OSA patients that exhibit daytime hypoxemia. However, it is possible that HPV-independent vasoconstrictor mechanisms additionally contribute to PH following IH. Indeed, vasoconstriction appears to play a central role in mediating PH associated with CH, evident by effects of vasodilators to dramatically lower pulmonary vascular resistance in CH rats that have been acutely returned to a normoxic environment [204, 205]. Recent studies from our group and others suggest that the vasoconstrictor response to sustained CH may be multifaceted, with contributions of both spontaneous pulmonary arterial tone [206] and enhanced agonist-dependent vasoconstriction [168, 207, 208]. Whereas IH also enhances pulmonary vasoreactivity to the thromboxane mimetic, U-46619, the magnitude of this response is markedly blunted compared to that observed with CH [168]. Additional research is needed to define the contribution of vasoconstriction to IH-induced pulmonary hypertension, and whether increased vasoreactivity in this setting is the result of altered VSM signal transduction or endothelial dysfunction, both of which occur following CH [205–207, 209, 210].
Previous studies from our group have identified ET-1 as a critical mediator of IH-induced systemic hypertension in rats [23, 68] (discussed above). Given the well established contribution of ET-1 to CH-induced PH [211–213], that circulating ET-1 levels are elevated in IH rats [23], and pre-pro ET-1 mRNA is up-regulated in lungs from IH mice [29] a distinct possibility is that ET-1 similarly contributes to IH-induced PH. Future studies are needed to assess the potential role of ET-1 in this response, and potential influences of ET-1 on the remodelling and vasoconstrictor responses to IH. In agreement with evidence that IH augments pulmonary vasoconstrictor sensitivity to U-46619 [168], preliminary studies from our group indicate that IH similarly augments VSM reactivity to ET-1 in pressurized small pulmonary arteries from rats through a PKC signalling pathway [214]. Interestingly, this mechanism of increased PKC-dependent vasoconstriction following IH stands in marked contrast to our findings of enhanced ρ kinase-mediated myofilament Ca2+ sensitivity in the pulmonary circulation of CH rats [207]. Whether such increases in pulmonary vasoconstrictor sensitivity to ET-1 contribute to IH-dependent PH remains to be established.
Considering the contribution of ROS to IH-induced systemic hypertension [63], and the well-established role of ROS in various models of hypoxic PH [215–217], a reasonable hypothesis is that ROS similarly contribute to the development of IH-induced pulmonary hypertension. NOX [215, 218–221] and xanthine oxidase [222, 223] have been the major sources of ROS implicated in the development of pulmonary hypertension. Indeed, a role for NOX in IH-dependent PH has recently been reported by Nisbet and colleagues [201], who found that the NOX subunits NOX4 and p22phox and ROS levels are increased in lungs from IH mice (1.3 min. cycles of 10%/21% O2, 8 hrs/day, 5 days/ week, 8 weeks). Evidence from this same study that IH-induced RV hypertrophy and arterial remodelling are abolished in NOX2 (gp91phox) knockout mice further supports a major contribution of NOX2-derived ROS in this pulmonary hypertensive response.
The mechanisms by which ROS mediate IH-dependent pulmonary hypertension are not understood, but may involve both enhanced pulmonary vasoconstrictor sensitivity [207, 214] and increases in gene expression secondary to activation of HIF-1 [179]. Consistent with the latter possibility and as reviewed above, studies using HIF-1α heterozygous mice suggest that HIF-1-mediated gene expression is central to not only the development of CH-induced PH [224], but to CB-mediated ROS generation and associated cardiorespiratory responses to IH as well [45]. Whether a functional link exists between NOX, HIF-1 activity and vasoconstrictor sensitivity in the development of IH-induced pulmonary hypertension remains to be determined.
Apnoeic episodes in patients with SDB are associated not only with hypoxemia but with hypercapnia as well [225]. Therefore, studies that employ a purely IH stimulus without provision of supplemental CO2 do not accurately reflect the blood gas changes that occur during apnoeic episodes in OSA patients. Rather, the hypoxic increases in ventilation resulting from IH alone lead to cyclical reductions in arterial Pco2 and consequent alkalemia [168]. To more closely mimic the arterial blood gas and pH changes observed in patients with sleep apnoea, studies in our group have begun to compare effects of IH with and without supplemental CO2 on indices of pulmonary hypertension, polycythemia and pulmonary vasoreactivity. Interestingly, we found that supplemental CO2 significantly reduces the arterial remodelling, polycythemic and right ventricular hypertrophic responses to IH [168]. These results are consistent with earlier studies demonstrating a protective influence of CO2 on the development of CH-induced pulmonary hypertension [226, 227]. Although the mechanisms by which CO2 reduces the severity of PH are not understood, it is possible that supplemental CO2 leads to suppression of oxidant stress [227] and hypoxia-inducible gene expression through an as yet unidentified mechanism.
In summary, animal models of SA involving chronic exposure to IH have provided new insight into the complex aetiology of SA-associated PH. Although these models of PH share some characteristics of those involving CH, some notable differences have been revealed with respect to the severity of PH and intracellular signalling pathways responsible for enhanced vasoconstrictor sensitivity. In addition, important new questions have been raised regarding the mechanisms that mediate both vasoconstrictor and arterial remodelling components of IH-dependent PH, including the roles of ET-1, ROS, PKC and HIF-1-induced gene expression. Neither is it understood how CO2 blunts the pulmonary hypertensive response to IH, nor whether a similar protective influence of CO2 exists in human beings. Animal models of PH will continue to improve our understanding of potential differences in the pathogenesis of CH versus IH-induced PH, which in turn is necessary to indentify effective therapeutic strategies to treat these conditions.
Conclusions
Rodent models of IH have provided a tremendous advance in our understanding of the cardiovascular alterations observed in patients with SDB.
After reviewing the literature on this field is clear that ROS plays a major role in the pathogenesis of the cardiovascular morbidities induced by IH and also present in SDB patients. We are proposing a working model where ROS is the activator of the main factors enhanced by IH but also is part of the intracellular signalling pathways initiated by these factors (Fig. 4), which contribute to inflammation, endothelial dysfunction, vascular remodelling and increased vasoconstriction leading to hypertension.
Fig 4.
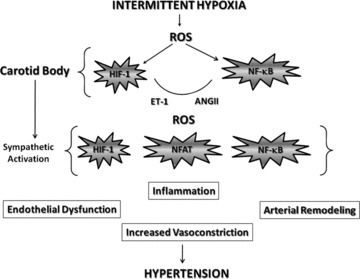
Working model of the mechanisms of IH-induced hypertension. ROS = reactive oxygen species; ET-1 = endothelin-1; ANG II = angiotensin II; HIF-1 = hypoxia inducible factor-1; NF-κB = nuclear factor-κ-light chain enhancer of activated B cells; NFAT = nuclear factor of activated T cells.
Acknowledgments
Funding sources: L.V.G.B. from AHA (0535347N SDG) and NHLBI (R01 HL088151); T.R. from NHLBI (R01 HL88192); B.W. from NHLBI (R01 HL 58124) and N.L.K. from EPA (STAR award 83186001) and NHLBI (R01 HL82799).
