

Abstract
Epsins have an important role in mediating clathrin-mediated endocytosis of ubiquitinated cell surface receptors. The potential role for epsins in tumorigenesis and cancer metastasis by regulating intracellular signaling pathways has largely not been explored. Epsins are reportedly upregulated in several types of cancer including human skin, lung, and canine mammary cancers. However, whether their expression is elevated in prostate cancer is unknown. In this study, we investigated the potential role of epsins in prostate tumorigenesis using the wild type or epsin-deficient human prostate cancer cells, LNCaP, in a human xenograft model, and the spontaneous TRAMP mouse model in wild type or epsin-deficient background. Here, we reported that the expression of epsins 1 and 2 is upregulated in both human and mouse prostate cancer cells and cancerous tissues. Consistent with upregulation of epsins in prostate tumors, we discovered that depletion of epsins impaired tumor growth in both the human LNCaP xenograft and the TRAMP mouse prostate. Furthermore, epsin depletion significantly prolonged survival in the TRAMP mouse model. In summary, our findings suggest that epsins may act as oncogenic proteins to promote prostate tumorigenesis and that depletion or inhibition of epsins may provide a novel therapeutic target for future prostate cancer therapies.
1. Introduction
Solid tumors, such as those in prostate cancer, contribute the majority of all cancers and result in significant distant tumor metastasis to vital organs such as the lungs, brain, and bones [1, 2]. Prostate cancer contributes significantly to the morbidity and mortality of men in the United States [3]. Advanced prostate cancer is associated with significant mortality because the cancer metastasizes and spreads throughout the body, making recovery nearly impossible [1, 4]. The high rates of prostate cancer metastasis are, in part, caused by aggressive primary tumor growth in prostate [5]. Prostate tumorigenesis is a result of several upregulated signaling pathways, including Notch, EGF, FGF, and Wnt signaling, which promote tumor cell proliferation [6–11]. Understanding the mechanisms responsible for upregulated signaling during early tumorigenesis is an important step in identifying key regulators and potential therapeutic targets. More importantly, targeting early stages of tumorigenesis will facilitate stabilization of rapid growing tumors, leading to effective surgical removal of primary tumors and inhibition of further tumor metastasis.
Epsins are endocytic adaptor proteins that regulate clathrin-mediated endocytosis of cell surface receptors by binding ubiquitin moieties on the receptor cytoplasmic tail [12–20]. Mammals express three epsins; epsins 1 and 2 are ubiquitously expressed in all tissues while epsin 3 is exclusively expressed in the stomach [14, 20, 21]. Epsins 1 and 2 likely play redundant functions in mediating endocytosis because a single deletion of either epsin 1 or 2 does not present with a significant phenotype [22, 23]. However, epsins 1 and 2 are essential during development because deletion of both alleles (DKO) resulted in embryonic lethality (E9.5) [22]. Embryonic lethality was due in large part to a defective vascular phenotype. We recently reported, using inducible epsin depletion, that postnatal loss of epsins 1 and 2 (iDKO) presented a normal phenotype, suggesting epsins play an important role in the development, but not the maintenance, of normal healthy tissues [23]. In contrast, endothelial cell-specific epsin depletion significantly impaired the growth of several tumor types as a result of aberrant tumor angiogenesis [23]. Solid tumorigenesis, including tumorigenesis of the prostate, is dependent on both tumor angiogenesis and tumor cell proliferation [5, 24]. Several signaling pathways involved in tumor cell proliferation involve clathrin-mediated receptor internalization and, therefore, may be mediated by epsins [11].
Elevated levels of epsins have been reported in human skin and lung cancers, as well as canine mammary cancers [23, 25–28]. Epsin overexpression promotes cancer cell invasion through the inhibition of Cdc42 and Rac1 GAP activity and by binding to RalBP1 [25, 27, 29–31]. Intriguingly, in this study, we have also discovered that expression of epsins is upregulated in both human and mouse prostate cancer cells and prostate cancer tissues. We have utilized both the human prostate LNCaP xenograft model [32] and the spontaneous TRAMP mouse model [33] to determine if epsin depletion affects prostate tumorigenesis. Our findings implicate epsins as oncogenic proteins whose expression is upregulated in prostate epithelium and facilitates growth and progression of prostate cancer. Furthermore, our data suggest that depletion or inhibition of epsins may provide novel therapeutic targets for solid tumor therapies.
2. Materials and Methods
2.1. Generation of Conditional Epn1fl/fl Mice and Tamoxifen-Inducible Epsin DKO
We recently reported a strategy for generating a global epsins 1 and 2 double knockout (DKO) mouse model [22]. C57BL/6 background mice with epsin 1 flanked by loxP sites (Epn1fl/fl) on a global epsin-2-deficient background (Epn1fl/fl, Epn2−/−) were used to create iDKO mice by crossing Epn1fl/fl, Epn2−/− mice with ERT2Cre deleter mice that express Cre Recombinase in all cell types upon tamoxifen injection (iDKO) [23].
2.2. Prostate Tumor Model
We crossed iDKO and WT mice with Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) mice on C57BL/6J background [33] to generate TRAMP-WT or TRAMP-iDKO mice. Ten-week-old, sex and genetic background matched TRAMP-iDKO or TRAMP-WT mice were IP-injected with 4-hydroxytamoxifen (150 μg per 30 g of body weight) for five to seven consecutive days to induce deletion of epsin 1. Mortality of TRAMP-WT and TRAMP-iDKO mice was recorded and survival rates plotted as a Kaplan-Meier plot. Mice were also dissected at weeks 20, 24, 28, 32, and 36 and checked for prostate tumors. Tumor growth in two groups of mice was monitored by measuring tumor size with digital calipers. We recognized tumors more than 2 mm in diameter as positive and calculated tumor volume based on the formula: 0.5326 (length [mm] × width [mm]2).
2.3. Antibodies and Reagents
Polyclonal rabbit antibodies for epsins 1 and 2 were obtained as previously described [14, 17]; goat anti-epsin 1 and mouse anti-GAPDH were obtained from Santa Cruz. 4-hydroxytamoxifen was from Sigma.
2.4. Cell Culture
Human LNCaP cells (ATCC) were cultured at 37°C with 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
2.5. Western Blot Analyses
Human LNCaP cells were treated with either control GFP lentivirus or epsins 1 and 2 shRNA lentivirus for 2 days then incubated with fresh media for an additional 2 days. Cells were lysed with RIPA Buffer (1%Triton X-100/0.1%SDS/0.5% sodium deoxycholic acid/5 mM tetrasodium pyrophosphate/50 mM sodium fluoride/5 mM EDTA/150 mM NaCl/25 mM Tris, pH 7.5/5 mM Na3VO4/5 mM N-ethylmaleimide, and protease inhibitor cocktail) then the deletion of epsins 1 and 2 was analyzed by western blot using goat anti-epsin 1 and rabbit anti-epsin 2. Deletion of epsins 1 and 2 in normal prostate tissues and prostate tissue with tumor was analyzed by western blotting homogenized tissues using rabbit anti-epsin 1 and rabbit anti-epsin 2 antibodies.
2.6. Tumor Implantation
To assess tumor growth, we subcutaneously implanted human prostate WT or epsins- 1- and 2-deficient LNCaP cells (3 × 106 cells/tumor) in twelve-week-old SCID mice. We estimated the time of tumor appearance and monitored the tumor growth in two groups of mice by measuring tumor size with digital calipers. We recognized tumors more than 2 mm in diameter as positive and calculated tumor volume based on the formula: 0.5326 (length [mm] × width [mm]2). SCID mice were obtained from The Jackson Laboratory, Maine, USA.
2.7. Immunohistochemistry and Immunofluorescence of Tissue Samples
Immunohistochemistry and immunofluorescence were performed as described with modifications [13, 22]. Human prostate frozen tissue samples with stage 2C tumors, and their corresponding surrounding nonneoplastic (normal) tissues, used for epsins 1 and 2 immunofluorescence studies were purchased as AccuMax array from ISU ABXIS. Images were collected and quantified using an Olympus Spinning Disc Confocal Microscope equipped with 20x objective and Slidebook 5.0 software.
2.8. Study Approval
All animal studies were performed in compliance with institutional guidelines and were approved by Institutional Animal Care and Use Committee (IACUC), Oklahoma Medical Research Foundation, Oklahoma City.
2.9. Statistical Analysis
Data were shown as mean ± SEM. Data was analyzed by the two-tailed student's t-test or ANOVA, where appropriate. The Wilcoxon signed-rank test was used to compare data that did not satisfy the student's t test or ANOVA. P value ≤0.05 was considered significant.