References
- 1.Brooks D, Horner RL, Kozar LF, et al. Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest. 1997;99:106–9. doi: 10.1172/JCI119120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jun J, Polotsky VY. Metabolic consequences of sleep-disordered breathing. ILAR J. 2009;50:289–306. doi: 10.1093/ilar.50.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasali E, Ip MSM. Obstructive sleep apnea and metabolic syndrome: alterations in glucose metabolism and inflammation. Proc Am Thorac Soc. 2008;5:207–17. doi: 10.1513/pats.200708-139MG. [DOI] [PubMed] [Google Scholar]
- 4.Kanagy NL. Vascular effects of intermittent hypoxia. ILAR J. 2009;50:282–8. doi: 10.1093/ilar.50.3.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dematteis M, Julien C, Guillermet C, et al. Intermittent hypoxia induces early functional cardiovascular remodeling in mice. Am J Respir Crit Care Med. 2008;177:227–35. doi: 10.1164/rccm.200702-238OC. [DOI] [PubMed] [Google Scholar]
- 6.Fletcher EC, Bao G. The rat as a model of chronic recurrent episodic hypoxia and effect upon systemic blood pressure. Sleep. 1996;19:S210–2. [PubMed] [Google Scholar]
- 7.Neubauer JA. Physiological and genomic consequences of intermittent hypoxia: invited review: physiological and pathophysiological responses to intermittent hypoxia. J Appl Physiol. 2001;90:1593–9. doi: 10.1152/jappl.2001.90.4.1593. [DOI] [PubMed] [Google Scholar]
- 8.Savransky V, Bevans S, Nanayakkara A, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–7. doi: 10.1152/ajpgi.00145.2007. [DOI] [PubMed] [Google Scholar]
- 9.Sica AL, Greenberg HE, Ruggiero DA, et al. Chronic-intermittent hypoxia: a model of sympathetic activation in the rat. Respir Physiol. 2000;121:173–84. doi: 10.1016/s0034-5687(00)00126-2. [DOI] [PubMed] [Google Scholar]
- 10.Tahawi Z, Orolinova N, Joshua IG, et al. Physiological and genomic consequences of intermittent hypoxia: selected contribution: altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol. 2001;90:2007–13. doi: 10.1152/jappl.2001.90.5.2007. [DOI] [PubMed] [Google Scholar]
- 11.Tarasiuk A, Scharf SM. Cardiovascular effects of periodic obstructive and central apneas in dogs. Am J Respir Crit Care Med. 1994;150:83–9. doi: 10.1164/ajrccm.150.1.8025778. [DOI] [PubMed] [Google Scholar]
- 12.Al Mobeireek AF, Al Kassimi FA, Al Majed SA, et al. Clinical profile of sleep apnea syndrome. A study at a university hospital. Saudi Med J. 2000;21:180–3. [PubMed] [Google Scholar]
- 13.Bananian S, Lehrman SG, Maguire GP. Cardiovascular consequences of sleep-related breathing disorders. Heart Dis. 2002;4:296–305. doi: 10.1097/00132580-200209000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Barthlen GM. Sleep disorders. Obstructive sleep apnea syndrome, restless legs syndrome, and insomnia in geriatric patients. Geriatrics. 2002;57:34–9. [PubMed] [Google Scholar]
- 15.Berger HA, Somers VK, Phillips BG. Sleep disordered breathing and hypertension. Curr Opin Pulm Med. 2001;7:386–90. doi: 10.1097/00063198-200111000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Fogel RB, White DP. Obstructive sleep apnea. Adv Intern Med. 2000;45:351–89. [PubMed] [Google Scholar]
- 17.Goodday RH, Percious DS, Morrison AD, et al. Obstructive sleep apnea syndrome: diagnosis and management. J Can Dent Assoc. 2001;67:652–8. [PubMed] [Google Scholar]
- 18.Waldhorn RE. Sleep apnea syndrome. Am Fam Physician. 1985;32:149–66. [PubMed] [Google Scholar]
- 19.Fletcher EC. Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol. 2001;90:1600–5. doi: 10.1152/jappl.2001.90.4.1600. [DOI] [PubMed] [Google Scholar]
- 20.Fletcher EC, Lesske J, Qian W, et al. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992;19:555–61. doi: 10.1161/01.hyp.19.6.555. [DOI] [PubMed] [Google Scholar]
- 21.Greenberg HE, Sica A, Batson D, et al. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- 22.Kraiczi H, Magga J, Sun XY, et al. Hypoxic pressor response, cardiac size, and natriuretic peptides are modified by long-term intermittent hypoxia. J Appl Physiol. 1999;87:2025–31. doi: 10.1152/jappl.1999.87.6.2025. [DOI] [PubMed] [Google Scholar]
- 23.Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension. 2001;37:511–5. doi: 10.1161/01.hyp.37.2.511. [DOI] [PubMed] [Google Scholar]
- 24.Tahawi Z, Orolinova N, Joshua IG, et al. Altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol. 2001;90:2007–13. doi: 10.1152/jappl.2001.90.5.2007. [DOI] [PubMed] [Google Scholar]
- 25.Peng Y, Yuan G, Overholt JL, et al. Systemic and cellular responses to intermittent hypoxia: evidence for oxidative stress and mitochondrial dysfunction. Adv Exp Med Biol. 2003;536:559–64. doi: 10.1007/978-1-4419-9280-2_71. [DOI] [PubMed] [Google Scholar]
- 26.Phillips SA, Olson EB, Jr, Morgan BJ, et al. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and sckeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2003;286:H388–93. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- 27.Campen MJ, Shimoda LA, O’Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J Appl Physiol. 2005;99:2028–35. doi: 10.1152/japplphysiol.00411.2005. [DOI] [PubMed] [Google Scholar]
- 28.Lai CJ, Yang CCH, Hsu YY, et al. Enhanced sympathetic outflow and decreased baroreflex sensitivity are associated with intermittent hypoxia-induced systemic hypertension in conscious rats. J Appl Physiol. 2006;100:1974–82. doi: 10.1152/japplphysiol.01051.2005. [DOI] [PubMed] [Google Scholar]
- 29.De Frutos S, Duling L, Alo D, et al. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H2382–90. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savransky V, Nanayakkara A, Li J, et al. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–7. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soukhova-O’Hare GK, Ortines RV, Gu Y, et al. Postnatal intermittent hypoxia and developmental programming of hypertension in spontaneously hypertensive rats: the role of reactive oxygen species and L-Ca2+ channels. Hypertension. 2008;52:156–62. doi: 10.1161/HYPERTENSIONAHA.108.110296. [DOI] [PubMed] [Google Scholar]
- 32.Tamisier R, Gilmartin GS, Launois SH, et al. A new model of chronic intermittent hypoxia in humans: effect on ventilation, sleep and blood pressure. J Appl Physiol. 2009;107:17–24. doi: 10.1152/japplphysiol.91165.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braga VA, Soriano RN, Machado BH. Sympathoexcitatory response to peripheral chemoreflex activation is enhanced in juvenile rats exposed to chronic intermittent hypoxia. Exp Physiol. 2006;91:1025–31. doi: 10.1113/expphysiol.2006.034868. [DOI] [PubMed] [Google Scholar]