3. Results
3.1. Upregulated Epsins 1 and 2 in Human Prostate Carcinoma
Epsins are reportedly upregulated in several types of cancer [23, 25, 26]. To begin investigating a potential correlation between epsins and the development of prostate cancer, we measured the protein levels of epsins 1 and 2 in nonneoplastic (normal) human prostate and tumorigenic human prostate tissues via immunofluorescent staining. We found that epsins 1 and 2 were present at low levels in epithelial cells of acini of normal human prostate tissue (Figure 1(a)). Furthermore, epithelial epsins 1 and 2 were significantly upregulated in the human prostate tumor tissues (Figures 1(a) and 1(b)). These data demonstrate a positive correlation between protein levels of epsins and prostate tumorigenesis.
Figure 1.
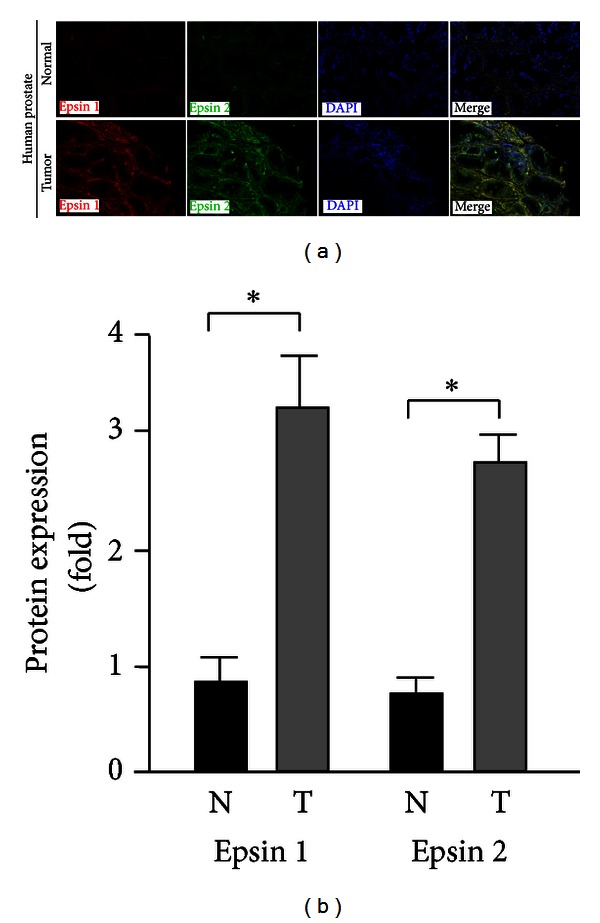
Expression of epsins 1 and 2 is increased in human prostate cancer tissues. Protein levels of epsins 1 and 2 were analyzed by immunostaining paired nonneoplastic (normal) and tumorigenic human prostate tissues. (a) Representative image from a single paired sample. (b) Quantification of fluorescence intensity from n = 6. *indicates P value <0.05.
3.2. Upregulated Epsins 1 and 2 Expression in Prostate Carcinoma of Spontaneous TRAMP Model
To investigate whether the increased proteins detected in human prostate cancer tissues were a result of increased epsins 1 and 2 gene expression, we employed the spontaneous Transgenic Adenocarcinoma of Mouse Prostate (TRAMP) mouse model [33–36]. We first confirmed, via western blotting and immunohistochemistry (IHC), that epsins 1 and/or 2 protein levels were upregulated in prostate tumors extracted from TRAMP mice (Figures 2(a)-2(b)). Then, we used quantitative real-time PCR (qRT-PCR) to investigate the gene expression of epsins 1 and 2. Consistent with increased protein detection, we found that the gene expression of epsins 1 and 2 increased by 3.5- or 2.5-fold, respectively, in extracted TRAMP prostate tumors, compared to normal prostate (Figure 2(c)). These data demonstrate that the expression of epsins 1 and 2 is upregulated in prostate tumors in vivo and further support a correlation between increased epsins 1 and 2 and prostate tumorigenesis.
Figure 2.
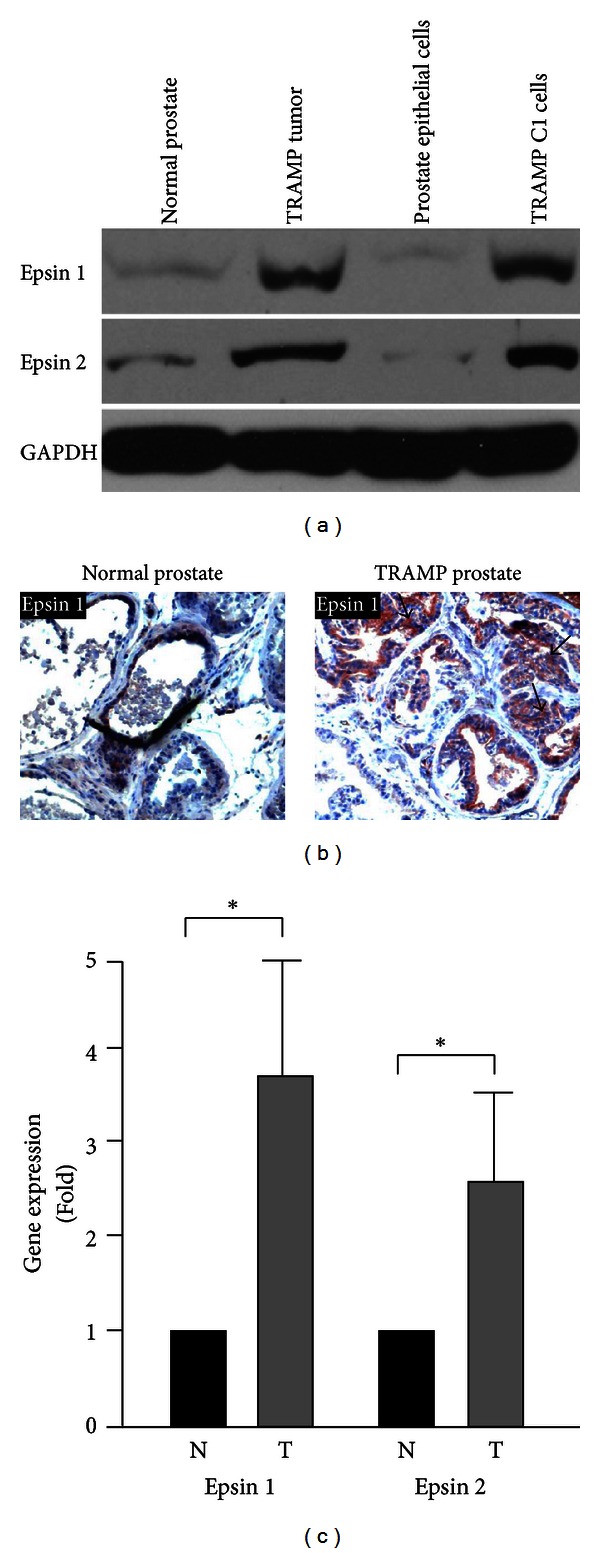
Expression of epsins 1 and 2 is increased in prostate tumor compared to normal prostate tissues in mice. (a) Protein levels of epsins 1 and 2 in the indicated normal mouse prostate or TRAMP prostate tumor tissues, as well as normal prostate epithelial cells and TRAMP C1 cells, were analyzed by western blotting. (b) IHC staining for epsin 1 in normal prostate and prostate tumor from TRAMP mice. Arrows indicate prostate cancer cells with upregulated epsin 1 protein expression. (c) qRT-PCR revealed increased expression of epsins 1 and 2 in normal (N) and prostate tumor (T) samples from TRAMP mice. n ≥ 5 per group in all panels. *indicates P value <0.05.
3.3. Epsins 1 and 2 Depletion Impairs Prostate Cancer Progression in Xenograft Model
Human prostate cancer LNCaP cells are a common model for prostate cancer progression because they share many characteristics of early prostate cancer progression including low tumorigenesis and androgen sensitivity [7, 32, 37, 38]. We transfected LNCaP cells with control GFP lentivirus or epsins 1 and 2 shRNA lentivirus and confirmed epsin depletion by western bot (Figure 3(a)). We subcutaneously inoculated control or epsin-depleted LNCaP cells into SCID mice to determine if epsin depletion would alter LNCaP tumorigenesis. LNCaP tumors were removed and measured from days 80 to 100 after-inoculation. We found that knockdown of epsins significantly impaired LNCaP tumor growth (Figures 3(b)–3(d)) and reduced tumor incidence (Figure 3(c)). These data suggest an oncogenic role for epsins in prostate tumorigenesis and cancer progression in a human xenograft model.
Figure 3.