- 34.Dick TE, Hsieh YH, Wang N, et al. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol. 2007;92:87–97. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- 35.Fletcher EC. Effect of episodic hypoxia on sympathetic activity and blood pressure. Respir Physiol. 2000;119:189–97. doi: 10.1016/s0034-5687(99)00114-0. [DOI] [PubMed] [Google Scholar]
- 36.Lesske J, Fletcher EC, Bao G, et al. Hypertension caused by chronic intermittent hypoxia–influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- 37.Zoccal DB, Bonagamba LGH, Oliveira FoRT, et al. Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol. 2007;92:79–85. doi: 10.1113/expphysiol.2006.035501. [DOI] [PubMed] [Google Scholar]
- 38.Zoccal DB, Simms AE, Bonagamba LGH, et al. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–65. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar GK, Rai V, Sharma SD, et al. Chronic intermittent hypoxia induces hypoxia-evoked catecholamine efflux in adult rat adrenal medulla via oxidative stress. J Physiol. 2006;575:229–39. doi: 10.1113/jphysiol.2006.112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bao G, Metreveli N, Li R, et al. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol. 1997;83:95–101. doi: 10.1152/jappl.1997.83.1.95. [DOI] [PubMed] [Google Scholar]
- 41.Kuri BA, Khan SA, Chan SA, et al. Increased secretory capacity of mouse adrenal chromaffin cells by chronic intermittent hypoxia: involvement of protein kinase C. J Physiol. 2007;584:313–9. doi: 10.1113/jphysiol.2007.140624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Souvannakitti D, Kumar GK, Fox A, et al. Neonatal intermittent hypoxia leads to long-lasting facilitation of acute hypoxia-evoked catecholamine secretion from rat chromaffin cells. J Neurophysiol. 2009;101:2837–46. doi: 10.1152/jn.00036.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soukhova-O’Hare GK, Cheng ZJ, Roberts AM, et al. Postnatal intermittent hypoxia alters baroreflex function in adult rats. Am J Physiol Heart Circ Physiol. 2006;290:H1157–64. doi: 10.1152/ajpheart.00767.2005. [DOI] [PubMed] [Google Scholar]
- 44.Peng YJ, Overholt JL, Kline D, et al. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci USA. 2003;100:10073–8. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peng YJ, Yuan G, Ramakrishnan D, et al. Heterozygous HIF-1{alpha} deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol Online. 2006;577:705–16. doi: 10.1113/jphysiol.2006.114033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenberg HE, Sica AL, Scharf SM, et al. Expression of c-fos in the rat brainstem after chronic intermittent hypoxia. Brain Res. 1999;816:638–45. doi: 10.1016/s0006-8993(98)01222-0. [DOI] [PubMed] [Google Scholar]
- 47.Machaalani R, Waters KA. Increased neuronal cell death after intermittent hypercapnic hypoxia in the developing piglet brainstem. Brain Res. 2003;985:127–34. doi: 10.1016/s0006-8993(03)03003-8. [DOI] [PubMed] [Google Scholar]
- 48.Sica AL, Greenberg HE, Scharf SM, et al. Immediate-early gene expression in cerebral cortex following exposure to chronic-intermittent hypoxia. Brain Res. 2000;870:204–10. doi: 10.1016/s0006-8993(00)02170-3. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Y, Fenik P, Zhan G, et al. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci. 2007;27:10060–71. doi: 10.1523/JNEUROSCI.0857-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogawa S, Kitao Y, Hori O. Ischemia-induced neuronal cell death and stress response. Antioxid Redox Signal. 2007;9:573–87. doi: 10.1089/ars.2006.1516. [DOI] [PubMed] [Google Scholar]
- 51.Zhu Y, Fenik P, Zhan G, et al. Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J Neurosci. 2008;28:2168–78. doi: 10.1523/JNEUROSCI.5232-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raghuraman G, Rai V, Peng Y, et al. Pattern-specific sustained activation of tyrosine hydroxylase by intermittent hypoxia: role of reactive oxygen species-dependent downregulation of protein phosphatase 2A and upregulation of protein kinases. Antioxid Redox Signal. 2009;11:1777–89. doi: 10.1089/ars.2008.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma SD, Raghuraman G, Lee MS, et al. Intermittent hypoxia activates peptidylglycine {alpha}-amidating monooxygenase in rat brain stem via reactive oxygen species-mediated proteolytic processing. J Appl Physiol. 2009;106:12–9. doi: 10.1152/japplphysiol.90702.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fletcher EC, Lesske J, Behm R, et al. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;72:1978–84. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- 55.Prabhakar NR, Peng YJ, Jacono FJ, et al. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol. 2005;32:447–9. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- 56.Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–9. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- 57.Peng Y, Nanduri J, Yuan G, et al. NADPH oxidase is required for the sensory plasticity of the carotid body by chronic intermittent hypoxia. J Neurosci. 2009;29:4903–10. doi: 10.1523/JNEUROSCI.4768-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rey S, Iturriaga R. Endothelins and nitric oxide: vasoactive modulators of carotid body chemoreception. Curr Neurovasc Res. 2004;1:465–73. doi: 10.2174/1567202043361857. [DOI] [PubMed] [Google Scholar]
- 59.Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 and endothelin A and B receptors to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Adv Exp Med Biol. 2008;605:228–32. doi: 10.1007/978-0-387-73693-8_40. [DOI] [PubMed] [Google Scholar]
- 60.Rey S, Del Rio R, Iturriaga R. Contribution of endothelin-1 to the enhanced carotid body chemosensory responses induced by chronic intermittent hypoxia. Brain Res. 2006;1086:152–9. doi: 10.1016/j.brainres.2006.02.082. [DOI] [PubMed] [Google Scholar]
- 61.Liu Y, Ji ES, Xiang S, et al. Exposure to cyclic intermittent hypoxia increases expression of functional NMDA receptors in the rat carotid body. J Appl Physiol. 2009;106:259–67. doi: 10.1152/japplphysiol.90626.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Row BW, Liu R, Xu W, et al. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med. 2003;167:1548–53. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- 63.Troncoso Brindeiro CM, Da Silva AQ, Allahdadi KJ, et al. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–6. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–14. doi: 10.1161/01.hyp.34.2.309. [DOI] [PubMed] [Google Scholar]
- 65.Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J Appl Physiol. 2002;92:627–33. doi: 10.1152/japplphysiol.00152.2001. [DOI] [PubMed] [Google Scholar]
- 66.Yuan ZM, Chen BY, Wang PX, et al. Changes of angiotensin II and its receptor during the development of chronic intermittent hypoxia-induced hypertension in rats. Zhonghua Jie He He Hu Xi Za Zhi. 2004;27:577–80. [PubMed] [Google Scholar]
- 67.Allahdadi KJ, Walker BR, Kanagy NL. Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension. 2005;45:705–9. doi: 10.1161/01.HYP.0000153794.52852.04. [DOI] [PubMed] [Google Scholar]
- 68.Allahdadi KJ, Cherng TW, Pai H, et al. Endothelin type A receptor antagonist normalizes blood pressure in rats exposed to eucapnic intermittent hypoxia. Am J Physiol Heart Circ Physiol. 2008;295:H434–40. doi: 10.1152/ajpheart.91477.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belaidi E, Joyeux-Faure M, Ribuot C, et al. Major role for hypoxia inducible factor-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. Journal of the American College of Cardiology. 2009;53:1309–17. doi: 10.1016/j.jacc.2008.12.050. [DOI] [PubMed] [Google Scholar]
- 70.Prabhakar NR, Kumar GK, Nanduri J, et al. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal. 2007;9:1397–403. doi: 10.1089/ars.2007.1732. [DOI] [PubMed] [Google Scholar]
- 71.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–9. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- 72.Sedeek M, Hebert RL, Kennedy CR, et al. Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr Opin Nephrol Hypertens. 2009;18:122–7. doi: 10.1097/MNH.0b013e32832923c3. [DOI] [PubMed] [Google Scholar]
- 73.Carriere A, Galinier A, Fernandez Y, et al. [Physiological and physiopathological consequences of mitochondrial reactive oxygen species] Med Sci. 2006;22:47–53. doi: 10.1051/medsci/200622147. [DOI] [PubMed] [Google Scholar]
- 74.Wang W, Fang H, Groom L, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–90. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ Res. 2008;103:873–80. doi: 10.1161/CIRCRESAHA.108.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shan X, Chi L, Ke Y, et al. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiology of Disease. 2007;28:206–15. doi: 10.1016/j.nbd.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim SY, Seo M, Kim Y, et al. Stimulatory heterotrimeric GTP-binding protein inhibits hydrogen peroxide-induced apoptosis by repressing BAK induction in SH-SY5Y human neuroblastoma cells. J Biol Chem. 2008;283:1350–61. doi: 10.1074/jbc.M702344200. [DOI] [PubMed] [Google Scholar]
- 78.Han KY, Yang D, Chang EJ, et al. Inhibition of osteoclast differentiation and bone resorption by sauchinone. Biochem Pharmacol. 2007;74:911–23. doi: 10.1016/j.bcp.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 79.Porta S, Serra SA, Huch M, et al. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Hum Mol Genet. 2007;16:1039–50. doi: 10.1093/hmg/ddm049. [DOI] [PubMed] [Google Scholar]
- 80.Frossi B, Rivera J, Hirsch E, et al. Selective activation of Fyn/PI3K and p38 MAPK regulates IL-4 production in BMMC under nontoxic stress condition. J Immunol. 2007;178:2549–55. doi: 10.4049/jimmunol.178.4.2549. [DOI] [PubMed] [Google Scholar]
- 81.Tu VC, Sun H, Bowden GT, et al. Involvement of oxidants and AP-1 in angiotensin II-activated NFAT3 transcription factor. Am J Physiol Cell Physiol. 2007;292:C1248–55. doi: 10.1152/ajpcell.00624.2005. [DOI] [PubMed] [Google Scholar]
- 82.Ke Q, Li J, Ding J, et al. Essential role of ROS-mediated NFAT activation in TNF-{alpha} induction by crystalline silica exposure. Am J Physiol Lung Cell Mol Physiol. 2006;291:L257–64. doi: 10.1152/ajplung.00007.2006. [DOI] [PubMed] [Google Scholar]
- 83.Phillips SA, Olson EB, Lombard JH, et al. Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J Appl Physiol. 2006;100:1117–23. doi: 10.1152/japplphysiol.00994.2005. [DOI] [PubMed] [Google Scholar]
- 84.Earley S, Nelin LD, Chicoine LG, et al. Hypoxia-induced pulmonary endothelin-1 expression is unaltered by nitric oxide. J Appl Physiol. 2002;92:1152–8. doi: 10.1152/japplphysiol.00829.2001. [DOI] [PubMed] [Google Scholar]
- 85.Gertler JP, Ocasio VH. Endothelin production by hypoxic human endothelium. J Vasc Surg. 1993;18:178–82. [PubMed] [Google Scholar]
- 86.Kagamu H, Suzuki T, Arakawa M, et al. Low oxygen enhances endothelin-1 (ET-1) production and responsiveness to ET-1 in cultured cardiac myocytes. Biochem Biophys Res Commun. 1994;202:1612–8. doi: 10.1006/bbrc.1994.2117. [DOI] [PubMed] [Google Scholar]
- 87.Kourembanas S, McQuillan LP, Leung GK, et al. Nitric oxide regulates the expression of vasoconstrictors and growth factors by vascular endothelium under both normoxia and hypoxia. J Clin Invest. 1993;92:99–104. doi: 10.1172/JCI116604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaehler J, Sill B, Koester R, et al. Endothelin-1 mRNA and protein in vascular wall cells is increased by reactive oxygen species. Clin Sci. 2002;103:176S–8S. doi: 10.1042/CS103S176S. [DOI] [PubMed] [Google Scholar]
- 89.Pawar A, Nanduri J, Yuan G, et al. Reactive oxygen species-dependent endothelin signaling is required for augmented hypoxic sensory response of the neonatal carotid body by intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R735–42. doi: 10.1152/ajpregu.90490.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Allahdadi KJ, Duling LC, Walker BR, et al. Eucapnic intermittent hypoxia augments endothelin-1 vasoconstriction in rats: role of PKC{delta} Am J Physiol Heart Circ Physiol. 2008;294:H920–7. doi: 10.1152/ajpheart.01264.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brezniceanu ML, Wei CC, Zhang SL, et al. Transforming growth factor-beta 1 stimulates angiotensinogen gene expression in kidney proximal tubular cells. Kidney Int. 2006;69:1977–85. doi: 10.1038/sj.ki.5000396. [DOI] [PubMed] [Google Scholar]
- 92.Hsieh TJ, Fustier P, Wei CC, et al. Reactive oxygen species blockade and action of insulin on expression of angiotensinogen gene in proximal tubular cells. J Endocrinol. 2004;183:535–50. doi: 10.1677/joe.1.05871. [DOI] [PubMed] [Google Scholar]
- 93.Hsu YH, Chen JJ, Chang NC, et al. Role of reactive oxygen species-sensitive extracellular signal-regulated kinase pathway in angiotensin II-induced endothelin-1 gene expression in vascular endothelial cells. J Vasc Res. 2004;41:64–74. doi: 10.1159/000076247. [DOI] [PubMed] [Google Scholar]
- 94.Knappe D, Sill B, Tharun B, et al. Endothelin-1 in humans is increased by oxygen-derived radicals ex vivo and in vivo. J Investig Med. 2007;55:306–14. doi: 10.2310/6650.2007.00013. [DOI] [PubMed] [Google Scholar]
- 95.Sedeek MH, Llinas MT, Drummond H, et al. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension. 2003;42:806–10. doi: 10.1161/01.HYP.0000084372.91932.BA. [DOI] [PubMed] [Google Scholar]
- 96.Hanna IR, Taniyama Y, Szocs K, et al. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002;4:899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 97.Weber DS, Rocic P, Mellis AM, et al. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;288:H37–42. doi: 10.1152/ajpheart.00638.2004. [DOI] [PubMed] [Google Scholar]