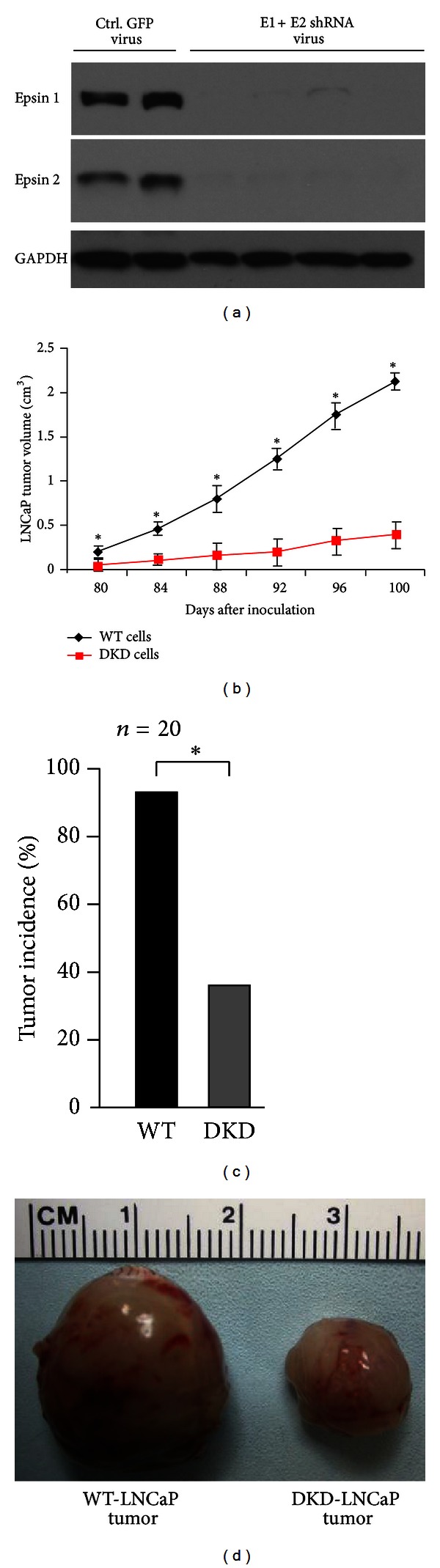
Epsins-1- and 2-deficient human prostate cells result in retarded tumor growth. (a) Lysates of LNCaP cells treated with either control GFP lentivirus or epsins-1- and 2-shRNA lentivirus were analyzed for epsins 1 and 2 depletion by western blotting. (b) WT or epsins-1- and 2-deficient LNCaP cells (DKD) were injected subcutaneously into SCID mice and LNCaP tumor size measured from days 80 to 100 afte-inoculation. (c) LNCaP tumor incidence. (d) Representative WT or epsins-1- and 2-deficient LNCaP tumors dissected out after 100 days of postinoculation. n = 5 in a, n = 20 in (b)–(d). *indicates P-value <0.05.
3.4. Epsins 1 and 2 Depletion Impairs Prostate Cancer Progression in Spontaneous TRAMP Model
To further investigate the role of epsins 1 and 2 in the development and progression of prostate cancer in a spontaneous cancer model, we generated an epsin-deficient TRAMP mouse model (Figure 4). First, we created mice with a conditional epsin 1 allele (Epn1fl/fl) (Figure 4(a)) then crossed them with epsin 2 null mice (Epn2−/−), thereby creating Epn1fl/fl, Epn2−/− mice [22]. To obtain epsins 1 and 2 inducible-double knockout mice (iDKO), the Epn1fl/fl, Epn2−/− mice were crossed with mice expressing tamoxifen-inducible Cre Recombinase (ERT2Cre) (Figure 4(b)). The Epn1fl/fl, Epn2−/−, ERT2Cre mice were further crossed with TRAMP mice to generate TRAMP, Epn1fl/fl, Epn2−/−, ERT2Cre mice. Ten-week old TRAMP, Epn1fl/fl, Epn2−/−, ERT2Cre mice, sex and genetic background matched to TRAMP-WT mice, were treated every day for two weeks with 5 mg of 4-hydroxytamoxifen per kg body weight via intraperitoneal injection (I.P.) to induce epsin 1 deletion (TRAMP-iDKO) (Figure 4(c)). To examine the effects of epsin deletion on spontaneous prostate tumor progression in the TRAMP background, we examined tumor development in the male urogenital tract of TRAMP-WT and TRAMP-iDKO mice. We found that similar to the LNCaP model, depletion of epsins impaired spontaneous prostate tumorigenesis (Figures 4(d)–4(g)). Furthermore, epsin deletion in TRAMP mice significantly improved prostate cancer survival rates (Figure 4(h)). In summary, our data demonstrates that epsins are upregulated in, and likely facilitate tumorigenesis of, the prostate. Furthermore, interfering with epsins may protect against the growth and progression of prostate cancer.
Figure 4.
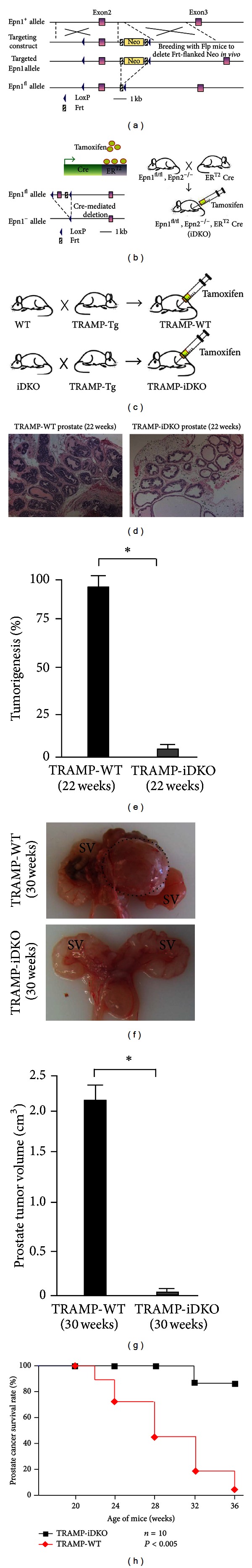
Loss of epsins 1 and 2 results in reduced tumor growth and increased survival rate in spontaneous TRAMP tumor model. (a) Generation of Epn1fl/fl. The diagram shows homologous recombination of the floxed gene-targeting vector at the Epsin 1 (Epn1) locus. Wild type Epn1 allele, top row; targeting construct, second row; targeted Epn1 allele, third row; and Epn1 floxed allele without Neo cassette (Epn1fl/fl), fourth row. (b) Strategy to generate tamoxifen inducible DKO (iDKO) by crossing Epn1fl/fl; Epn2−/− with ERT2Cre deleter mice. (c) Strategy to generate TRAMP-WT and TRAMP-iDKO mice by crossing TRAMP-tg mice with WT or Epn1fl/fl; and Epn2−/−; ERT2Cre (iDKO) mice. (d) H&E staining of prostate tissue from TRAMP-WT and TRAMP-iDKO mice dissected at 22 weeks. (e) Percentage of prostate tumorigenesis in TRAMP-WT and TRAMP-iDKO mice dissected at 22 weeks. (f) Male urogenital tract of TRAMP-WT and TRAMP-iDKO mice show tumor in prostate of TRAMP-WT but not in TRAMP-iDKO. Black dotted lines indicate tumors. Mice were sacrificed at 30 weeks. (g) Quantification of prostate tumor volume from TRAMP-WT and TRAMP-iDKO mice dissected at 30 weeks. (h) Percentage of male mice surviving in presence and absence of epsins 1 and 2 as a Kaplan-Meier plot. n = 20 in (d)–(g) and n = 10 in (h). *indicates P-value <0.05.
4. Discussion
Studies investigating the physiological and pathological roles of epsins have revealed their importance in many biological processes [16, 20, 22, 23, 26]. However, the full extent with which epsins are involved in the pathological development of diseases, such as cancer, or their tissue and cell type-specific contributions is still largely unknown. In this study, we investigated the potential role of epsins in the development and progression of prostate cancer. Our findings suggest that epsins act as oncogenic proteins to promote prostate tumor growth and progression. Furthermore, our results suggest that inhibition of epsins may provide a mechanism to impair the onset and progression of prostate cancer.
We chose to study the effects of epsin depletion in two common models of prostate cancer, subcutaneous xenograft of the LNCaP human prostate cancer cells and the genetic TRAMP mouse model. In both models we found that depletion of epsins impaired the growth of prostate tumors. In addition, using the TRAMP mouse model, which closely mimics the spontaneous growth and progression of prostate tumors in human prostate cancer [35, 36, 39], we were able to show that depletion of epsins significantly prolonged prostate cancer survival. Increased survival is likely a result of lower tumor incidence, impaired tumor growth, and reduced metastasis.
Interestingly, both human prostate tumors and TRAMP mouse prostate tumors exhibited elevated epsin proteins. Furthermore, using the TRAMP model, we determined that increased epsin proteins were due to significant increases in the gene expression of epsins 1 and 2 in tumor tissue. It is not clear from these results whether upregulated epsin expression is a cause or consequence of tumorigenesis. However, the fact that loss of epsins protected against tumor growth suggests the former, rather than the latter, is likely. The transcriptional regulation of epsins is not currently understood but this data suggests it may provide important information about what mediates early prostate cancer progression.