- 98.Wilcox CS. Reactive oxygen species: roles in blood pressure and kidney function. Curr Hypertens Rep. 2002;4:160–6. doi: 10.1007/s11906-002-0041-2. [DOI] [PubMed] [Google Scholar]
- 99.Moller DS, Lind P, Strunge B, et al. Abnormal vasoactive hormones and 24-hour blood pressure in obstructive sleep apnea. Am J Hypertens. 2003;16:274–80. doi: 10.1016/s0895-7061(02)03267-3. [DOI] [PubMed] [Google Scholar]
- 100.Takahashi S, Nakamura Y, Nishijima T, et al. Essential roles of angiotensin II in vascular endothelial growth factor expression in sleep apnea syndrome. Respiratory Medicine. 2005;99:1125–31. doi: 10.1016/j.rmed.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 101.Gjorup PH, Sadauskiene L, Wessels J, et al. Abnormally increased endothelin-1 in plasma during the night in obstructive sleep apnea: relation to blood pressure and severity of disease. Am J Hypertens. 2007;20:44–52. doi: 10.1016/j.amjhyper.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 102.Benjamin JA, Moller M, Ebden P, et al. Serum angiotensin converting enzyme and the obstructive sleep apnea hypopnea syndrome. J Clin Sleep Med. 2008;4:325–31. [PMC free article] [PubMed] [Google Scholar]
- 103.Gjorup PH, Sadauskiene L, Wessels J, et al. Increased nocturnal sodium excretion in obstructive sleep apnoea. Relation to nocturnal change in diastolic blood pressure. Scand J Clin Lab Invest. 2008;68:11–21. doi: 10.1080/00365510701352020. [DOI] [PubMed] [Google Scholar]
- 104.Kizawa T, Nakamura Y, Takahashi S, et al. Pathogenic role of angiotensin II and oxidized LDL in obstructive sleep apnoea. Eur Respir J. 2009 doi: 10.1183/09031936.00009709. ; [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 105.Sen R, Baltimore D. Inducibility of [kappa] immunoglobulin enhancer-binding protein NF-[kappa]B by a posttranslational mechanism. Cell. 1986;47:921–8. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 106.Fraser CC. G Protein-Coupled Receptor Connectivity to NF-kB in Inflammation and Cancer. Int Rev Immunol. 2008;27:320–50. doi: 10.1080/08830180802262765. [DOI] [PubMed] [Google Scholar]
- 107.Garvey JF, Taylor CT, McNicholas WT. Cardiovascular disease in obstructive sleep apnoea syndrome: the role of intermittent hypoxia and inflammation. Eur Respir J. 2009;33:1195–205. doi: 10.1183/09031936.00111208. [DOI] [PubMed] [Google Scholar]
- 108.Ryan S, Taylor CT, McNicholas WT. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation. 2005;112:2660–7. doi: 10.1161/CIRCULATIONAHA.105.556746. [DOI] [PubMed] [Google Scholar]
- 109.Ryan S, McNicholas WT, Taylor CT. A critical role for p38 map kinase in NF-[kappa]B signaling during intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun. 2007;355:728–33. doi: 10.1016/j.bbrc.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 110.Ryan S, Taylor CT, McNicholas WT. Predictors of elevated nuclear factor-{kappa}b-dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med. 2006;174:824–30. doi: 10.1164/rccm.200601-066OC. [DOI] [PubMed] [Google Scholar]
- 111.Greenberg H, Ye X, Wilson D, et al. Chronic intermittent hypoxia activates nuclear factor-[kappa]B in cardiovascular tissues in vivo. Biochem Biophys Res Commun. 2006;343:591–6. doi: 10.1016/j.bbrc.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 112.Htoo A, Greenberg H, Tongia S, et al. Activation of nuclear factor kB in obstructive sleep apnea: a pathway leading to systemic inflammation. Sleep Breath. 2006;10:43–50. doi: 10.1007/s11325-005-0046-6. [DOI] [PubMed] [Google Scholar]
- 113.Foncea R, Carvajal C, Almarza C, et al. Endothelial cell oxidative stress and signal transduction. Biol Res. 2000;33:89–96. doi: 10.4067/s0716-97602000000200008. [DOI] [PubMed] [Google Scholar]
- 114.Lavie L. Sleep-disordered breathing and cerebrovascular disease: a mechanistic approach. Neurologic Clinics. 2005;23:1059–75. doi: 10.1016/j.ncl.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 115.Hill-Eubanks DC, Gomez MF, Stevenson AS, et al. NFAT Regulation in Smooth Muscle. Trends Cardiovasc Med. 2003;13:56–62. doi: 10.1016/s1050-1738(02)00212-8. [DOI] [PubMed] [Google Scholar]
- 116.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–47. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 117.Decker EL, Skerka C, Zipfel PF. The early growth response protein (EGR-1) regulates interleukin-2 transcription by synergistic interaction with the nuclear factor of activated T cells. J Biol Chem. 1998;273:26923–30. doi: 10.1074/jbc.273.41.26923. [DOI] [PubMed] [Google Scholar]
- 118.Decker EL, Nehmann N, Kampen E, et al. Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. 2003;31:911–21. doi: 10.1093/nar/gkg186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen L, Glover JN, Hogan PG, et al. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–8. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- 120.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–89. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 121.Sweetser MT, Hoey T, Sun YL, et al. The roles of nuclear factor of activated T cells and ying-yang 1 in activation-induced expression of the interferon-gamma promoter in T cells. J Biol Chem. 1998;273:34775–83. doi: 10.1074/jbc.273.52.34775. [DOI] [PubMed] [Google Scholar]
- 122.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 123.McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–7. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 124.Olson EN, Williams RS. Calcineurin signaling and muscle remodeling. Cell. 2000;101:689–92. doi: 10.1016/s0092-8674(00)80880-6. [DOI] [PubMed] [Google Scholar]
- 125.Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wada H, Hasegawa K, Morimoto T, et al. Calcineurin-GATA-6 pathway is involved in smooth muscle-specific transcription. J Cell Biol. 2002;156:983–91. doi: 10.1083/jcb.200106057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hogan PG, Chen L, Nardone J, et al. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–32. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 128.Amberg GC, Rossow CF, Navedo MF, et al. NFATc3 regulates Kv2.1 expression in arterial smooth muscle. J Biol Chem. 2004;279:47326–34. doi: 10.1074/jbc.M408789200. [DOI] [PubMed] [Google Scholar]
- 129.Chen J, Amasaki Y, Kamogawa Y, et al. Role of NFATx (NFAT4/NFATc3) in expression of immunoregulatory genes in murine peripheral CD4+ T cells. J Immunol. 2003;170:3109–17. doi: 10.4049/jimmunol.170.6.3109. [DOI] [PubMed] [Google Scholar]