Lastly, epsins are characterized as endocytic adaptors that regulate the internalization of several ubiquitinated receptors [20]. Although not addressed herein, it is tempting to speculate that the overexpression of epsins may play an important role in upregulating signaling pathways dependent on receptor internalization. Alternatively, the multidomain nature of epsins may facilitate the stabilization of cell surface receptor complexes, thereby providing a mechanism to prolong signals that promote tumor growth. In summary, epsins are upregulated in human and mouse prostate carcinoma and this upregulation positively correlates with tumor growth. Loss of epsins impaired prostate tumor progression and promoted survival in the TRAMP mouse model. Our results suggest targeted inhibition of epsins may provide novel therapeutics to combat prostate cancer.
Authors' Contribution
K. L. Tessneer and S. Pasula contributed equally to this work.
Conflict of Interests
The authors have declared no conflict of interests.
Acknowledgments
The authors thank the Oklahoma Medical Research Foundation Imaging Core for help with the histological studies. This work was supported in part by NIH Grants R01HL-093242 P20 RR018758, a Grant from the Oklahoma Center for Advanced Science Technology (OCAST) HR09-116, and a Grant from Department of Defense W81XWH-11-1-00226 to H. Chen, and a Grant from Oklahoma Center for Advanced Science and Technology (OCAST) AR11-043 to Y. Dong.
References
- 1.Jung Y, Shiozawa Y, Wang J, et al. Prevalence of prostate cancer metastases after intravenous inoculation provides clues into the molecular basis of dormancy in the bone marrow microenvironment. Neoplasia. 2012;14(5):429–439. doi: 10.1596/neo.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y, Scher HI. Prostate cancer in 2011: hitting old targets better and identifying new targets. Nature Reviews Clinical Oncology. 2012;9(2):70–72. doi: 10.1038/nrclinonc.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer Journal for Clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 4.Khamis ZI, Iczkowski KA, Sang QXA. Metastasis suppressors in human benign prostate, intraepithelial neoplasia, and invasive cancer: their prospects as therapeutic agents. Medicinal Research Reviews. 2011;32(5):1026–1077. doi: 10.1002/med.20232. [DOI] [PubMed] [Google Scholar]
- 5.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature Genetics. 2003;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Li Y, Banerjee S, et al. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-κB signaling pathways. Journal of Cellular Biochemistry. 2010;109(4):726–736. doi: 10.1002/jcb.22451. [DOI] [PubMed] [Google Scholar]
- 7.Oosterhoff JK, Grootegoed JA, Blok LJ. Expression profiling of androgen-dependent and -independent LNCaP cells: EGF versus androgen signalling. Endocrine-Related Cancer. 2005;12(1):135–148. doi: 10.1677/erc.1.00897. [DOI] [PubMed] [Google Scholar]
- 8.Oosterhoff JK, Kühne LC, Grootegoed JA, Blok LJ. EGF signalling in prostate cancer cell lines is inhibited by a high expression level of the endocytosis protein REPS2. International Journal of Cancer. 2005;113(4):561–567. doi: 10.1002/ijc.20612. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Yu W, Cai Y, Ren C, Ittmann MM. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression. Neoplasia. 2008;10(8):847–856. doi: 10.1593/neo.08450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta S, Iljin K, Sara H, et al. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Research. 2010;70(17):6735–6745. doi: 10.1158/0008-5472.CAN-10-0244. [DOI] [PubMed] [Google Scholar]
- 11.Bache KG, Slagsvold T, Stenmark H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO Journal. 2004;23(14):2707–2712. doi: 10.1038/sj.emboj.7600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barriere H, Nemes C, Lechardeur D, Khan-Mohammad M, Fruh K, Lukacs GL. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in mammalian cells. Traffic. 2006;7(3):282–297. doi: 10.1111/j.1600-0854.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, De Camilli P. The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(8):2766–2771. doi: 10.1073/pnas.0409719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen H, Fre S, Slepnev VI, et al. Epsin is an EH-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 1998;394(6695):793–797. doi: 10.1038/29555. [DOI] [PubMed] [Google Scholar]
- 15.Ford MGJ, Mills IG, Peter BJ, et al. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419(6905):361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 16.Hawryluk MJ, Keyel PA, Mishra SK, Watkins SC, Heuser JE, Traub LM. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic. 2006;7(3):262–281. doi: 10.1111/j.1600-0854.2006.00383.x. [DOI] [PubMed] [Google Scholar]
- 17.Rosenthal JA, Chen H, Slepnev VI, et al. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. Journal of Biological Chemistry. 1999;274(48):33959–33965. doi: 10.1074/jbc.274.48.33959. [DOI] [PubMed] [Google Scholar]
- 18.Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and Vps27p/Hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nature Cell Biology. 2002;4(5):389–393. doi: 10.1038/ncb790. [DOI] [PubMed] [Google Scholar]
- 19.Sugiyama S, Kishida S, Chayama K, Koyama S, Kikuchi A. Ubiquitin-interacting motifs of epsin are involved in the regulation of insulin-dependent endocytosis. Journal of Biochemistry. 2005;137(3):355–364. doi: 10.1093/jb/mvi044. [DOI] [PubMed] [Google Scholar]
- 20.Wendland B. Epsins: adaptors in endocytosis? Nature Reviews Molecular Cell Biology. 2002;3(12):971–977. doi: 10.1038/nrm970. [DOI] [PubMed] [Google Scholar]
- 21.Ko G, Paradise S, Chen H, et al. Selective high-level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21511–21516. doi: 10.1073/pnas.1016390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Ko G, Zatti A, et al. Embryonic arrest at midgestation and disruption of Notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13838–13843. doi: 10.1073/pnas.0907008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasula S, Cai X, Dong Y, et al. Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. Journal of Clinical Investigation. 2012;122(12):4424–4438. doi: 10.1172/JCI64537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerbel RS. Tumor angiogenesis. New England Journal of Medicine. 2008;358(19):2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coon BG, Burgner J, Camonis JH, Aguilar RC. The Epsin family of endocytic adaptors promotes fibrosarcoma migration and invasion. Journal of Biological Chemistry. 2010;285(43):33073–33081. doi: 10.1074/jbc.M110.124123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coon BG, DiRenzo DM, Konieczny SF, Aguilar RC. Epsins’ novel role in cancer cell invasion. Communicative & Integrative Biology. 2011;4(1):95–97. doi: 10.4161/cib.4.1.14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee D, Coon BG, Edwards DF, et al. The yeast endocytic protein Epsin 2 functions in a cell-division signaling pathway. Journal of Cell Science. 2009;122(14):2453–2463. doi: 10.1242/jcs.041137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanos B, Rodriguez-Boulan E. The epithelial polarity program: machineries involved and their hijacking by cancer. Oncogene. 2008;27(55):6939–6957. doi: 10.1038/onc.2008.345. [DOI] [PubMed] [Google Scholar]
- 29.Rossé C, L’Hoste S, Offner N, Picard A, Camonis J. RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate Epsin during the switch off of endocytosis in mitosis. Journal of Biological Chemistry. 2003;278(33):30597–30604. doi: 10.1074/jbc.M302191200. [DOI] [PubMed] [Google Scholar]
- 30.Aguilar RC, Longhi SA, Shaw JD, et al. Epsin N-terminal homology domains perform an essential function regulating Cdc42 through binding Cdc42 GTPase-activating proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(11):4116–4121. doi: 10.1073/pnas.0510513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mollberg NM, Steinert G, Aigner M, et al. Overexpression of RalBP1 in colorectal cancer is an independent predictor of poor survival and early tumor relapse. Cancer Biology and Therapy. 2012;13(8):695–701. doi: 10.4161/cbt.20087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horoszewicz JS, Leong SS, Kawinski E. LNCaP model of human prostatic carcinoma. Cancer Research. 1983;43(4):1809–1818. [PubMed] [Google Scholar]
- 33.Greenberg NM, DeMayo F, Medina D, et al. Prostate cancer in a transgenic mouse. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(8):3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Research. 1997;57(21):4687–4691. [PubMed] [Google Scholar]
- 35.Gingrich JR, Barrios RJ, Morton RA, et al. Metastatic prostate cancer in a transgenic mouse. Cancer Research. 1996;56(18):4096–4102. [PubMed] [Google Scholar]
- 36.Gingrich JR, Greenberg NM. A transgenic mouse prostate cancer model. Toxicologic Pathology. 1996;24(4):502–504. doi: 10.1177/019262339602400414. [DOI] [PubMed] [Google Scholar]
- 37.Thalmann GN, Anezinis PE, Chang SM, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Research. 1994;54(10):2577–2581. [PubMed] [Google Scholar]
- 38.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LWK. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. International Journal of Cancer. 1994;57(3):406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 39.Gingrich JR, Barrios RJ, Foster BA, Greenberg NM. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer and Prostatic Diseases. 1999;2(2):70–75. doi: 10.1038/sj.pcan.4500296. [DOI] [PubMed] [Google Scholar]