- 130.Han S, Lu J, Zhang Y, et al. Recruitment of histone deacetylase 4 by transcription factors represses interleukin-5 transcription. Biochem J. 2006;400:439–48. doi: 10.1042/BJ20061085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nieves-Cintron M, Amberg GC, Nichols CB, et al. Activation of NFATC3 down-regulates the beta 1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J Biol Chem. 2006;282:3231–40. doi: 10.1074/jbc.M608822200. [DOI] [PubMed] [Google Scholar]
- 132.Stevenson AS, Gomez MF, Hill-Eubanks DC, et al. NFAT4 movement in native smooth muscle. A role for differential Ca(2+) signaling. J Biol Chem. 2001;276:15018–24. doi: 10.1074/jbc.M011684200. [DOI] [PubMed] [Google Scholar]
- 133.Gomez MF, Stevenson AS, Bonev AD, et al. Opposing actions of inositol 1,4,5-trisphosphate and ryanodine receptors on nuclear factor of activated T-cells regulation in smooth muscle. J Biol Chem. 2002;277:37756–64. doi: 10.1074/jbc.M203596200. [DOI] [PubMed] [Google Scholar]
- 134.Gomez MF, Gonzalez Bosc LV, Stevenson AS, et al. Constitutively elevated nuclear export activity opposes Ca2+-dependent NFATc3 nuclear accumulation in vascular smooth muscle: role of JNK2 and Crm-1. J Biol Chem. 2003;278:46847–53. doi: 10.1074/jbc.M304765200. [DOI] [PubMed] [Google Scholar]
- 135.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, et al. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation – Role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem. 2004;279:10702–9. doi: 10.1074/jbc.M312920200. [DOI] [PubMed] [Google Scholar]
- 136.Nilsson J, Nilsson LM, Chen YW, et al. High glucose activates nuclear factor of activated t cells in native vascular smooth muscle. Arterioscler Thromb Vasc Biol. 2006;26:794–800. doi: 10.1161/01.ATV.0000209513.00765.13. [DOI] [PubMed] [Google Scholar]
- 137.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001;11:505–12. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- 138.Graef IA, Chen F, Chen L, et al. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001;105:863–75. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- 139.Ohkawa Y, Hayashi K, Sobue K. Calcineurin-mediated pathway involved in the differentiated phenotype of smooth muscle cells. Biochem Biophys Res Commun. 2003;301:78–83. doi: 10.1016/s0006-291x(02)02965-0. [DOI] [PubMed] [Google Scholar]
- 140.Gonzalez Bosc LV, Layne J, Nelson MT, et al. Nuclear factor of activated T-cells and serum response factor cooperatively regulate an α-actin intronic enhancer. J Biol Chem. 2004;280:26113–20. doi: 10.1074/jbc.M411972200. [DOI] [PubMed] [Google Scholar]
- 141.Layne JJ, Werner ME, Hill-Eubanks DC, et al. NFATc3 regulates BK channel function in murine urinary bladder smooth muscle. Am J Physiol Cell Physiol. 2008;295:C611–23. doi: 10.1152/ajpcell.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boss V, Abbott KL, Wang XF, et al. The cyclosporin A-sensitive nuclear factor of activated T cells (NFAT) proteins are expressed in vascular smooth muscle cells. Differential localization of NFAT isoforms and induction of NFAT-mediated transcription by phospholipase C-coupled cell surface receptors. J Biol Chem. 1998;273:19664–71. doi: 10.1074/jbc.273.31.19664. [DOI] [PubMed] [Google Scholar]
- 143.De Frutos S, Spangler R, Alo D, et al. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha -actin up-regulation. J Biol Chem. 2007;282:15081–9. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaminuma O, Kitamura F, Kitamura N, et al. Differential contribution of NFATc2 and NFATc1 to TNF-alpha gene expression in T cells. J Immunol. 2008;180:319–26. doi: 10.4049/jimmunol.180.1.319. [DOI] [PubMed] [Google Scholar]
- 145.Nilsson LM, Sun ZW, Nilsson J, et al. Novel blocker of NFAT activation inhibits IL-6 production in human myometrial arteries and reduces vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C1167–78. doi: 10.1152/ajpcell.00590.2005. [DOI] [PubMed] [Google Scholar]
- 146.Weigmann B, Lehr HA, Yancopoulos G, et al. The transcription factor NFATc2 controls IL-6-dependent T cell activation in experimental colitis. J Exp Med. 2008;205:2099–110. doi: 10.1084/jem.20072484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nilsson LM. Nuclear Factor of Activated T-Cells is involved in the regulation of osteopontin expression in vascular smooth muscle. FASEB J. 2007;21 : 905.20. [Google Scholar]
- 148.Alberti A, Sarchielli P, Gallinella E, et al. Plasma cytokine levels in patients with obstructive sleep apnea syndrome: a preliminary study. J Sleep Res. 2003;12:305–11. doi: 10.1111/j.1365-2869.2003.00361.x. [DOI] [PubMed] [Google Scholar]
- 149.Yudkin JS, Kumari M, Humphries SE, et al. Inflammation, obesity, stress and coronary heart disease: is interleukin-6 the link. Atherosclerosis. 2000;148:209–14. doi: 10.1016/s0021-9150(99)00463-3. [DOI] [PubMed] [Google Scholar]
- 150.Lindmark E, Diderholm E, Wallentin L, et al. Relationship between interleukin 6 and mortality in patients with unstable coronary artery disease: effects of an early invasive or noninvasive strategy. JAMA. 2001;286:2107–13. doi: 10.1001/jama.286.17.2107. [DOI] [PubMed] [Google Scholar]
- 151.Ridker PM, Rifai N, Stampfer MJ, et al. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–72. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 152.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–9. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 153.Graf K, Do YS, Ashizawa N, et al. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation. 1997;96:3063–71. doi: 10.1161/01.cir.96.9.3063. [DOI] [PubMed] [Google Scholar]
- 154.Kurata M, Okura T, Watanabe S, et al. Osteopontin and carotid atherosclerosis in patients with essential hypertension. Clin Sci. 2006;111:319–24. doi: 10.1042/CS20060074. [DOI] [PubMed] [Google Scholar]
- 155.Ohmori R, Momiyama Y, Taniguchi H, et al. Plasma osteopontin levels are associated with the presence and extent of coronary artery disease. Atherosclerosis. 2003;170:333–7. doi: 10.1016/s0021-9150(03)00298-3. [DOI] [PubMed] [Google Scholar]
- 156.Uenoyama M, Ogata S, Nakanishi K, et al. Osteopontin expression in normal and hypobaric hypoxia-exposed rats. Acta physiologica. 2008;193:291–301. doi: 10.1111/j.1748-1716.2008.01844.x. [DOI] [PubMed] [Google Scholar]
- 157.Denhardt DT, Giachelli CM, Rittling SR. Role of osteopontin in cellular signaling and toxicant injury. Annu Rev Pharmacol Toxicol. 2001;41:723–49. doi: 10.1146/annurev.pharmtox.41.1.723. [DOI] [PubMed] [Google Scholar]
- 158.Denhardt DT, Noda M, O’Regan AW, et al. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–61. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nau GJ, Guilfoile P, Chupp GL, et al. A chemoattractant cytokine associated with granulomas in tuberculosis and silicosis. Proc Natl Acad Sci USA. 1997;94:6414–9. doi: 10.1073/pnas.94.12.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vaschetto R, Nicola S, Olivieri C, et al. Serum levels of osteopontin are increased in SIRS and sepsis. Intensive Care Med. 2008;34:2176–84. doi: 10.1007/s00134-008-1268-4. [DOI] [PubMed] [Google Scholar]
- 161.Muteliefu G, Enomoto A, Jiang P, et al. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051–8. doi: 10.1093/ndt/gfn757. [DOI] [PubMed] [Google Scholar]
- 162.Hu T, Luan R, Zhang H, et al. Hydrogen peroxide enhances the osteopontin expression and matrix metalloproteinase activity in aortic vascular smooth muscle cells. Clin Exp Pharmacol Physiol. 2008;36:626–30. doi: 10.1111/j.1440-1681.2008.05124.x. [DOI] [PubMed] [Google Scholar]
- 163.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–54. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Semenza GL, Agani F, Booth G, et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Int. 1997;51:553–5. doi: 10.1038/ki.1997.77. [DOI] [PubMed] [Google Scholar]
- 165.Ivan M, Haberberger T, Gervasi DC, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA. 2002;99:13459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Papandreou I, Cairns RA, Fontana L, et al. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabolism. 2006;3:187–97. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 167.Fandrey J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol. 2004;286:R977–88. doi: 10.1152/ajpregu.00577.2003. [DOI] [PubMed] [Google Scholar]
- 168.Snow JB, Kitzis V, Norton CE, et al. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol. 2008;104:110–8. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- 169.Hoffstein V, Herridge M, Mateika S, et al. Hematocrit levels in sleep apnea. Chest. 1994;106:787–91. doi: 10.1378/chest.106.3.787. [DOI] [PubMed] [Google Scholar]
- 170.Cramer T, Yamanishi Y, Clausen BrE, et al. HIF-1[alpha] is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Imagawa S, Yamaguchi Y, Higuchi M, et al. Levels of vascular endothelial growth factor are elevated in patients with obstructive sleep apnea-hypopnea syndrome. Blood. 2001;98:1255–7. doi: 10.1182/blood.v98.4.1255. [DOI] [PubMed] [Google Scholar]
- 172.Cummins E, Taylor C. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–71. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 173.Yuan G, Nanduri J, Bhasker CR, et al. Ca2+/Calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem. 2005;280:4321–8. doi: 10.1074/jbc.M407706200. [DOI] [PubMed] [Google Scholar]
- 174.Toffoli SÃ, Roegiers A, Feron O, et al. Intermittent hypoxia is an angiogenic inducer for endothelial cells: role of HIF-1. Angiogenesis. 2009;12:47–67. doi: 10.1007/s10456-009-9131-y. [DOI] [PubMed] [Google Scholar]
- 175.Hu J, Discher DJ, Bishopric NH, et al. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun. 1998;245:894–9. doi: 10.1006/bbrc.1998.8543. [DOI] [PubMed] [Google Scholar]
- 176.Minchenko A, Caro J. Regulation of endothelin-1 gene expression in human microvascular endothelial cells by hypoxia and cobalt: role of hypoxia responsive element. Mol Cell Biochem. 2000;208:53–62. doi: 10.1023/a:1007042729486. [DOI] [PubMed] [Google Scholar]
- 177.Yamashita K, Discher DJ, Hu J, et al. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. J Biol Chem. 2001;276:12645–53. doi: 10.1074/jbc.M011344200. [DOI] [PubMed] [Google Scholar]
- 178.Lam SY, Tipoe GL, Liong EC, et al. Hypoxia-inducible factor (HIF)-1alpha and endothelin-1 expression in the rat carotid body during intermittent hypoxia. Adv Exp Med Biol. 2006;580:21–7. doi: 10.1007/0-387-31311-7_4. [DOI] [PubMed] [Google Scholar]
- 179.Yuan G, Nanduri J, Khan S, et al. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:674–85. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bonello S, Zahringer C, Belaiba RS, et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arterioscler Thromb Vasc Biol. 2007;27:755–61. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 181.Gorlach A, Bonello S. The cross-talk between NF-kappaB and HIF-1: further evidence for a significant liaison. Biochem J. 2008;412:e17–9. doi: 10.1042/BJ20080920. [DOI] [PubMed] [Google Scholar]
- 182.Rius J, Guma M, Schachtrup C, et al. NF-[κ]B links innate immunity to the hypoxic response through transcriptional regulation of HIF-1[α] Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- 184.Ema M, Taya S, Yokotani N, et al. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA. 1997;94:4273–8. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Holmquist-Mengelbier L, Fredlund E, Löfstedt T, et al. Recruitment of HIF-1[alpha] and HIF-2[alpha] to common target genes is differentially regulated in neuroblastoma: HIF-2[alpha] promotes an aggressive phenotype. Cancer Cell. 2006;10:413–23. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 186.Nanduri J, Wang N, Yuan G, et al. Intermittent hypoxia degrades HIF-2+ via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA. 2009;106:1199–204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sajkov D, Mcevoy RD. Obstructive sleep apnea and pulmonary hypertension. Prog Cardiovasc Dis. 2009;51:363–70. doi: 10.1016/j.pcad.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 188.Schafer H, Hasper E, Ewig S, et al. Pulmonary haemodynamics in obstructive sleep apnoea: time course and associated factors. Eur Respir J. 1998;12:679–84. doi: 10.1183/09031936.98.12030679. [DOI] [PubMed] [Google Scholar]
- 189.Weitzenblum E, Chaouat A, Kessler R, et al. Overlap syndrome: obstructive sleep apnea in patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:237–41. doi: 10.1513/pats.200706-077MG. [DOI] [PubMed] [Google Scholar]
- 190.Somers VK, White DP, Amin R, et al. Sleep apnea and cardiovascular disease. An American Heart Association/American College of Cardiology Foundation Scientific Statement From the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing Council. Circulation. 2008;118:1080–111. doi: 10.1161/CIRCULATIONAHA.107.189375. [DOI] [PubMed] [Google Scholar]
- 191.Sajkov D, Wang T, Saunders NA, et al. Continuous positive airway pressure treatment improves pulmonary hemodynamics in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2002;165:152–8. doi: 10.1164/ajrccm.165.2.2010092. [DOI] [PubMed] [Google Scholar]
- 192.Arias MA, Garcia-Rio F, Alonso-Fernandez A, et al. Pulmonary hypertension in obstructive sleep apnoea: effects of continuous positive airway pressure: a randomized, controlled cross-over study. Eur Heart J. 2006;27:1106–13. doi: 10.1093/eurheartj/ehi807. [DOI] [PubMed] [Google Scholar]
- 193.Alchanatis M, Tourkohoriti G, Kakouros S, et al. Daytime pulmonary hypertension in patients with obstructive sleep apnea: the effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration. 2001;68:566–72. doi: 10.1159/000050574. [DOI] [PubMed] [Google Scholar]
- 194.Zielinski J. Effects of intermittent hypoxia on pulmonary haemodynamics: animal models versus studies in humans. Eur Respir J. 2005;25:173–80. doi: 10.1183/09031936.04.00037204. [DOI] [PubMed] [Google Scholar]
- 195.Sanner BM, Doberauer C, Konermann M, et al. Pulmonary hypertension in patients with obstructive sleep apnea syndrome. Arch Intern Med. 1997;157:2483–7. [PubMed] [Google Scholar]
- 196.Bady E, Achkar A, Pascal S, et al. Pulmonary arterial hypertension in patients with sleep apnoea syndrome. Thorax. 2000;55:934–9. doi: 10.1136/thorax.55.11.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163:19–25. doi: 10.1164/ajrccm.163.1.2001008. [DOI] [PubMed] [Google Scholar]
- 198.Golbin JM, Somers VK, Caples SM. Obstructive sleep apnea, cardiovascular disease, and pulmonary hypertension. Proc Am Thorac Soc. 2008;5:200–6. doi: 10.1513/pats.200708-143MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bradford A. Effects of chronic intermittent asphyxia on haematocrit, pulmonary arterial pressure and skeletal muscle structure in rats. Exp Physiol. 2004;89:44–52. doi: 10.1113/expphysiol.2003.002656. [DOI] [PubMed] [Google Scholar]
- 200.Fagan KA. Selected contribution: pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol. 2001;90:2502–7. doi: 10.1152/jappl.2001.90.6.2502. [DOI] [PubMed] [Google Scholar]
- 201.Nisbet RE, Graves AS, Kleinhenz DJ, et al. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am J Respir Cell Mol Biol. 2009;40:601–9. doi: 10.1165/rcmb.2008-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Fletcher EC, Bao G, Miller CC., III Effect of recurrent episodic hypocapnic, eucapnic, and hypercapnic hypoxia on systemic blood pressure. J Appl Physiol. 1995;78:1516–21. doi: 10.1152/jappl.1995.78.4.1516. [DOI] [PubMed] [Google Scholar]
- 203.Resta TC, Chicoine LG, Omdahl JL, et al. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol. 1999;276:H699–708. doi: 10.1152/ajpheart.1999.276.2.H699. [DOI] [PubMed] [Google Scholar]
- 204.Nagaoka T, Fagan KA, Gebb SA, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med. 2005;171:494–9. doi: 10.1164/rccm.200405-637OC. [DOI] [PubMed] [Google Scholar]
- 205.Nagaoka T, Morio Y, Casanova N, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–72. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 206.Broughton BR, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol. 2008;294:L797–806. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 207.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2008;295:L515–29. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Jernigan NL, Walker BR, Resta TC. Endothelium-derived reactive oxygen species and endothelin-1 attenuate NO-dependent pulmonary vasodilation following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2004;287:L801–8. doi: 10.1152/ajplung.00443.2003. [DOI] [PubMed] [Google Scholar]
- 209.Paffett ML, Naik JS, Resta TC, et al. Reduced store-operated Ca2+ entry in pulmonary endothelial cells from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1135–42. doi: 10.1152/ajplung.00432.2006. [DOI] [PubMed] [Google Scholar]
- 210.Murata T, Sato K, Hori M, et al. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem. 2002;277:44085–92. doi: 10.1074/jbc.M205934200. [DOI] [PubMed] [Google Scholar]
- 211.Dicarlo VS, Chen SJ, Meng QC, et al. ETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in rat. Am J Physiol. 1995;269:L690–7. doi: 10.1152/ajplung.1995.269.5.L690. [DOI] [PubMed] [Google Scholar]
- 212.Bonvallet ST, Zamora MR, Hasunuma K, et al. BQ123, an ETA-receptor antagonist, attenuates hypoxic pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 1994;266:H1327–31. doi: 10.1152/ajpheart.1994.266.4.H1327. [DOI] [PubMed] [Google Scholar]
- 213.Chen SJ, Chen YF, Meng QC, et al. Endothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in rats. J Appl Physiol. 1995;79:2122–31. doi: 10.1152/jappl.1995.79.6.2122. [DOI] [PubMed] [Google Scholar]
- 214.Snow J, Kanagy N, Walker B, et al. Intermittent hypoxia increases PKCα/β- and reactive oxygen species dependent myofilament Ca2+ sensitization in small pulmonary arteries. FASEB J. 2009;23 : 770.6. [Google Scholar]
- 215.Liu JQ, Zelko IN, Erbynn EM, et al. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox) Am J Physiol Lung Cell Mol Physiol. 2006;290:L2–10. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 216.Elmedal B, De Dam MY, Mulvany MJ, et al. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol. 2004;141:105–13. doi: 10.1038/sj.bjp.0705580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Matsui H, Shimosawa T, Itakura K, et al. Adrenomedullin can protect against pulmonary vascular remodeling induced by hypoxia. Circulation. 2004;109:2246–51. doi: 10.1161/01.CIR.0000127950.13380.FD. [DOI] [PubMed] [Google Scholar]
- 218.Grobe AC, Wells SM, Benavidez E, et al. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1069–77. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- 219.Fresquet F, Pourageaud F, Leblais V, et al. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol. 2006;148:714–23. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Brennan LA, Steinhorn RH, Wedgwood S, et al. Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res. 2003;92:683–91. doi: 10.1161/01.RES.0000063424.28903.BB. [DOI] [PubMed] [Google Scholar]
- 221.Fike CD, Slaughter JC, Kaplowitz MR, et al. Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;295:L881–8. doi: 10.1152/ajplung.00047.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hoshikawa Y, Ono S, Suzuki S, et al. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol. 2001;90:1299–306. doi: 10.1152/jappl.2001.90.4.1299. [DOI] [PubMed] [Google Scholar]
- 223.Jankov RP, Kantores C, Pan J, et al. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–45. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- 224.Shimoda LA, Manalo DJ, Sham JSK, et al. Partial HIF-1{alpha} deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–8. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- 225.Alford NJ, Fletcher EC, Nickeson D. Acute oxygen in patients with sleep apnea and COPD. Chest. 1986;89:30–8. doi: 10.1378/chest.89.1.30. [DOI] [PubMed] [Google Scholar]
- 226.Ooi H, Cadogan E, Sweeney M, et al. Chronic hypercapnia inhibits hypoxic pulmonary vascular remodeling. Am J Physiol Heart Circ Physiol. 2000;278:H331–8. doi: 10.1152/ajpheart.2000.278.2.H331. [DOI] [PubMed] [Google Scholar]
- 227.Kantores C, McNamara PJ, Teixeira L, et al. Therapeutic hypercapnia prevents chronic hypoxia-induced pulmonary hypertension in the newborn rat. Am J Physiol Lung Cell Mol Physiol. 2006;291:L912–22. doi: 10.1152/ajplung.00480.2005. [DOI] [PubMed] [Google Scholar]